Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity


1
Department of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117600, Singapore
2
Food Science and Technology Program, Department of Chemistry, National University of Singapore, Singapore 117600, Singapore
3
National University of Singapore (Suzhou) Research Institute, Suzhou 215123, China
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(2), 313; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020313
Submission received: 16 December 2018
/
Revised: 8 January 2019
/
Accepted: 10 January 2019
/
Published: 14 January 2019
(This article belongs to the Special Issue Therapeutic Aspects of Gasotransmitters in Cardiovascular and Renal Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Though historically known as a toxic gas, hydrogen sulfide (H2S) has displayed a new face as the third endogenous gaseous signaling molecule after nitric oxide (NO) and carbon monoxide (CO). Here in this review, we survey the role and therapeutic potential of H2S in cisplatin-induced nephrotoxicity. Specifically, reduction of H2S by cystathionine γ-lyase (CSE) downregulation upon cisplatin treatment may contribute to cisplatin-induced renal cell injury, possibly by augmentation of endogenous reactive oxygen species (ROS) production, while H2S donation may prevent subsequent renal dysfunction by inhibiting NADPH oxidase activation. Intriguingly, H2S slow-releasing compound GYY4137 seems to increase the anticancer activity of cisplatin, at least in several cancer cell lines, and this is probably due to its own anticancer effect. However, the efficacy of H2S donors in tumor-bearing animals remains to be tested in terms of renal protection and cancer inhibition after receiving cisplatin. Furthermore, accumulative evidence regarding usage of polysulfide, a novel H2S derived molecule, in the therapy of cisplatin-induced nephrotoxicity, was also summarized.
1. Introduction
Hydrogen sulfide (H2S) was historically known as a toxic gas with a rotten-egg smell [1]; however, extensive studies in the last two decades have unveiled its far-reaching effects in mammalian physiology and resulted in its recognition as the third endogenous gaseous signaling molecule after nitric oxide (NO) and carbon monoxide (CO) [2,3,4].
In the renal system, the abundance of H2S is clearly evidenced by the presence of all four biosynthesizing pathways, including cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), 3-mercaptopyruvate sulfurtransferase (3-MST) coupled with cysteine aminotransferase (CAT), and 3-MST coupled with d-amino acid oxidase (DAO). Not surprisingly, compelling data has suggested the modulatory effect of H2S in renal physiology and its protection in several kidney diseases [3,5,6]. Interestingly, emerging studies in recent years have indicated the possible involvement of H2S in cisplatin-induced nephrotoxicity [7,8]. In this review, we aim to survey the potential roles and protective mechanisms of H2S in cisplatin-induced nephrotoxicity. Additionally, progress in the use of polysulfide, an H2S derived endogenous molecule, for the disease is also reviewed.
2. Biosynthesis of H2S and Hydrogen Polysulfides in the Kidney
2.1. Biosynthesis of H2S in the Kidney
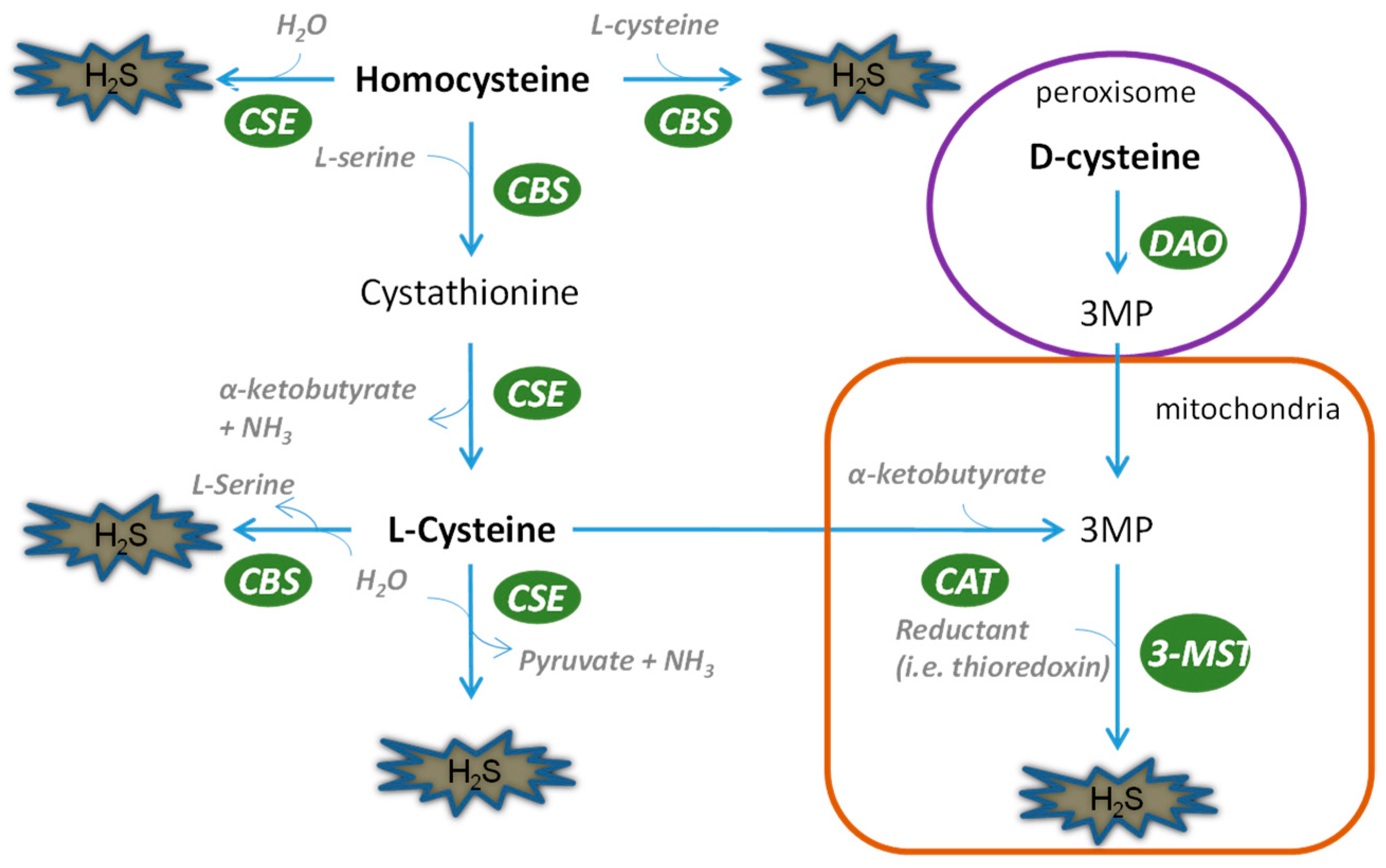
The kidney possesses all four of the H2S biosynthesis pathways (Figure 1), indicating the abundance of this gaseous molecule in the organ. Briefly, CSE leads to the generation of H2S by a two-step reaction, first dimerizing l-cysteine into thiocytsteine and then using it to react with other thiols to produce H2S. CBS can directly catalyze l-cysteine and homocystenin into H2S along with cystathinine. In contrast, 3-mercaptopyruvate has to be generated by CAT, after which 3-MST can use it as a substrate to produce pyruvate and H2S. Unlike CSE and CBS, 3-MST is mainly located in mitochondria and therefore regulates mitochondrial homeostasis of H2S. A fourth H2S generation pathway was recently discovered by Kimura’s group, in which d-cysteine serves as a substrate to produce H2S by d-amino acid oxidase coupled with 3-MST. For a detailed portrayal of H2S biosynthesis, it is advisable to refer to several excellent reviews published elsewhere [9,10,11]. It is worth mentioning that compared with its counterparts, the role of 3-MST is currently not well appreciated in either kidney physiology or disease in spite of its special role in producing H2S in mitochondria. This is an intriguing area warranting further exploration.
