Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions


Abstract
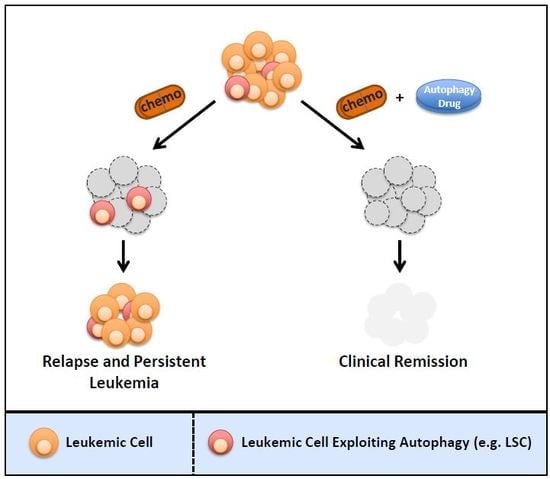
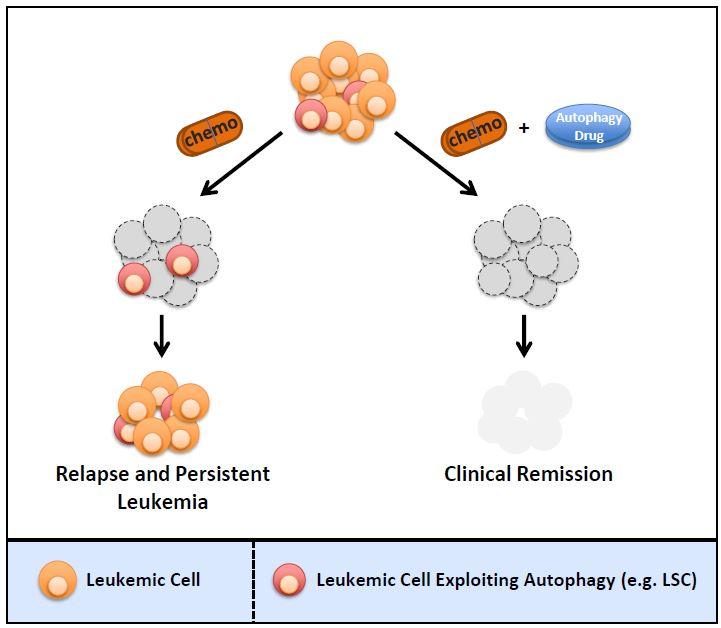
:
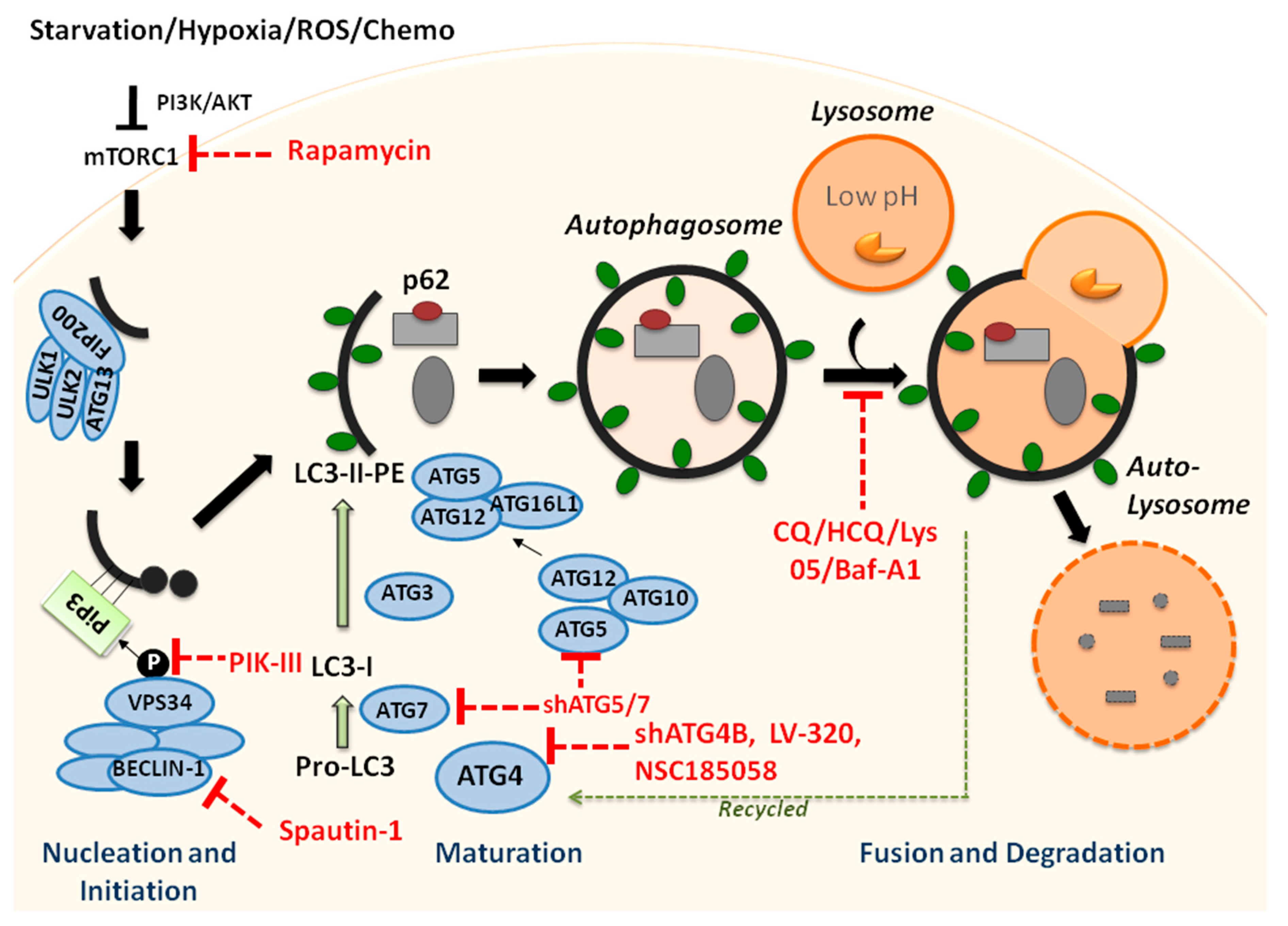
1. Macroautophagy Is a Cellular Recycling Process and Is Tightly Regulated
2. Autophagy Is Critical in the Maintenance of Hematopoietic Stem Cells
3. Autophagy Plays Context-Dependent Roles in Leukemia Initiation, Progression, and Drug Resistance
3.1. The Molecular and Functional Roles of Autophagy in CML
3.2. The Molecular and Functional Roles of Autophagy in AML
3.3. The Molecular and Functional Roles of Autophagy in Lymphocytic Leukemia
4. Targeting Autophagy Is Critical to Eradicate Drug-Resistant and Cancer Stem Cells in Human Leukemia
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. Fip200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegue, E. Foxo3a directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein atg4b is a potential biomarker and therapeutic target in cml stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2018. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mtorc1 association with the ulk1-atg13-fip200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. Ulk-atg13-fip200 complexes mediate mtor signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian atg14 and uvrag. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, M.; Zhang, H.; Wang, Z.; Yu, Y.; Xie, M.; Zhao, M.; Liu, L.; Cao, L. S100a8-targeting sirna enhances arsenic trioxide-induced myeloid leukemia cell death by down-regulating autophagy. Int. J. Mol. Med. 2012, 29, 65–72. [Google Scholar]
- Yang, M.; Zeng, P.; Kang, R.; Yu, Y.; Yang, L.; Tang, D.; Cao, L. S100a8 contributes to drug resistance by promoting autophagy in leukemia cells. PLoS ONE 2014, 9, e97242. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The atg8 and atg12 ubiquitin-like conjugation systems in macroautophagy. ’Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Kim, J.; Dalton, V.M.; Eggerton, K.P.; Scott, S.V.; Klionsky, D.J. Apg7p/cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell 1999, 10, 1337–1351. [Google Scholar] [CrossRef]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kda apg12-apg5.Apg16 multimeric complex, mediated by apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. Lc3, gabarap and gate16 localize to autophagosomal membrane depending on form-ii formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of apg8/aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Taherbhoy, A.M.; Tait, S.W.; Kaiser, S.E.; Williams, A.H.; Deng, A.; Nourse, A.; Hammel, M.; Kurinov, I.; Rock, C.O.; Green, D.R.; et al. Atg8 transfer from atg7 to atg3: A distinctive e1-e2 architecture and mechanism in the autophagy pathway. Mol. Cell 2011, 44, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational soluble n-ethylmaleimide-sensitive factor attachment protein receptor proteins vamp8 and vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of atg4b-lc3 complex reveals the mechanism of lc3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. Atg7 regulates energy metabolism, differentiation and survival of philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. 