Acute Exposure to Indoxyl Sulfate Impairs Endothelium-Dependent Vasorelaxation in Rat Aorta


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
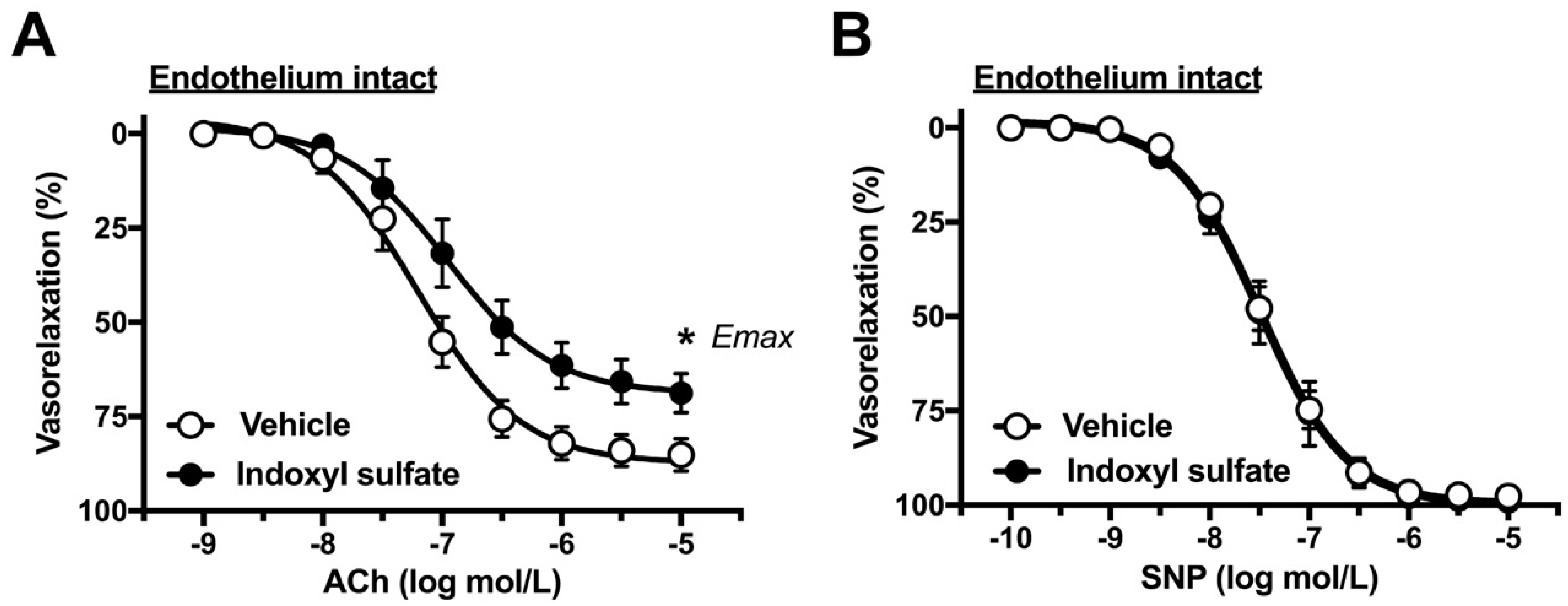
2.1. Effects of Indoxyl Sulfate on Vasorelaxations Induced by ACh and SNP
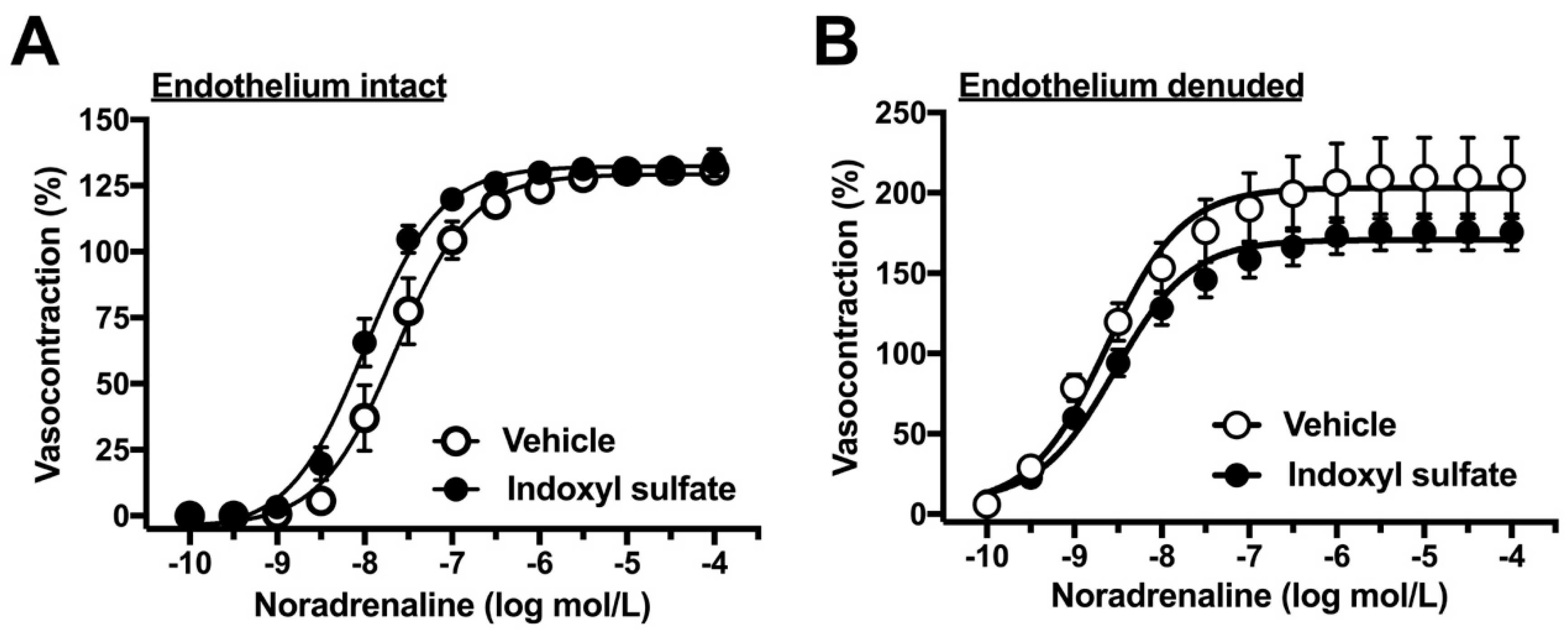
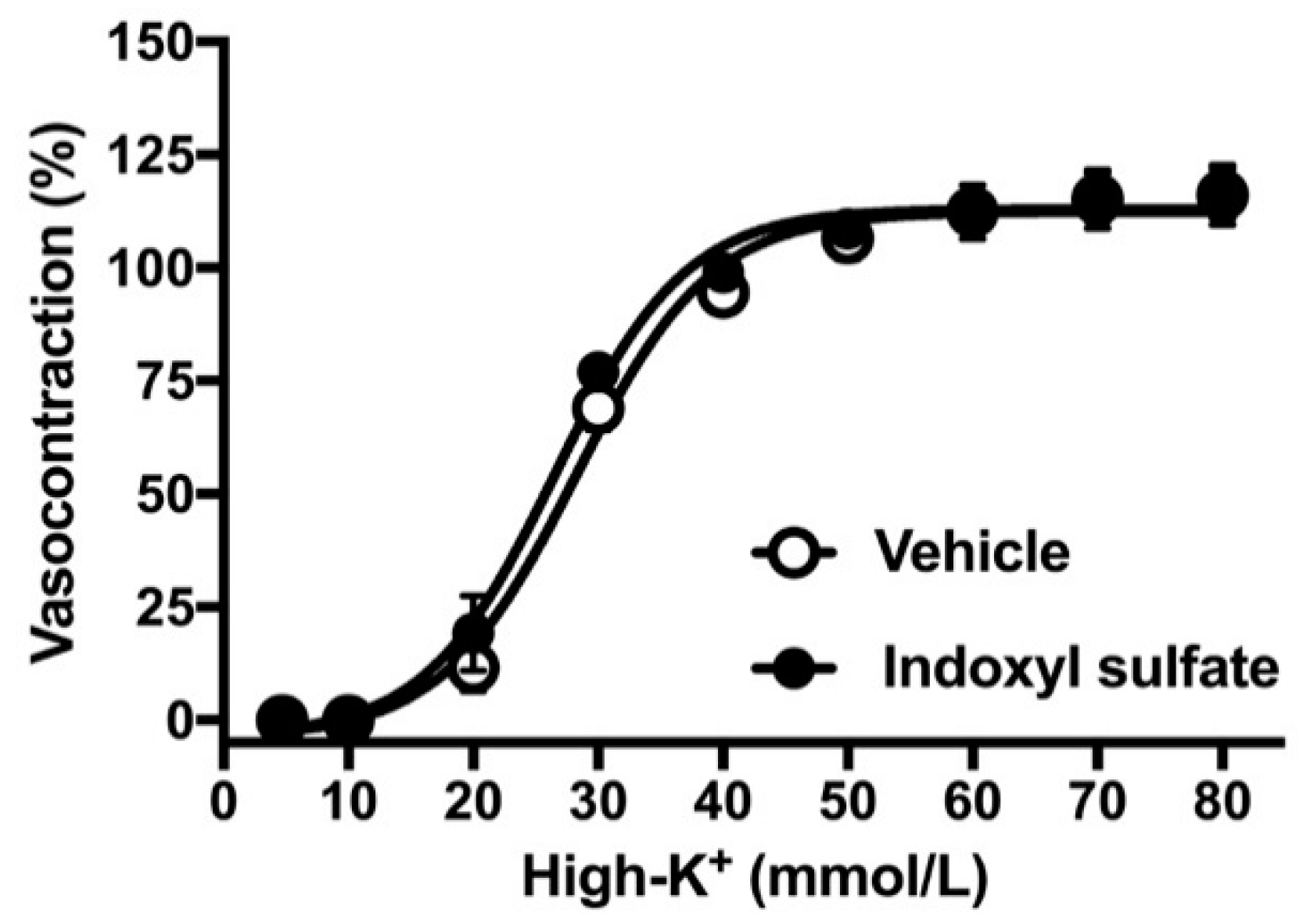
2.2. Effects of Indoxyl Sulfate on Vasocontraction Induced by Noradrenaline and Isotonic High-K+
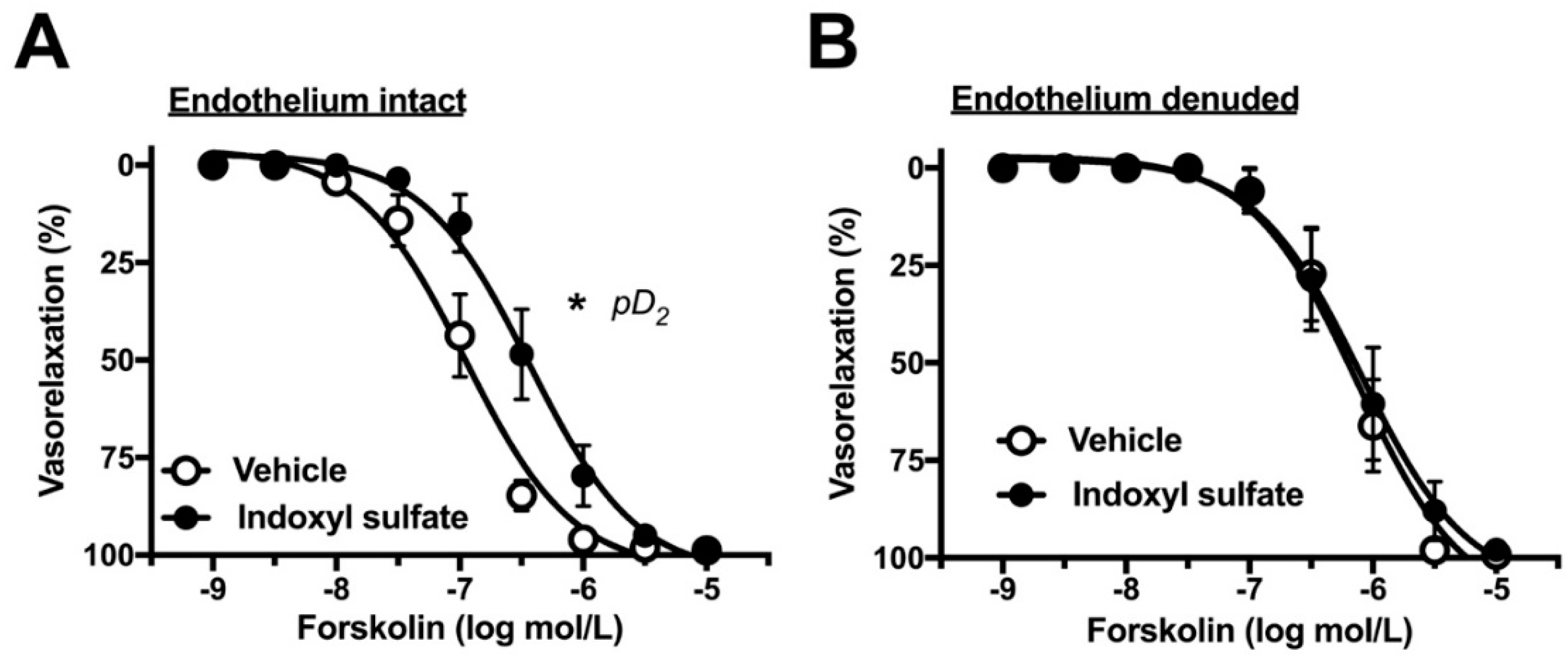
2.3. Effects of Indoxyl Sulfate on Adenylyl Cyclase Activator-Induced Vasorelaxation
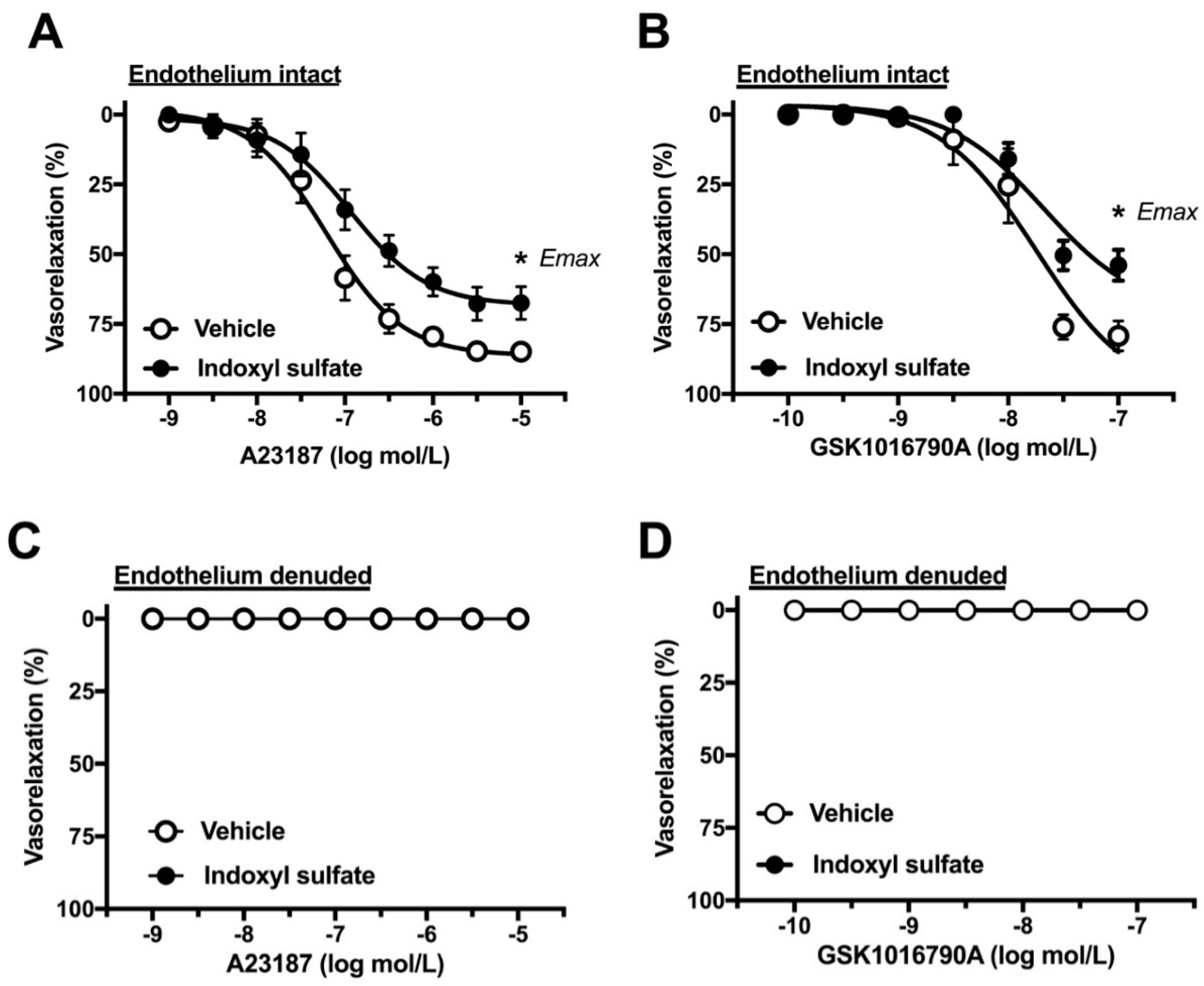
2.4. Effect of Indoxyl Sulfate on Calcium Ionophore- or TRPV4 Agonist-Induced Vasorelaxation
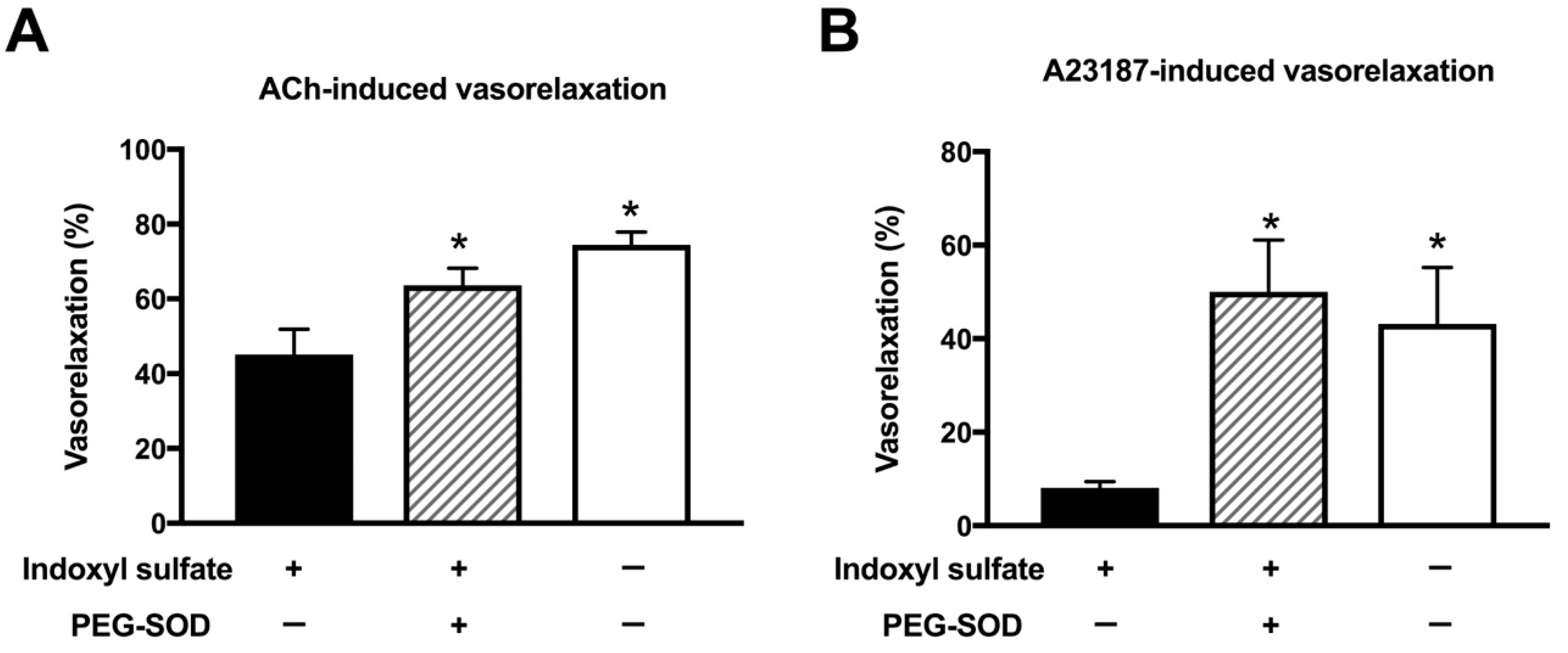
2.5. Effect of Cell-Permeant SOD on ACh- or A23187-Induced Vasorelaxation
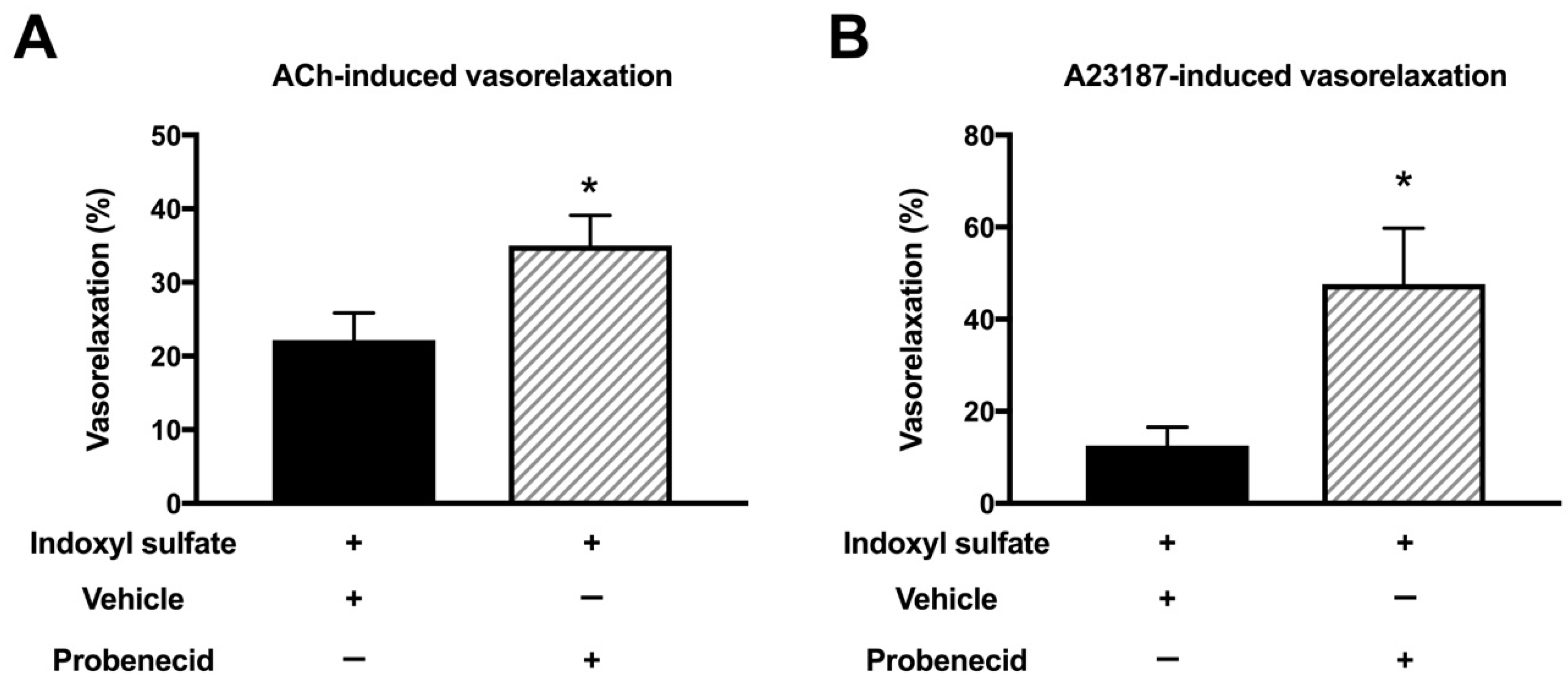
2.6. Effect of Organic Anion Transporter Inhibitor on ACh- or A23187-Induced Vasorelaxation
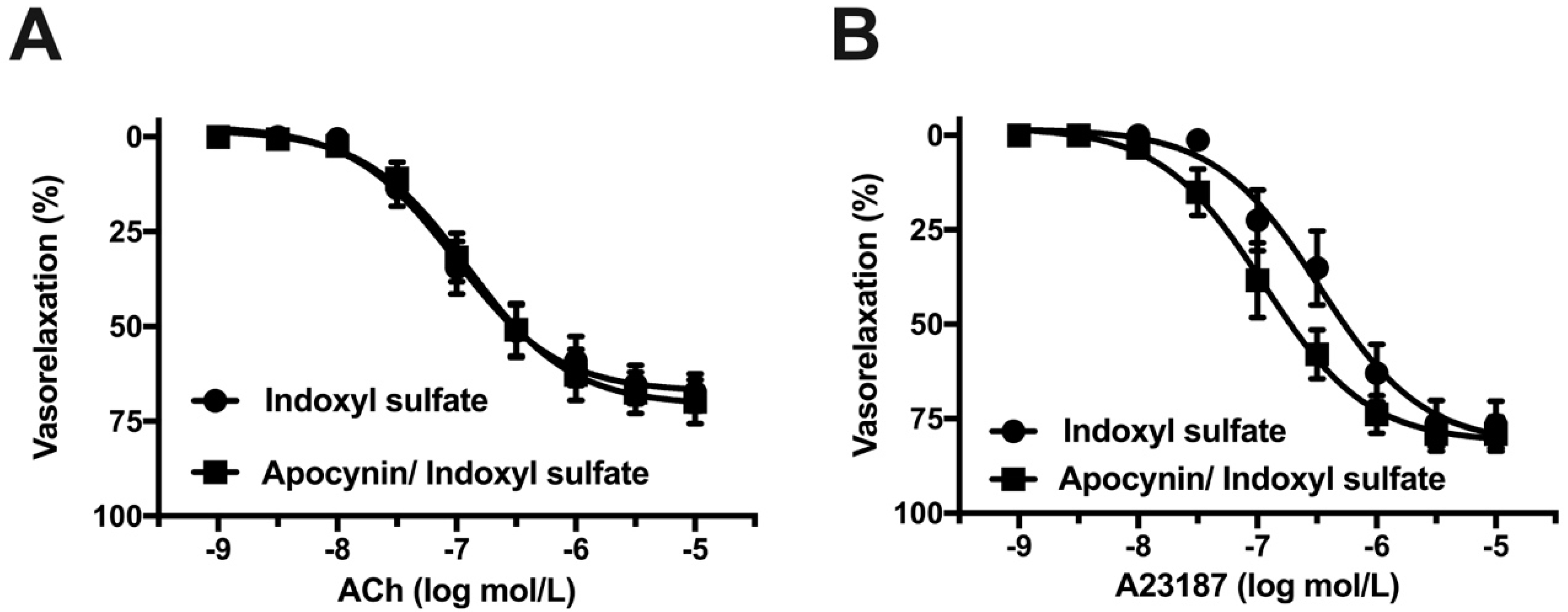
2.7. Effect of NADPH Oxidase Inhibitor on Vasorelaxation in Indoxyl Sulfate-Treated Aorta
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Vascular Function Study
4.3. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Karlsson, F.; Tremaroli, V.; Nielsen, J.; Backhed, F. Assessing the human gut microbiota in metabolic diseases. Diabetes 2013, 62, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Spor, A.; Felin, J.; Fak, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 4592–4598. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and alter weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut microbiota in cardiovascular health and disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Weir, T.L.; Gentile, C.L. The gut microbiota as a novel regulator of cardiovascular function and disease. J. Nutr. Biochem. 2018, 56, 1–15. [Google Scholar] [CrossRef]