2.2. Biosynthesis of Hydrogen Polysulfide in the Kidney
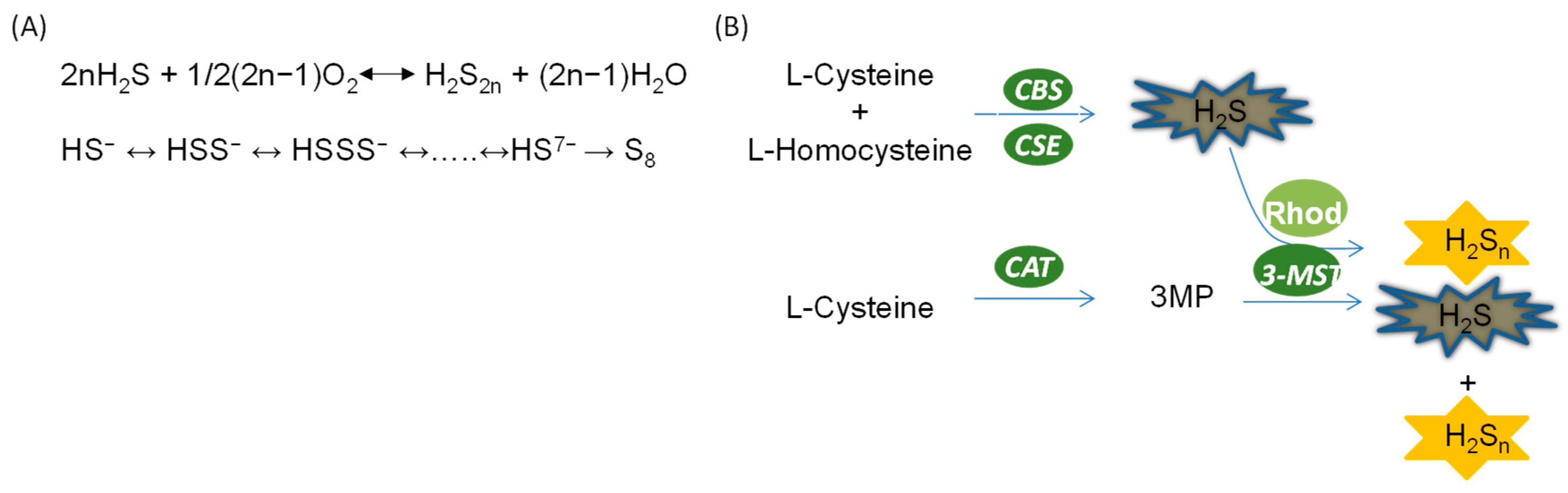
It is suggested that, similar to H2S, polysulfide is produced in mammals through both non-enzymatic pathways and enzymatic pathways (Figure 2). However, the proportion of endogenous polysulfide generated by these pathways remains elusive. Additionally, the regulation of polysulfide production is not well understood.
2.2.1. Non-Enzymatic Generation of Polysulfide
In the presence of oxygen, polysulfide is generated from H2S as described in the following equation: 2n H2S + 1/2 (2n−1) O2 = H2S2n + (2n−1) H2S [12]. Nevertheless, the reaction shown here with O2 is very slow unless it is catalyzed by a transition metal, and this may not occur much inside the cell. Moreover, HS− is also able to react with sulfur, S0, to generate polysulfide containing various numbers of sulfurs [13]. When the number of sulfurs reaches eight, the sulfur molecule cyclizes and becomes stable. Recently, another reaction was identified in which polysulfide serves as a product of the interaction between H2S and NO [14]. Importantly, these reactions should be partially responsible for the production of polysulfide in mammalian systems as they can occur in physiological conditions [14].
2.2.2. Enzymatic Generation of Polysulfide
Intriguingly, Kimura and others [15] found that hydrogen polysulfide, H2Sn, can also be produced by an enzymatic reaction catalyzed by H2S-producing enzyme 3-MST. In the study, it was found that 3MP promoted the production of H2Sn in brain cells of wide-type mice but not 3-MST-deficient mice, suggesting that H2Sn may be exclusively produced by 3-MST. Moreover, purified recombinant 3-MST also facilitates the production of H2Sn in the presence of 3MP. However, mutation of the cysteine residue at the catalytic site of 3-MST leads to failure in the production of H2S and H2Sn. Additionally, H2S can be used as a direct substrate for the production of H2Sn by 3-MST in the cooperation of rhodanese [15]. It is believed that polysulfide can also be generated by these pathways in other mammalian systems due to the universal presence of 3-MST [16].
3. Cisplatin-Induced Nephrotoxicity
Cisplatin is a simple compound, containing only a central platinum atom surrounded by two chlorine atoms and two ammonia groups in a cis configuration. Nevertheless, it is an extremely effective chemotherapeutic drug for cancer treatment. For example, it is widely used for the treatment of many solid tumors from areas such as head, neck, lung, testis, ovary, and breast [17]. However, its usage in cancer therapy is largely compromised due to its numerous adverse effects, which include nephrotoxicity, hearing loss, neurotoxicity, severe nausea, and myelosuppression [18]. Among these adverse effects, nephrotoxicity is the most prevalent; evidence shows that over 30% of patients show symptoms of acute kidney injury following the administration of cisplatin [19].
3.1. Clinical Features of Cisplatin-Induced Nephrotoxicity
Cisplatin-induced nephrotoxicity was initially described in clinical trials of cisplatin for cancer therapy [20], followed by similar observations in a number of species such as mice, rats, and dogs [21]. Typically, renal insufficiency manifests as increases in serum creatinine and blood urea nitrogen levels several days after the administration of cisplatin, along with a reduction of serum magnesium and potassium levels [22]. Meanwhile, urine output is normally preserved, and it usually contains glucose and small amounts of protein, indicating dysfunction of the proximal tubule [22]. Recovery of renal function usually occurs over a period of 2–4 weeks, provided cisplatin administration is discontinued; however, progressive and permanent nephrotoxicity may appear even with preventive measures [23,24].
3.2. Risk Factors of Cisplatin-Induced Nephrotoxicity
Several risk factors associated with cisplatin-induced nephrotoxicity have been identified. Accumulative evidence suggests that cumulative and/or high doses of cisplatin increase the rate of nephrotoxicity [25,26]. Besides this, other factors, such as smoking, older age, female sex, and pre-existing renal dysfunction have also been found to be associated with increased incidence of nephrotoxicity [27,28]. However, whether pre-existence of chronic kidney disease influences the occurrence of cisplatin nephrotoxicity remains elusive, due to limited data reported. In contrast, diabetes was reported to lower the risk of cisplatin-induced nephrotoxicity in rats [29]; however, this effect was not observed in human clinical trials [30,31]. Moreover, certain mutations in the gene of organic cation transporter 2 (OCT2) may be associated with a lower risk of nephrotoxicity, probably because OCT2 regulates the transportation of platinum into kidney cells [32,33].