2009, 84, 431–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 (Suppl. 2), 1542–1552. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of torc1 by rag gtpases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The tor and camp-dependent protein kinase signaling pathways coordinately control autophagy in saccharomyces cerevisiae. Autophagy 2010, 6, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Lowenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Uyar, A.; Young, K.; Kuffler, L.; Waldron-Francis, K.; Marquez, E.; Ucar, D.; Trowbridge, J.J. Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat. Commun. 2016, 7, 12166. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Fialkow, P.J.; Jacobson, R.J.; Papayannopoulou, T. Chronic myelocytic leukemia: Clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am. J. Med. 1977, 63, 125–130. [Google Scholar] [CrossRef]
- Sloma, I.; Jiang, X.; Eaves, A.C.; Eaves, C.J. Insights into the stem cells of chronic myeloid leukemia. Leukemia 2010, 24, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in philadelphia chromosome-positive cells, including primary cml stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Britschgi, A.; Schlafli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low autophagy (atg) gene expression is associated with an immature aml blast cell phenotype and can be restored during aml differentiation therapy. Oxid. Med. Cell. Lonzgev. 2018, 2018, 1482795. [Google Scholar] [CrossRef] [PubMed]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Buhler, C.; Marcantonio, D.D.; Martinez, E.; Gollner, S.; Wickenhauser, C.; et al. Ret-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N., Jr.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am. J. Hematol. 2014, 89, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Saw, K.M.; Eaves, A.; Eaves, C. Instability of bcr-abl gene in primary and cultured chronic myeloid leukemia stem cells. J. Natl. Cancer Inst. 2007, 99, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.; Peng, C.; Chen, Y.; Li, D.; Li, S. Targeted therapy of chronic myeloid leukemia. Biochem. Pharmacol. 2010, 80, 584–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Erbilgin, Y.; Eskazan, A.E.; Hatirnaz Ng, O.; Salihoglu, A.; Elverdi, T.; Firtina, S.; Tasar, O.; Mercan, S.; Sisko, S.; Khodzhaev, K.; et al. Deep sequencing of bcr-abl1 kinase domain mutations in chronic myeloid leukemia patients with resistance to tyrosine kinase inhibitors. Leuk. Lymphoma 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Kim, H.J.; Kim, Y.K.; Kwak, J.Y.; Yhim, H.Y.; Kim, S.H.; Do, Y.R.; Oh, S.; Lee, S.E.; et al. Comparison of frequency and sensitivity of bcr-abl1 kinase domain mutations in asian and white patients with imatinib-resistant chronic-phase chronic myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, e391–e399. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of cml in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre stop imatinib (stim) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Bartley, P.A.; Slader, C.; Field, C.; Dang, P.; Filshie, R.J.; et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA pcr. Leukemia 2010, 24, 1719–1724. [Google Scholar] [CrossRef]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (bms-354825) targets an earlier progenitor population than imatinib in primary cml but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in cd34+ cml cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999, 94, 2056–2064. [Google Scholar]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to sti571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, Y.; Smith, C.; Gasparetto, M.; Turhan, A.; Eaves, A.; Eaves, C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to bcr-abl targeted therapies. Leukemia 2007, 21, 926–935. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of bcr-abl activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of cml stem and progenitor cells from tyrosine kinase inhibitors through n-cadherin and wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined stat3 and bcr-abl1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Elzinga, B.M.; O’Sullivan, G.C.; McKenna, S.L. Autophagy induction by bcr-abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment. Am. J. Hematol. 2011, 86, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Rothe, K.; Yen, R.; Fruhstorfer, C.; Maetzig, T.; Chen, M.; Forrest, D.L.; Humphries, R.K.; Jiang, X. A novel ahi-1-bcr-abl-dnm2 complex regulates leukemic properties of primitive cml cells through enhanced cellular endocytosis and ros-mediated autophagy. Leukemia 2017, 31, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microrna-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef]
- Drullion, C.; Tregoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.; Auberger, P.; Mahon, F.X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced k562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of ampk-mtor-ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. Bcr-abl suppresses autophagy through atf5-mediated regulation of mtor transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef]
- Vlahakis, A.; Powers, T. A role for tor complex 2 signaling in promoting autophagy. Autophagy 2014, 10, 2085–2086. [Google Scholar] [CrossRef]
- Carayol, N.; Vakana, E.; Sassano, A.; Kaur, S.; Goussetis, D.J.; Glaser, H.; Druker, B.J.; Donato, N.J.; Altman, J.K.; Barr, S.; et al. Critical roles for mtorc2- and rapamycin-insensitive mtorc1-complexes in growth and survival of bcr-abl-expressing leukemic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12469–12474. [Google Scholar] [CrossRef]