- Cosola, C.; Rocchetti, M.T.; Cupisti, A.; Gesualdo, L. Microbiota metabolites: Pivotal players of cardiovascular damage in chronic kidney disease. Pharmacol. Res. 2018, 130, 132–142. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Vanholder, R.; Brunet, P. Protein-bound toxins—Update 2009. Semin. Dial. 2009, 22, 334–339. [Google Scholar] [CrossRef]
- Gao, H.; Liu, S. Role of uremic toxin indoxyl sulfate in the progression of cardiovascular disease. Life Sci. 2017, 185, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/β-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Adelibieke, Y.; Yisireyili, M.; Ng, H.Y.; Saito, S.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces IL-6 expression in vascular endothelial and smooth muscle cells through OAT3-mediated uptake and activation of AhR/NF-κB pathway. Nephron. Exp. Nephrol. 2014, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J. Artheroscler. Thromb. 2016, 60, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Bolati, W.; Lee, C.T.; Chien, Y.S.; Yisireyili, M.; Saito, S.; Pei, S.N.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates Mas receptor via aryl hydrocarbon receptor/nuclear factor-kappa B, and induces cell proliferation and tissue factor expression in vascular smooth muscle cells. Nephron 2016, 133, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yisireyili, M.; Saito, S.; Abudureyimu, S.; Adelibieke, Y.; Ng, H.Y.; Nishijima, F.; Takeshita, K.; Murohara, T.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS ONE 2014, 9, e109268. [Google Scholar] [CrossRef]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl hydrocarbon receptor mediates indoxyl sulfate-induced cellular senescence in human umbilical vein endothelial cells. J. Artheroscler. Thromb. 2014, 21, 904–916. [Google Scholar] [CrossRef]