3.3. Disease Pathophysiology of Cisplatin-Induced Nephrotoxicity
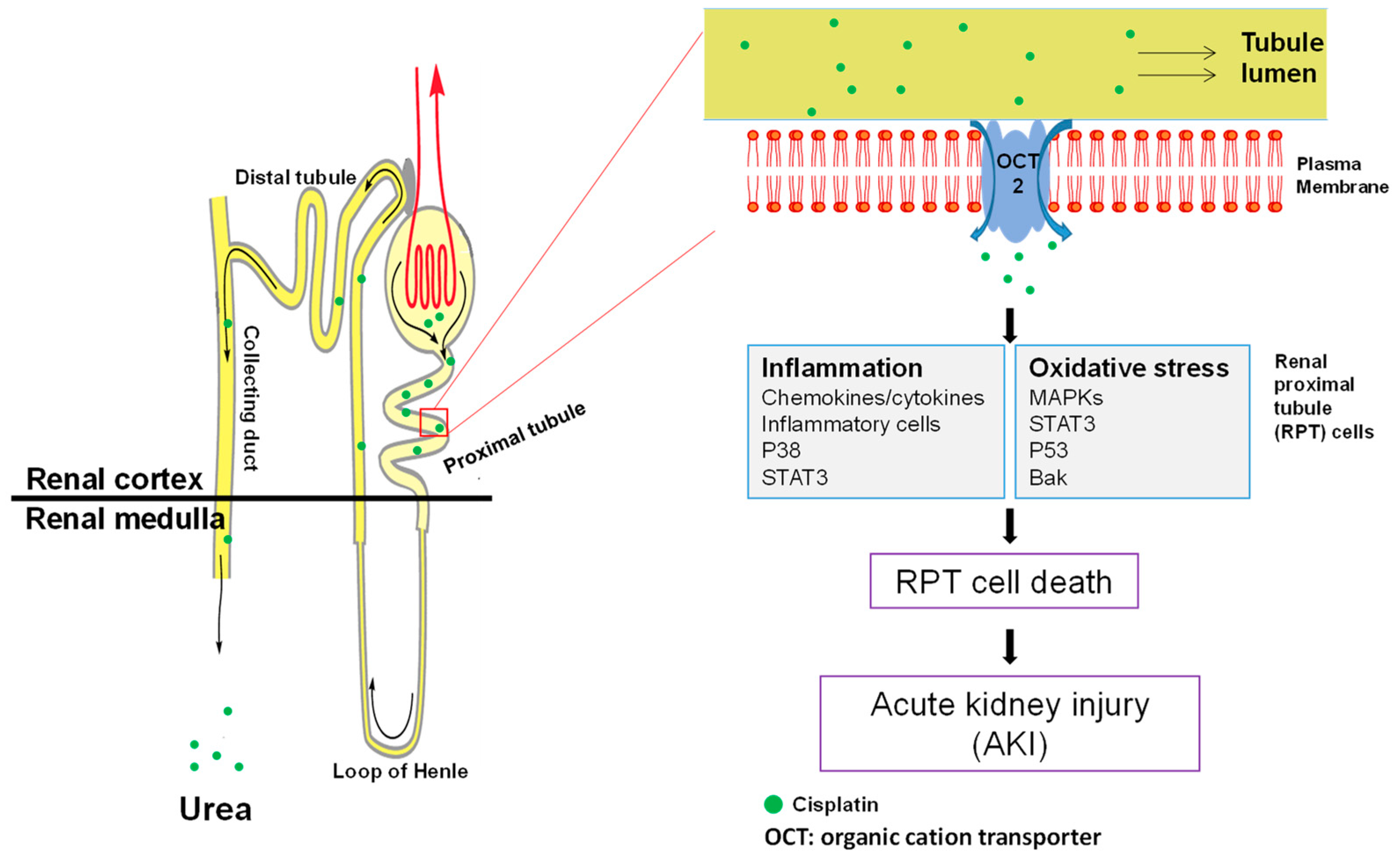
After decades of studies, the pathophysiology underlying cisplatin nephrotoxicity has gradually been elicited. In this section, the possible signaling and molecular mechanisms underlying the disease pathophysiology are summarized and illustrated in Figure 3.
3.3.1. Accumulation of Cisplatin in Kidney Cells
The clearance of cisplatin in mammals mainly occurs in the kidney by both glomerular filtration and tubular secretion [34]. In line with this, the concentration of cisplatin in the kidney largely exceeds its concentration in plasma, indicating an accumulation of the drug in renal cells [34]. This has been demonstrated with kidney slices and cultured renal proximal tubule segments [35,36]. In recent years, two different transporters, namely copper transporter 1 (Ctr1) and organic cation transporter 2 (OCT2), have been identified as responsible for the active transportation of cisplatin into mammalian cells. Ctr1 is highly expressed in both adult kidney and mammalian cells such as ovarian cancer cells [37]. Downregulation of Ctr1 in kidney cells attenuates cisplatin accumulation and subsequent toxicity [38], indicating that Ctr1 at least partially mediates the accumulation of cisplatin into kidney cells. However, whether Ctr1 plays a part in cisplatin-induced nephrotoxicity in vivo has not been studied. Unlike the universal expression of Ctr1, OCT2 is mainly located in renal proximal tubule cells [39]. Initially, it was found that cisplatin suppressed the transport of other OCT2 substrates into renal cells [39]. Conversely, an OCT2 substrate cimetidine decreases cisplatin uptake, and therefore cytotoxicity, in vitro [40], and also decreases nephrotoxicity in animals [33], suggesting a role of OCT2 in cisplatin-induced nephrotoxicity. This notion is strongly supported by a recent study which showed that OCT2 knockout mice exhibited higher resistance to cisplatin nephrotoxicity and lower urinary cisplatin excretion [32,33]. Consistently, certain mutations of the OCT2 gene are associated with reduced risk of cisplatin-induced nephrotoxicity in patients [32].
3.3.2. Cell Death in Cisplatin-Induced Nephrotoxicity: Types and Location
Cisplatin-induced nephrotoxicity is characterized by massive tubular cell death, consisting of both necrosis and apoptosis [41]. Recent studies have suggested that dosage of cisplatin may determine whether cells undergo necrosis or apoptosis [42]. For example, millimolar concentrations of cisplatin cause tubular cell necrosis in hours, while lower concentrations of cisplatin (micromolar) lead to apoptosis in cultured tubular cells [42]. Nevertheless, it is also likely that the necrosis may be a result of apoptosis, termed secondary necrosis. It has been well recognized that renal tubules are the major site of cell death; however, it is not until recently that the type of cells injured by cisplatin were identified. By using proximal and distal tubule-specific lectins, it was observed that most apoptotic cells could be stained with a proximal tubule-binding lectin, namely phytohemagglutinin, while only a very small proportion of apoptotic cells were stained by peanut lectin agglutinin, which specifically stains distal tubule [43]. This result indicates that cisplatin mainly causes proximal tubule cell death, which may lead to subsequent renal dysfunction.
3.3.3. Oxidative Stress in Cisplatin-Induced Nephrotoxicity
Oxidative stress has long been recognized as an important factor contributing to cisplatin nephrotoxicity [44]. Numerous studies have observed the massive production of reactive oxygen species (ROS) upon cisplatin treatment in cultured renal tubular cells, kidney slices, and in vivo animals [45,46]. Further studies have suggested three possible mechanisms that may account for the generation of ROS. First, cisplatin may cause mitochondrial ROS generation by suppressing the mitochondrial respiratory chain. For example, the activity of complex I-IV is reduced by 15–55% in 20 min after cisplatin treatment in tubule cells, which may result in ROS production [47]. However, one should bear in mind that ROS generation in this situation actually relies on residual electron flow through the mitochondrial respiratory chain; therefore, inhibition of the respiratory chain may block ROS accumulation [47]. Second, cisplatin may result in ROS production by activating NADPH oxidase. In agreement with this, pharmacological inhibition of NADPH oxidase protects renal cells in cultured proximal tubule cells and in vivo animals [48,49,50]. Finally, cisplatin may lead to ROS formation in microsomes via the cytochrome P450 (CYP) system. This is supported by evidence that knockout of CYP2E1 not only attenuates ROS accumulation but also alleviates cisplatin-induced renal injury [51]. In line with these findings, numerous antioxidants have been extensively studied in cisplatin nephrotoxicity and some of them are undergoing clinical trials [52].