- Chou, C.A.; Ng, H.Y.; Kuo, W.H.; Chiou, T.Y.; Pei, S.N.; Li, L.C.; Lee, Y.T.; Lee, C.T. Rosiglitazone attenuates indoxyl sulphate-induced endothelial dysfunction. Clin. Exp. Pharmacol. Physiol. 2015, 42, 287–292. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular incompetence in dialysis patients—Protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef]
- MacAllister, R.J.; Whitley, G.S.; Vallance, P. Effects of guanidine and uremic compounds on nitric oxide pathways. Kidney Int. 1994, 45, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Al-Zobaidy, M.J.; Craig, J.; Martin, W. Differential sensitivity of basal and acetylcholine-induced activity of nitric oxide to blockade by asymmetric dimethylarginine in the rat aorta. Br. J. Pharmacol. 2010, 160, 1476–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, S.R.; Sobey, C.G.; Piegors, D.J.; Bhopatkar, M.Y.; Faraci, F.M.; Malinow, M.R.; Heistad, D.D. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 24–29. [Google Scholar] [CrossRef]
- Emsley, A.M.; Jeremy, J.Y.; Gomes, G.N.; Angelini, G.D.; Plane, F. Investigation of the inhibitory effects of homocysteine and copper on nitric oxide-mediated relaxation of rat isolated aorta. Br. J. Pharmacol. 1999, 126, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Kredan, M.B.; Moat, S.J.; Hussain, S.A.; Powell, C.A.; Ballamy, M.F.; Powers, H.J.; Lewis, M.J. Homocysteine-induced inhibition of endothelium-dependent relaxation in rabbit aorta: Role for superoxide anions. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.M.; Wang, Y.; Ma, X.Z.; Wang, N.P.; Deng, X.L. Advanced glycation end products impair K(Ca)3.1- and K(Ca)2.3-mediated vasodilatation via oxidative stress in rat mesenteric arteries. Pflugers Arch. 2014, 466, 307–317. [Google Scholar] [CrossRef]
- Su, Y.; Mao, N.; Li, M.; Dong, X.; Lin, F.Z.; Xu, Y.; Li, Y.B. KB-R7943 restores endothelium-dependent relaxation induced by advanced glycosylation end products in rat aorta. J. Diabetes Complicat. 2013, 27, 6–10. [Google Scholar] [CrossRef]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef]
- Six, I.; Gross, P.; Remond, M.C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious vascular effects of indoxyl sulfate and reversal by oral adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Yu, M.; Lee, S.; Ryu, D.R.; Kim, S.J.; Kang, D.H.; Choi, K.B. AST-120 improves microvascular endothelial dysfunction in end-stage renal disease patients receiving hemodialysis. Yonsei Med. J. 2016, 57, 942–949. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta. Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Kobayashi, T. Changes in superoxide dismutase mRNA expression by streptozotocin-induced diabetes. Br. J. Pharmacol. 1996, 119, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faveretto, G.; Souza, L.M.; Gregorio, P.C.; Cunha, R.S.; Maciel, R.A.P.; Sassaki, G.L.; Toledo, M.G.; Pecoits-Filho, R.; Souza, W.M.; Stinghen, A.E.M. Role of organic anion transporters in the uptake of protein-bound uremic toxins by human endothelial cells and monocyte chemoattractant protein-1 expression. J. Vasc. Res. 2017, 54, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T. Indoxyl sulfate is a nephron-vascular toxin. J. Ren. Nutr. 2010, 20, S2–S6. [Google Scholar] [CrossRef]
- Lassegue, B.; San Martin, A.; Griedendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Virdis, A.; Gesi, M.; Taddei, S. Impact of apocynin on vascular disease in hypertension. Vascul. Pharmacol. 2016, 87, 1–5. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Drummond, G.R.; Sobey, C.G.; De Silva, T.M.; Kemp-Harper, B.K. The opposing roles of NO and oxidative stress in cardiovascular disease. Pharmacol. Res. 2017, 116, 57–69. [Google Scholar] [CrossRef]
- Lee, C.T.; Lee, Y.T.; Ng, H.Y.; Chiou, T.T.; Cheng, C.I.; Kuo, C.C.; Wu, C.H.; Chi, P.J.; Lee, W.C. Lack of modulatory effect of simvastatin on indoxyl sulfate-induced activation of cultured endothelial cells. Life Sci. 2012, 90, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hirose, Y.; Nishijima, F.; Tsubakihara, Y.; Miyazaki, H. ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am. J. Physiol. Cell Physiol. 2009, 297, C389–C396. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Nakayama, N.; Ishida, K.; Kobayashi, T.; Kamata, K. Eicosapentaenoic acid improves imbalance between vasodilator and vasoconstrictor actions of endothelium-derived factors in mesenteric arteries from rats at chronic stage of type 2 diabetes. J. Pharmacol. Exp. Ther. 2009, 329, 324–334. [Google Scholar] [CrossRef]
- Ando, M.; Matsumoto, T.; Taguchi, K.; Kobayashi, T. Poly (I:C) impairs NO donor-induced relaxation by overexposure to NO via the NF-kappa B/iNOS pathway in rat superior mesenteric arteries. Free Radic. Biol. Med. 2017, 112, 553–566. [Google Scholar] [CrossRef]