3.3.4. MAPK Activation in Cisplatin-Induced Nephrotoxicity
The MAPK (mitogen-activated protein kinase)-signaling pathways consist of the ERK, p38, and JNK pathways. The activation of MAPKs plays critical roles in the regulation of proliferation, differentiation, and apoptosis [53]. Emerging evidence suggests the activation of MAPKs in various experimental models of cisplatin-induced nephrotoxicity. For example, Nowak et al. first described that the activation of ERK led to its accumulation in mitochondria upon cisplatin treatment in primary renal tubule cells [54]. Pharmacological inhibition of ERK by PD98059 or U0126 largely ameliorates cisplatin-induced apoptosis [55]. In line with this, overexpression of constitutively active MEK1 leads to enhanced apoptosis, while dominant negative MEK1 decreases cisplatin-induced apoptosis in renal tubular cells. Importantly, ERK inhibitor U0126 attenuates in vivo cisplatin-induced nephrotoxicity [56], suggesting its involvement in cisplatin-induced renal toxicity. Similarly, p38 and JNK were also reported to be activated upon cisplatin treatment in animals [55]. Inhibition of either of the two kinases was reported to be protective in cisplatin-induced nephrotoxicity [57,58]. Besides, p38 may also take part in the regulation of TNFα expression and subsequent inflammatory response during cisplatin nephrotoxicity [57,58].
3.3.5. Inflammation in Cisplatin-Induced Nephrotoxicity
Cisplatin-induced nephrotoxicity is associated with apparent inflammatory responses [59]. Massive expression of cytokines such as TNFα, IL-2, and MCP-1 has been reported in cisplatin-treated kidneys [60]. Deng et al. first showed that IL-10 can attenuate cisplatin-induced renal tissue injury and tubular apoptosis, suggesting that inflammatory response contributes to cisplatin nephrotoxicity [60]. Recent studies suggest that TNFα appears to be the key regulator of inflammatory response induced by cisplatin [60,61,62]. For example, TNFR2-deficient mice show higher resistance to cisplatin-induced nephrotoxicity compared with controls [61]. Moreover, Reeves and others showed that TNFα is crucial for the recruitment of inflammatory cells and upregulation of other proinflammatory factors because blockage of TNFα largely diminishes the effects mentioned above [62]. Recently, the initial production of TNFα during cisplatin nephrotoxicity was ascribed to proximal tubular cells instead of infiltrated inflammatory cells [63]. This further demonstrates the pivotal role of renal RPT cell in the pathogenesis of cisplatin-induced nephrotoxicity. Importantly, Faubel and colleagues showed that IL-1β, IL-18, and IL-6 may not significantly contribute to cisplatin-induced renal toxicity [64]. However, the involvement of other cytokines remains to be determined.
3.4. Prevention of Cisplatin-Induced Nephrotoxicity
Volume expansion by hydration has shown some success in the prevention of cisplatin nephrotoxicity [65]. However, the use of diuretics such as mannitol or furosemide fails to provide any beneficial effects in the scenario of cisplatin-induced nephrotoxicity in spite of their inclusion in many hydration regimens [66]. In fact, one comparative study observed aggravated nephrotoxicity in patients who received mannitol plus saline in comparison to those who received saline alone [23]. As a result, a recently issued clinical guideline suggests volume expansion with 0.9% saline and avoidance of diuretics [67]. Another approach aiming to prevent cisplatin-induced nephrotoxicity is to synthesize and screen novel analogues of cisplatin with lower toxicity to normal cells. In this direction, carboplatin, an analogue of cisplatin, has been approved for clinical usage of multiple cancers [68]. Compared with cisplatin, carboplatin presents less risk of gastrointestinal, renal and neurologic toxicity [68]; however, carboplatin appears to be less potent in terms of therapeutic effectiveness, at least in germ cell tumors, bladder cancers, and head and neck cancers, according to a clinical study [69].
4. Protective Effect of Hydrogen Sulfide in Cisplatin-Induced Nephrotoxicity
4.1. Role of Endogenous H2S in Cisplatin-Induced Nephrotoxicity
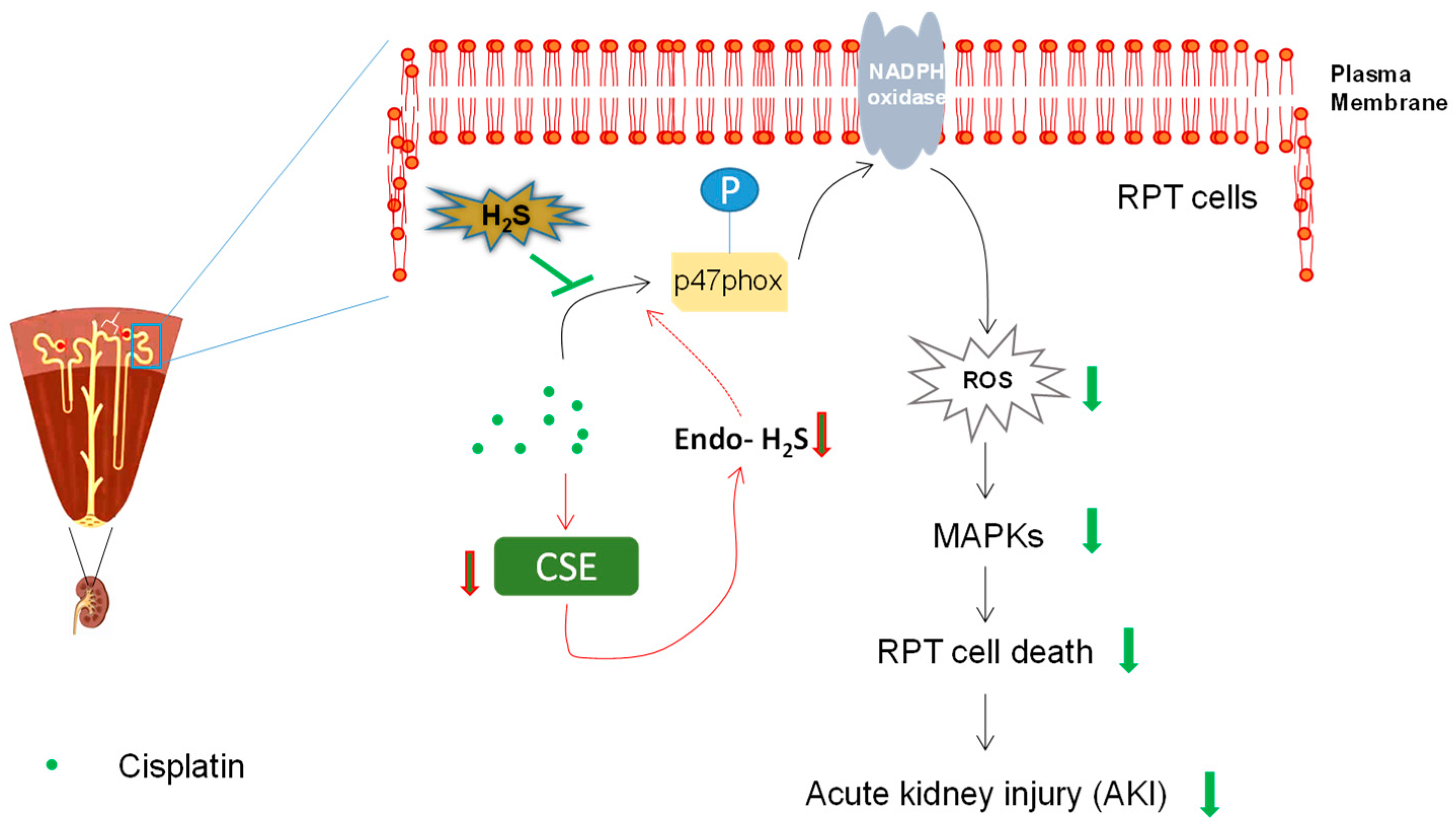
The production of H2S is precisely controlled in the kidney by an enzymatic system. Nevertheless, changed expression levels of these H2S-producing enzymes were often found in various pathological conditions, which leads to altered levels of endogenous H2S [9,70]. Intriguingly, the alteration contributes to the progression of diseases such as renal ischemic–reperfusion injuries and diabetic nephropathy [9,70], perhaps due to significant roles of H2S not only in renal physiology but also in the maintenance of cellular homeostasis [14,71]. Although previous evidence suggests the reduced mRNA levels of CSE and CBS [72], whether and how this reduction influences the pathogenesis remains largely unexplored. We recently demonstrated the reduction of endogenous H2S levels due to the reduced protein level of CSE, but not CBS, upon cisplatin treatment [73]. Interestingly, antioxidant-like N-acetylcysteine (NAC) can almost completely abolish cisplatin-mediated CSE downregulation, consistent with a previous study showing that oxidative stress results in reduction of CSE [3]. Furthermore, the data also found that production of endogenous H2S is protective against cisplatin-induced RPT cell death, by overexpression of CSE or addition of H2S-producing substrates in RPT cells [73]. These results suggest that reduction of endogenous H2S may contribute to cisplatin-induced RPT cell injury (Figure 4). Nevertheless, further studies, such as examination of whether CSE deficiency worsens cisplatin-induced nephrotoxicity in animals, may be required to consolidate this conclusion. Moreover, how the reduction of endogenous H2S influences the disease progression may also need exploration in the future.