- Garcia-Morales, V.; Cuinas, A.; Elies, J.; Campos-Toimil, M. PKA and Epac activation mediates cAMP-induced vasorelaxation by increasing endothelial NO production. Vascul. Pharmacol. 2014, 60, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.; Evans, M.C.; Miner, A.S.; Berg, K.M.; Ward, K.R.; Ratz, P.H. Convergence of Ca2+-desensitizing mechanisms activated by forskolin and phenylephrine pretreatment, but not 8-bromo-cGMP. Am. J. Physiol. Cell Physiol. 2006, 290, C1552–C1559. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Morales, V.; Luaces-Regueira, M.; Campos-Toimil, M. The cAMP effectors PKA and Epac activate endothelial NO synthase through PI3K/Akt pathway in human endothelial cells. Biochem. Pharmacol. 2017, 145, 94–101. [Google Scholar] [CrossRef]
- Namkoong, S.; Kim, C.K.; Cho, Y.L.; Kim, J.H.; Lee, H.; Ha, K.S.; Choe, J.; Kim, P.H.; Won, M.H.; Kwon, Y.G.; et al. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell Signal. 2009, 21, 906–915. [Google Scholar] [CrossRef]
- Kobayashi, T.; Matsumoto, T.; Kamata, K. Mechanisms underlying the chronic pravastatin treatment-induced improvement in the impaired endothelium-dependent aortic relaxation seen in streptozotocin-induced diabetic rats. Br. J. Pharmacol. 2000, 131, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Kobayashi, S.; Ando, M.; Watanabe, S.; Iguchi, M.; Taguchi, K.; Kobayashi, T. Impaired endothelium-derived hyperpolarization-type relaxation in superior mesenteric arteries isolated from female Otsuka Long-Evans Tokushima Fatty rats. Eur. J. Pharmacol. 2017, 807, 151–158. [Google Scholar] [CrossRef]