4.2. Donation of H2S Protects Against Cisplatin-Induced Nephrotoxicity
Although H2S donation exhibits broad protective effects in multiple organs, the therapeutic value of the strategy is controversial in the context of cisplatin-induced nephrotoxicity. For example, a study from Ahangarpour et al. showed that an H2S donor, NaSH, mitigated cisplatin-induced renal dysfunction and damage in rats [7]. In contrast to this study, a recent study reported that H2S slow releaser GYY4137 aggravated cisplatin-induced renal damage by increasing inflammatory response [72], which is controversial to the well-known role of H2S donors as anti-inflammatory substances [74,75,76,77]. However, several defects in the use of GYY4137 can be clearly identified in the study. For example, GYY4137 was prepared and stored in DMSO. This has been shown to be detrimental and accelerate GYY4137 decomposition [78]. Therefore, it is possible that the decomposed product of GYY4137 aggravated cisplatin-induced renal damage when dissolved in DMSO. Additionally, the study used a rather low dose of GYY4137 (21 mg/kg). It is likely that H2S might not be adequately produced considering the releasing property of H2S by the compound [79,80,81,82,83].
To resolve this disparity, we recently used several distinct H2S donors, namely H2S acute releaser NaSH, H2S slow releaser GYY4137, and H2S mitochondrial releaser AP39, in parallel to examine the effect of H2S in cisplatin-induced nephrotoxicity [73]. Our data indicated a strong protective effect of NaSH and GYY4137 in alleviating the cellular injuries caused by cisplatin treatment with a porcine-derived RPT cell line LLC-PK1. However, we did not observe any cytoprotective effect of AP39 (0.1–1 µM) in the same cell model [73]. Considering the main effect of AP39 in sequestering mitochondrial ROS [84,85], this could be explained by the lack of mitochondrial ROS generation in our cell model [73]. Actually, Takahashi et al. recently observed no mitochondrial ROS production in primarily cultured RPT cells [86], reflecting our results, which showed that NADPH oxidase might be the main ROS producer upon cisplatin treatment in RPT cells. The efficacy of NaSH and GYY4237 was later investigated in cisplatin-treated mice. A relatively high, yet commonly used, dose of GYY4137 (100 mg/kg; prepared in PBS) was used in the animal study [81,83]. Our data showed that H2S donors significantly mitigated cisplatin-induced increase of blood creatinine and urea nitrogen level, indicating the amelioration of renal function [73]. Consistently, the apoptotic level of renal cortex in mice receiving H2S donors but not vehicle was reduced. Taken together, the study may suggest a novel pharmaceutical usage for H2S in cisplatin-induced nephrotoxicity (Figure 4).
4.3. H2S Exhibited Anti-Oxidant Effect
Due to the role of ROS in the pathogenesis of cisplatin-induced nephrotoxicity [52], we mainly examined the antioxidant effects mediated by H2S. The results indicate that H2S significantly suppressed cisplatin-induced ROS generation by inhibiting the activation of NADPH oxidase. In fact, emerging evidence suggests that NADPH oxidase is a potential target of H2S [9]. Mechanistically, it seems that NaSH and GYY4137 led to significant S-sulfhydration of p47phox, indicating a direct interaction between H2S and p47phox. Similarly, it was shown that S-nitrosylation, mediated by NO and nitroxyl (a product of NO and H2S interaction), of crucial thiol residues on p47phox inhibits the activity of NADPH oxidase [87,88]. These results may imply a novel mechanism underlying H2S-mediated suppression of NADPH oxidase activity by directly S-sulfhydrating p47phox, although further studies are absolutely required to establish a definitive causality between the two events and to identify the cysteine residues targeted by H2S. Due to its suppressive effect of H2S on NADPH oxidase activation, it blunted the downstream ROS generation and MAPKs activation. Besides, the data also indicate the involvement of nucleus translocation of Nrf2 in H2S-mediated antioxidant effect [73]. This is not surprising, as a previous study demonstrated that H2S-mediated S-sulfhydration of Keap1 at cysteine-151 triggers the release and nucleus translocation of Nrf2 [89]. All of these may contribute to the protective effect of H2S in cisplatin-induced renal toxicity (Figure 2). In line with these findings, antioxidants have been extensively studied in cisplatin nephrotoxicity and some of them, such as N-acetylcysteine and silymarin, are undergoing clinical trials [42].
4.4. H2S Exhibited Anti-Apoptotic Effect
The most prominent feature of cisplatin-treated kidney is the apoptosis of cortical tissues, particularly in the region of RPT cells [42], which is obviously attenuated upon the treatment with H2S donors such as NaHS and GYY4137 in mice [73]. Consistently, cell apoptosis of cultured RPT cells is also mitigated upon cisplatin treatment when H2S is supplemented [73], indicating a strong antiapoptotic effect of H2S in cisplatin-induced RPT cell injury. In our cell model, we found that cisplatin-mediated activation of MAPKs contribute to subsequent RPT apoptosis because pharmacological inhibitors of MAPKs, particularly ERK and JNK, lessened cell apoptosis, while pretreatment with H2S donors largely prevented the phosphorylation of MAPKs [73]. This suggests that H2S-mediated antiapoptotic effect can be at least partially ascribed to its suppressive effect on MAPKs activation. Additionally, other antiapoptotic effects mediated by H2S may include prevention of cytochrome C release [90], activation of ATP potassium channels [75], etc.
As H2S exhibits pleiotropic activity in biology, it is highly likely that more potential mechanisms may underlie its therapeutic effect on cisplatin-induced nephrotoxicity. For example, H2S may inhibit inflammatory responses [74,77], which were shown to contribute cisplatin-induced renal injuries [59], and H2S has also been demonstrated to activate cell survival pathways that may counteract cisplatin-induced cytotoxicity [90,91]. These potential mechanisms in the context of cisplatin-induced nephrotoxicity have been summarized by Dugbartey et al. in a recent review paper [92].