- Stinghen, A.E.; Chillon, J.M.; Massy, Z.A.; Boullier, A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bEnd.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Oral sorbent AST-120 increases renal NO synthesis in uremic rats. J. Ren. Nutr. 2008, 18, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Atoh, K.; Itoh, H.; Haneda, M. Serum indoxyl sulfate levels in patients with diabetic nephropathy: Relation to renal function. Diabetes Res. Clin. Pract. 2009, 83, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.A.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chung, F.M.; Tsai, I.T.; Yang, C.Y.; Hsu, C.C.; Lu, Y.C.; Wang, C.P.; et al. Increased levels of total P-Cresylsulphate and indoxyl sulphate are associated with coronary artery disease in patients with diabetic nephrophathy. Rev. Diabet. Stud. 2010, 7, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Tarng, D.C. Diet, gut microbiome and indoxyl sulfate in chronic kidney disease patients. Nephrology 2018, 23 (Suppl. 4), 16–20. [Google Scholar] [CrossRef]
- Guo, J.; Lu, L.; Huang, K.; Wang, I.; Huang, L.; Fu, Q.; Chen, A.; Chan, P.; Fan, H.; Liu, Z.M.; et al. Vasculopathy in the setting of cardiorenal syndrome: Roles of protein-bound uremic toxins. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1–H13. [Google Scholar] [CrossRef]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar]
- Niwa, T.; Miyazaki, T.; Tsukushi, S.; Maeda, K.; Tsubakihara, Y.; Owada, A.; Shiigai, T. Accumulation of indoxyl-beta-D-glucuronide in uremic serum: Suppression of its production by oral sorbent and efficient removal by hemodialysis. Nephron 1996, 74, 72–78. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl sulfate impairs endothelial progenitor cells and might contribute to vascular dysfunction in patients with chronic kidney disease. Kidney Blood Press Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Kobayashi, S.; Ando, M.; Iguchi, M.; Takayanagi, K.; Kojima, M.; Taguchi, K.; Kobayashi, T. Alteration of vascular responsiveness to uridine adenosine tetraphosphate in aortas isolated from male diabetic Otsuka Long-Evans Tokushima Fatty rats: The involvement of prostanoids. Int. J. Mol. Sci. 2017, 18, E2378. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Watanabe, S.; Kobayashi, S.; Ando, M.; Taguchi, K.; Kobayashi, T. Age-related reduction of contractile responses to urotensin II is seen in aortas from Wistar rats but not from type 2 diabetic Goto-Kakizaki rats. Rejuvenation Res. 2017, 20, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Matsumoto, T.; Ando, M.; Iguchi, M.; Watanabe, S.; Taguchi, K.; Kobayashi, T. UDP-induced relaxation is enhanced in aorta from female obese Otsuka Long-Evans Tokushima Fatty rats. Purinergic Signal. 2018, 14, 91–96. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, T.; Takayanagi, K.; Kojima, M.; Taguchi, K.; Kobayashi, T. Acute Exposure to Indoxyl Sulfate Impairs Endothelium-Dependent Vasorelaxation in Rat Aorta. Int. J. Mol. Sci. 2019, 20, 338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020338
Matsumoto T, Takayanagi K, Kojima M, Taguchi K, Kobayashi T. Acute Exposure to Indoxyl Sulfate Impairs Endothelium-Dependent Vasorelaxation in Rat Aorta. International Journal of Molecular Sciences. 2019; 20(2):338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020338
Chicago/Turabian StyleMatsumoto, Takayuki, Keisuke Takayanagi, Mihoka Kojima, Kumiko Taguchi, and Tsuneo Kobayashi. 2019. "Acute Exposure to Indoxyl Sulfate Impairs Endothelium-Dependent Vasorelaxation in Rat Aorta" International Journal of Molecular Sciences 20, no. 2: 338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020338