4.5. Can H2S Enhance the Anti-Cancer Effect of Cisplatin?
The best approach to treat cisplatin-induced nephrotoxicity will probably be the one that can prevent renal dysfunction while at the same time enhance (or at least, not compromise) the anticancer activity of cisplatin. Intriguingly, H2S has been demonstrated, in recent years, as a novel anticancer molecule [93]. Therefore, it is conceivable to ask how H2S donors could influence the anticancer effect of H2S. It was noticed that NaSH, at the concentration that alleviated cisplatin-induced RPT cell injuries, did not affect the anticancer activity of cisplatin in cancer cell lines like MCF7 and HepG2 [73]. Interestingly, GYY4137 can add more anticancer activity to cisplatin, which is probably due to its anticancer effect, shown previously [94,95]. These data indicate a promising potential of H2S donors, particularly GYY4137, in prevention of cisplatin-induced renal toxicity. Nevertheless, whether H2S donors would affect the antitumor effect of cisplatin in tumor-bearing animals remains unknown.
5. Therapeutic Potential of Polysulfide in Cisplatin-Induced Nephrotoxicity
Polysulfides are a category of chemical compounds comprising chains of sulfur atoms. There are two main classes of polysulfide reported, namely anions and organic polysulfides. Anions have the general formula Sn2−, which is the chemical basis of hydrogen polysulfide (H2Sn) and sodium polysulfide (Na2Sn). Organic polysulfides, such as garlic-derived diallyl disulfides (DADS) and diallyl trisulfides (DATS), usually possess a formula of RSnR, where R is either an alkyl or aryl group. Since anions such as sodium polysulfide (Na2Sn) solely provide Sn2− in aqueous solutions, they should be able to more closely mimic endogenously produced polysulfide, and therefore are commonly used to explore the biological activity of polysulfide.
Similar to the effect of H2S, recent data showed that polysulfide also exhibited strong renal protective effects in cisplatin-induced nephrotoxicity by preventing renal dysfunction and apoptosis [96]. For example, it attenuates the membrane translocation of p47phox and therefore suppresses the activation of NADPH oxidase upon cisplatin treatment. Likewise, Akt activation was observed after addition of polysulfide, which results in the translocation of Nrf2 [96]. Remarkably, it was observed that that the number of sulfur atoms in polysulfide well reflects the efficacy of these molecules, not only in cell protection but also in cancer inhibition [96]. This may serve as a guide for further development of polysulfide donors for pharmaceutical usage. Nevertheless, the study only used the acute releaser of polysulfide, namely sodium polysulfide. Therefore, the development and study of more types of polysulfide donors might be warranted in the future.
Although evidence suggests that hydrogen polysulfide can be synthesized by H2S-producing enzyme 3-MST, the proportion of polysulfide derived from this pathway remains unclear. Moreover, the measurement of endogenous hydrogen polysulfide in biological samples is extremely difficult due to the abundance of other types of polysulfide such as organic polysulfide [15]. Therefore, the role of endogenous hydrogen polysulfide has not yet been explored. Nevertheless, the reduction of endogenous H2S implies that cisplatin may also reduce the level of endogenous hydrogen polysulfide as H2S at least partially serves as a source of hydrogen polysulfide in mammals [97,98]. This needs to be demonstrated in the future.
6. H2S and Polysulfide as A Remedy for Cisplatin-Mediated Toxicity in Other Organs?
Besides renal toxicity, administration of cisplatin often leads to other adverse effects such as ototoxicity, neutropenia, neurotoxicity, and liver toxicity. Interestingly, the disease mechanisms seem to be similar to that of nephrotoxicity (i.e., accumulation of cisplatin leads to excessive production of ROS and inflammatory responses which cause tissue damage). For example, recent studies demonstrated a critical role of NADPH oxidase in cisplatin-mediated neurotoxicity [99,100], which is very consistent with our findings in the renal system [73]. Thus, it is conceivable to speculate that H2S and polysulfide are likely to alleviate the toxicities caused by cisplatin in these organs. An examination of H2S and polysulfide effects on cisplatin-mediated antitumor activity and toxicities in different organs in the same tumor-bearing animals is definitely warranted, as it will provide us an answer as to whether H2S and polysulfide can serve as an effective combination therapy with cisplatin.
7. Future Perspectives and Conclusions
In spite many years’ effort, nephrotoxicity remains the main factor limiting the use of cisplatin in the treatment of cancer. Substances with anticancer activity probably serve as an ideal agent to treat cisplatin-induced nephrotoxicity as they may preserve and/or enhance the anticancer activity of cisplatin while limiting its toxic effects. Evidence is accumulated to suggest certain H2S donors are one such agent. However, there are key questions remaining to be addressed, such as (1) whether H2S donors can limit the renal toxicity but enhance the antitumor activity of cisplatin in tumor-bearing animals; (2) testing of drugs such as H2S donors, particularly those in clinical trials [93], as GYY4137 displays efficacy only at high concentration (in vitro: 400 µM and above; in vivo: 100 mg/kg); and (3) further mechanistic portrayal of H2S-mediated cytoprotective effects in RPT cells and inhibitory effects in cancer cells. The understanding of these issues will largely facilitate the translation of H2S into a novel therapy for cisplatin-induced nephrotoxicity.
Funding
This work was supported by Ministry of Education of Singapore Tier 2 Research grant (MOE2017-T2-2-029) and Jiangsu Nature Science Foundation, China (BK20181185).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| H2S | Hydrogen sulfide |
| NO | Nitric oxide |
| CO | Carbon monoxide |
| CSE | Cystathionine γ-lyase |
| CBS | Cystathionine β-synthase |
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| CAT | Cysteine aminotransferase |
| DAO | d-amino acid oxidase |
| OCT2 | Organic cation transporter 2 |
| Ctrl1 | Copper transporter 1 |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| NAC | N-Acetylcysteine |
| RPT | Renal proximal tubule |
| DADS | Diallyl disulfides |
| DATS | Diallyl trisulfides |
References
- Smith, R.P.; Gosselin, R.E. Hydrogen sulfide poisoning. J. Occup. Med. Off. Publ. Ind. Med. Assoc. 1979, 21, 93–97. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’sa crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhang, Y.; Yang, M.; Wang, S.; Jiang, Z.; Li, Z. Exogenous hydrogen sulfide prevents kidney damage following unilateral ureteral obstruction. Neurourol. Urodyn. 2014, 33, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Ren. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef] [PubMed]
- Ahangarpour, A.; Abdollahzade Fard, A.; Gharibnaseri, M.K.; Jalali, T.; Rashidi, I. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. Vet. Res. Forum Int. Q. J. 2014, 5, 121–127. [Google Scholar]
- Karimi, A.; Absalan, F.; Khorsandi, L.; Valizadeh, A.; Mansouri, E. Sodium hydrogen sulfide (NaHS) ameliorates alterations caused by cisplatin in filtration slit diaphragm and podocyte cytoskeletal in rat kidney. J. Nephropathol. 2017, 6, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Bian, J.S. The Role of Hydrogen Sulfide in Renal System. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef]
- Dugbartey, G.J. The smell of renal protection against chronic kidney disease: Hydrogen sulfide offers a potential stinky remedy. Pharmacol. Rep. 2018, 70, 196–205. [Google Scholar] [CrossRef]
- Kasinath, B.S.; Feliers, D.; Lee, H.J. Hydrogen sulfide as a regulatory factor in kidney health and disease. Biochem. Pharmacol. 2018, 149, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Winterbourn, C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Toyofuku, Y.; Koike, S.; Shibuya, N.; Nagahara, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci. Rep. 2015, 5, 14774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Ries, F.; Klastersky, J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am. J. Kidney Dis. 1986, 8, 368–379. [Google Scholar] [CrossRef]
- Hill, J.; Speer, R. Organo-platinum complexes as antitumor agents (review). Anticancer Res. 1981, 2, 173–186. [Google Scholar]
- Kociba, R.J.; Sleight, S. Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemother. Rep. Part 1 1971, 55, 1–8. [Google Scholar]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed]
- Santoso, J.T.; Lucci, J.A., III; Coleman, R.L.; Schafer, I.; Hannigan, E.V. Saline, mannitol, and furosemide hydration in acute cisplatin nephrotoxicity: A randomized trial. Cancer Chemother. Pharmacol. 2003, 52, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Nazneen, A.; Abid, M.; Razzaque, M. Cisplatin-associated nephrotoxicity and pathological events. In Cellular Stress Responses in Renal Diseases; Karger Publishers: Basel, Switzerland, 2005; Volume 148, pp. 107–121. [Google Scholar]
- Reece, P.A.; Stafford, I.; Russell, J.; Khan, M.; Gill, P. Creatinine clearance as a predictor of ultrafilterable platinum disposition in cancer patients treated with cisplatin: Relationship between peak ultrafilterable platinum plasma levels and nephrotoxicity. J. Clin. Oncol. 1987, 5, 304–309. [Google Scholar] [CrossRef]
- Madias, N.E.; Harrington, J.T. Platinum nephrotoxicity. Am. J. Med. 1978, 65, 307–314. [Google Scholar] [CrossRef]
- De Jongh, F.E.; Verweij, J.; Loos, W.J.; de Wit, R.; de Jonge, M.J.; Planting, A.S.; Nooter, K.; Stoter, G.; Sparreboom, A. Body-surface area–based dosing does not increase accuracy of predicting cisplatin exposure. J. Clin. Oncol. 2001, 19, 3733–3739. [Google Scholar] [CrossRef]
- De Jongh, F.; Van Veen, R.; Veltman, S.; de Wit, R.; Van der Burg, M.; Van den Bent, M.; Planting, A.; Graveland, W.; Stoter, G.; Verweij, J. Weekly high-dose cisplatin is a feasible treatment option: Analysis on prognostic factors for toxicity in 400 patients. Br. J. Cancer 2003, 88, 1199–1206. [Google Scholar] [CrossRef]
- Scott, L.A.; Madan, E.; Valentovic, M.A. Attenuation of cisplatin nephrotoxicity by streptozotocin-induced diabetes. Fundam. Appl. Toxicol. 1989, 12, 530–539. [Google Scholar] [CrossRef]
- Gogas, H.; Shapiro, F.; Aghajanian, C.; Fennelly, D.; Almadrones, L.; Hoskins, W.; Spriggs, D. The impact of diabetes mellitus on the toxicity of therapy for advanced ovarian cancer. Gynecol. Oncol. 1996, 61, 22–26. [Google Scholar] [CrossRef]
- Stewart, D.J.; Dulberg, C.S.; Mikhael, N.Z.; Redmond, M.D.; Montpetit, V.A.; Goel, R. Association of cisplatin nephrotoxicity with patient characteristics and cisplatin administration methods. Cancer Chemother. Pharmacol. 1997, 40, 293–308. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of Organic Cation Transporter 2 (OCT2) to Cisplatin-Induced Nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H. Organic cation transporter 2 mediates cisplatin-induced oto-and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Safirstein, R.; Miller, P.; Guttenplan, J.B. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984, 25, 753–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, R.J.; Ghazi, M.A.; Barfuss, D.W. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl-l-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother. Pharmacol. 2003, 51, 132–138. [Google Scholar] [PubMed]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol.-Ren. Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, T.; Riethmüller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. American journal of physiology. Ren. Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef]
- Lieberthal, W.; Triaca, V.; Levine, J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: Apoptosis vs. necrosis. Am. J. Physiol. 1996, 270, F700–F708. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baliga, R.; Ueda, N.; Walker, P.D.; Shah, S.V. Oxidant mechanisms in toxic acute renal failure. Drug Metab. Rev. 1999, 31, 971–997. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Safirstein, R.L. Cisplatin Nephrotoxicity, Seminars in Nephrology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 460–464. [Google Scholar]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Rashed, L.A.; Hashem, R.M.; Soliman, H.M. Oxytocin inhibits NADPH oxidase and P38 MAPK in cisplatin-induced nephrotoxicity. Biomed. Pharmacother. 2011, 65, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.; Molina-Jijon, E.; Medina-Campos, O.N.; Rodriguez-Munoz, R.; Reyes, J.L.; Barrera, D.; Pedraza-Chaverri, J. Superoxide anion production and expression of gp91(phox) and p47(phox) are increased in glomeruli and proximal tubules of cisplatin-treated rats. J. Biochem. Mol. Toxicol. 2015, 29, 149–156. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Pan, H.; Huang, W.; Wang, X.; Wen, H.; Shen, K.; Jin, B. Pharmacological inhibition of NADPH oxidase protects against cisplatin induced nephrotoxicity in mice by two step mechanism. Food Chem. Toxicol. 2015, 83, 251–260. [Google Scholar] [CrossRef]
- Liu, H.; Baliga, R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Megyesi, J.K.; Kaneto, H.; Price, P.M.; Safirstein, R.L. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. American journal of physiology. Ren. Physiol. 2004, 287, F543–F549. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, H.J.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, J.M. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J. Appl. Toxicol. 2005, 25, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Baba, A.; Matsuo, M.; Itoh, Y.; Oishi, R. Protective effect of cyclic AMP against cisplatin-induced nephrotoxicity. Free Radic. Biol. Med. 2006, 40, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. American journal of physiology. Ren. Physiol. 2005, 289, F166–F174. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int. 2004, 65, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Brian Reeves, W. Cisplatin increases TNF-alpha mRNA stability in kidney proximal tubule cells. Ren. Fail. 2006, 28, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, T.L.; Reed, E. Nephrotoxicity and hydration management for cisplatin, carboplatin, and ormaplatin. Gynecol. Oncol. 1993, 50, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lehane, D.; Winston, A.; Gray, R.; Daskal, Y. The effect of diuretic pre-treatment on clinical, morphological and ultrastructural cis-platinum induced nephrotoxicity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1393–1399. [Google Scholar] [CrossRef]
- Launay-Vacher, V.; Rey, J.B.; Isnard-Bagnis, C.; Deray, G.; Daouphars, M. Prevention of cisplatin nephrotoxicity: State of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother. Pharmacol. 2008, 61, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, J.; Plante, M.; Vergote, I.; du Bois, A.; Hirte, H.; Lacave, A.J.; Wagner, U.; Stahle, A.; Stuart, G.; Kimmig, R. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: An intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [Google Scholar] [CrossRef]
- Lokich, J.; Anderson, N. Carboplatin versus cisplatin in solid tumors: An analysis of the literature. Ann. Oncol. 1998, 9, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Jia, Z.; Sun, Y.; Zhang, A.; Yang, T. A H 2 S Donor GYY4137 Exacerbates Cisplatin-Induced Nephrotoxicity in Mice. Mediat. Inflamm. 2016, 2016, 8145785. [Google Scholar] [CrossRef]
- Cao, X.; Xiong, S.; Zhou, Y.; Wu, Z.; Ding, L.; Zhu, Y.; Wood, M.E.; Whiteman, M.; Moore, P.K.; Bian, J.S. Renal Protective Effect of Hydrogen Sulfide in Cisplatin-Induced Nephrotoxicity. Antioxid. Redox Signal. 2018, 29, 455–470. [Google Scholar] [CrossRef]
- Cao, X.; Bian, J.-S. The Signaling Interaction Systems of in NO Biology and H2S and Medicine. Gasotransmitters 2018, 12, 145. [Google Scholar]
- Cao, X.; Cao, L.; Ding, L.; Bian, J.S. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, Z.; Xiong, S.; Cao, L.; Sethi, G.; Bian, J.S. The role of hydrogen sulfide in cyclic nucleotide signaling. Biochem. Pharmacol. 2018, 149, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, Z.; Cao, X.; Ding, L.; Wen, Z.; Bian, J.S. HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-kappaB and p38 MAPK pathways. Pharmacol. Res. 2016, 111, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Perry, A.; Zhou, Z.; Bucci, M.; Papapetropoulos, A.; Cirino, G.; Wood, M.E. Phosphinodithioate and phosphoramidodithioate hydrogen sulfide donors. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide; Springer: Berlin, Germany, 2015; pp. 337–363. [Google Scholar]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, J.; Tang, C.S.; Geng, B. Effect of synthesized GYY4137, a slowly releasing hydrogen sulfide donor, on cell viability and distribution of hydrogen sulfide in mice. Health Sci. 2010, 42, 493–497. [Google Scholar]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [Green Version]
- Meng, G.; Zhu, J.; Xiao, Y.; Huang, Z.; Zhang, Y.; Tang, X.; Xie, L.; Chen, Y.; Shao, Y.; Ferro, A.; et al. Hydrogen Sulfide Donor GYY4137 Protects against Myocardial Fibrosis. Oxid. Med. Cell. Longev. 2015, 2015, 691070. [Google Scholar] [CrossRef]
- Lin, S.; Visram, F.; Liu, W.; Haig, A.; Jiang, J.; Mok, A.; Lian, D.; Wood, M.E.; Torregrossa, R.; Whiteman, M.; et al. GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J. Urol. 2016, 196, 1778–1787. [Google Scholar] [CrossRef]
- Ikeda, K.; Marutani, E.; Hirai, S.; Wood, M.E.; Whiteman, M.; Ichinose, F. Mitochondria-targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide Biol. Chem. 2015, 49, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H2 S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Maxwell, K.F.; Chrissobolis, S.; Bullen, M.L.; Ku, J.M.; Michael De Silva, T.; Selemidis, S.; Hooker, E.U.; Drummond, G.R.; Sobey, C.G.; et al. Nitroxyl (HNO) suppresses vascular Nox2 oxidase activity. Free Radic. Biol. Med. 2013, 60, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Dusting, G.J.; Peshavariya, H.; Kemp-Harper, B.K.; Drummond, G.R. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 2007, 75, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Ding, L.; Wu, Z.; Cao, X.; Zhang, Q.; Lin, L.; Bian, J.S. Hydrogen sulfide reduces RAGE toxicity through inhibition of its dimer formation. Free Radic. Biol. Med. 2017, 104, 262–271. [Google Scholar] [CrossRef]
- Luan, H.F.; Zhao, Z.B.; Zhao, Q.H.; Zhu, P.; Xiu, M.Y.; Ji, Y. Hydrogen sulfide postconditioning protects isolated rat hearts against ischemia and reperfusion injury mediated by the JAK2/STAT3 survival pathway. Braz. J. Med. Biol. Res. 2012, 45, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Dugbartey, G.J.; Bouma, H.R.; Lobb, I.; Sener, A. Hydrogen sulfide: A novel nephroprotectant against cisplatin-induced renal toxicity. Nitric Oxide 2016, 57, 15–20. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef]
- Kashfi, K. Anti-cancer activity of new designer hydrogen sulfide-donating hybrids. Antioxid. Redox Signal. 2014, 20, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Nie, X.; Xiong, S.; Cao, L.; Wu, Z.; Moore, P.K.; Bian, J.S. Renal protective effect of polysulfide in cisplatin-induced nephrotoxicity. Redox Biol. 2018, 15, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen Sulfide and Polysulfide Signaling. Antioxid. Redox Signal. 2017, 27, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Mukherjea, D.; Jajoo, S.; Kaur, T.; Sheehan, K.E.; Ramkumar, V.; Rybak, L.P. Transtympanic administration of short interfering (si) RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid. Redox Signal. 2010, 13, 589–598. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Kim, S.-J.; Oh, G.S.; Moon, H.-D.; Kwon, K.-B.; Park, C.; Park, B.H.; Lee, H.-K.; Chung, S.-Y. Roles of NADPH oxidases in cisplatin-induced reactive oxygen species generation and ototoxicity. J. Neurosci. 2010, 30, 3933–3946. [Google Scholar] [CrossRef]
Figure 1.
Biosynthesis of H2S in the kidney. The biological production of H2S in the kidney is mediated by four pathways. CSE and CBS can use l-cysteine and homocysteine as substrates to generate H2S in cytosol, while they can translocate into mitochondria in hypoxic states. l-cysteine and d-cysteine have to be catalyzed into 3MP before they can be utilized by 3-MST for the production of H2S. 3-MST mediates H2S production, mainly in mitochondria.
Figure 1.
Biosynthesis of H2S in the kidney. The biological production of H2S in the kidney is mediated by four pathways. CSE and CBS can use l-cysteine and homocysteine as substrates to generate H2S in cytosol, while they can translocate into mitochondria in hypoxic states. l-cysteine and d-cysteine have to be catalyzed into 3MP before they can be utilized by 3-MST for the production of H2S. 3-MST mediates H2S production, mainly in mitochondria.

Figure 2.
Generation of hydrogen polysulfides in the kidney. (A) Non-enzymatic production of hydrogen polysulfide; (B) enzymatic production of hydrogen polysulfide.
Figure 2.
Generation of hydrogen polysulfides in the kidney. (A) Non-enzymatic production of hydrogen polysulfide; (B) enzymatic production of hydrogen polysulfide.

Figure 3.
Disease pathophysiology of cisplatin nephrotoxicity. When passing through renal tubules, cisplatin is actively accumulated into RPT cells due to the abundance of OCT2 on the cell membrane of RPT cells. This leads to the massive production of intracellular ROS and inflammatory responses, both of which contribute to RPT cell death and subsequent acute kidney injury. RPT: renal proximal tubule; AKI: acute kidney injury; OCT: organic cation transporter.
Figure 3.
Disease pathophysiology of cisplatin nephrotoxicity. When passing through renal tubules, cisplatin is actively accumulated into RPT cells due to the abundance of OCT2 on the cell membrane of RPT cells. This leads to the massive production of intracellular ROS and inflammatory responses, both of which contribute to RPT cell death and subsequent acute kidney injury. RPT: renal proximal tubule; AKI: acute kidney injury; OCT: organic cation transporter.

Figure 4.
Protective effect of H2S in cisplatin-induced nephrotoxicity. Cisplatin led to the reduction of endogenous H2S by downregulating the expression of H2S-producing enzyme cystathionine γ-lyase (CSE), which may be involved in the subsequent renal proximal tubule (RPT) cell death and nephrotoxicity. Furthermore, H2S donors such as NaSH and GYY4137 ameliorated cisplatin-induced renal toxicity in vitro and in vivo, probably by suppression of NADPH oxidase activation, downstream reactive oxygen species (ROS) generation, and mitogen-activated protein kinases (MAPKs) activation.
Figure 4.
Protective effect of H2S in cisplatin-induced nephrotoxicity. Cisplatin led to the reduction of endogenous H2S by downregulating the expression of H2S-producing enzyme cystathionine γ-lyase (CSE), which may be involved in the subsequent renal proximal tubule (RPT) cell death and nephrotoxicity. Furthermore, H2S donors such as NaSH and GYY4137 ameliorated cisplatin-induced renal toxicity in vitro and in vivo, probably by suppression of NADPH oxidase activation, downstream reactive oxygen species (ROS) generation, and mitogen-activated protein kinases (MAPKs) activation.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cao, X.; Zhang, W.; Moore, P.K.; Bian, J. Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020313
AMA Style
Cao X, Zhang W, Moore PK, Bian J. Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity. International Journal of Molecular Sciences. 2019; 20(2):313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020313
Chicago/Turabian StyleCao, Xu, Wencan Zhang, Philip K. Moore, and Jinsong Bian. 2019. "Protective Smell of Hydrogen Sulfide and Polysulfide in Cisplatin-Induced Nephrotoxicity" International Journal of Molecular Sciences 20, no. 2: 313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020313
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.