- Pengo, N.; Agrotis, A.; Prak, K.; Jones, J.; Ketteler, R. A reversible phospho-switch mediated by ulk1 regulates the activity of autophagy protease atg4b. Nat. Commun. 2017, 8, 294. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. Mst4 phosphorylation of atg4b regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef]
- Ianniciello, A.; Dumas, P.Y.; Drullion, C.; Guitart, A.; Villacreces, A.; Peytour, Y.; Chevaleyre, J.; de la Grange, P.B.; Vigon, I.; Desplat, V.; et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 2017, 8, 96984–96992. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the european leukemianet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 16010. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Basilico, S.; Gottgens, B. Dysregulation of haematopoietic stem cell regulatory programs in acute myeloid leukaemia. J. Mol. Med. 2017, 95, 719–727. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Vedi, A.; Santoro, A.; Dunant, C.F.; Dick, J.E.; Laurenti, E. Molecular landscapes of human hematopoietic stem cells in health and leukemia. Ann. N. Y. Acad. Sci. 2016, 1370, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Wunderlich, M.; Mulloy, J.C. Xenograft models for normal and malignant stem cells. Blood 2015, 125, 2630–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Ailles, L.E.; Gerhard, B.; Hogge, D.E. Detection and characterization of primitive malignant and normal progenitors in patients with acute myelogenous leukemia using long-term coculture with supportive feeder layers and cytokines. Blood 1997, 90, 2555–2564. [Google Scholar]
- Blair, A.; Hogge, D.E.; Ailles, L.E.; Lansdorp, P.M.; Sutherland, H.J. Lack of expression of thy-1 (cd90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 1997, 89, 3104–3112. [Google Scholar]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of lmpp-like and gmp-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.J.; Doondeea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically distinct leukemic stem cells in human cd34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J. Exp. Med. 2016, 213, 1513–1535. [Google Scholar] [CrossRef]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in nod/scid/il2rgammac-deficient mice. J. Clin. InvestIG. 2011, 121, 384–395. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Vargaftig, J.; Miraki-Moud, F.; Griessinger, E.; Sharrock, K.; Luke, T.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the cd34− fraction. Blood 2010, 115, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.P.; Pettirossi, V.; Thiede, C.; Bonifacio, E.; Mezzasoma, F.; Cecchini, D.; Pacini, R.; Tabarrini, A.; Ciurnelli, R.; Gionfriddo, I.; et al. Cd34+ cells from aml with mutated npm1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood 2010, 116, 3907–3922. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The mll recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Park, T.S.; Wan, T.S. Recurrent cytogenetic abnormalities in acute myeloid leukemia. Methods Mol. Biol. 2017, 1541, 223–245. [Google Scholar]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Yohe, S. Molecular genetic markers in acute myeloid leukemia. J. Clin. Med. 2015, 4, 460–478. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Yamaguchi, H.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; Kobayashi, Y.; et al. Prognostic impact of low allelic ratio flt3-itd and npm1 mutation in acute myeloid leukemia. Blood Adv. 2018, 2, 2744–2754. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Chavez-Valencia, V. Flt3-itd and its current role in acute myeloid leukaemia. Med. Oncol. 2017, 34, 114. [Google Scholar] [CrossRef]
- Al-Issa, K.; Nazha, A. Molecular landscape in acute myeloid leukemia: Where do we stand in 2016. Cancer Biol. Med. 2016, 13, 474–482. [Google Scholar]
- Ayatollahi, H.; Shajiei, A.; Sadeghian, M.H.; Sheikhi, M.; Yazdandoust, E.; Ghazanfarpour, M.; Shams, S.F.; Shakeri, S. Prognostic importance of c-kit mutations in core binding factor acute myeloid leukemia: A systematic review. Hematol./Oncol. Stem Cell Ther. 2017, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. Idh1/2 but not dnmt3a mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the acute leukemia french association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Neuberg, D.S.; Wander, S.A.; Sadrzadeh, H.; Ballen, K.K.; Amrein, P.C.; Attar, E.; Hobbs, G.S.; Chen, Y.B.; Perry, A.; et al. Isocitrate dehydrogenase 1 and 2 mutations, 2-hydroxyglutarate levels, and response to standard chemotherapy for patients with newly diagnosed acute myeloid leukemia. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2017, 31, 505–510. [Google Scholar] [CrossRef]
- Walter, M.J.; Payton, J.E.; Ries, R.E.; Shannon, W.D.; Deshmukh, H.; Zhao, Y.; Baty, J.; Heath, S.; Westervelt, P.; Watson, M.A.; et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc. Natl. Acad. Sci. USA 2009, 106, 12950–12955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic flt3-itd supports autophagy via atf4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an mll-af9-driven mouse model. Cell Death Dis. 2016, 7, e2361. [Google Scholar] [CrossRef]
- Porter, A.H.; Leveque-El Mouttie, L.; Vu, T.; Bruedigam, C.; Sutton, J.; Jacquelin, S.; Hill, G.R.; MacDonald, K.P.A.; Lane, S.W. Acute myeloid leukemia stem cell function is preserved in the absence of autophagy. Haematologica 2017, 102, e344–e347. [Google Scholar] [CrossRef]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for kmt2a/mll-mllt3/af9 aml maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jin, J.; Pan, J.; Yao, R.; Li, X.; Huang, X.; Ma, Z.; Huang, S.; Yan, X.; Jin, J.; et al. The change of nuclear lc3 distribution in acute myeloid leukemia cells. Exp. Cell Res. 2018, 369, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Ramsay, A.J.; Rodriguez, D.; Puente, X.S.; Campo, E.; Lopez-Otin, C. The genomic landscape of chronic lymphocytic leukemia: Clinical implications. BMC Med. 2013, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Ridout, K.; Robbe, P.; Vavoulis, D.; Klintman, J.; Stamatopoulos, B.; Bruce, D.; Burns, A.; Cavalieri, D.; Dreau, H.; Schuh, A. The genomic landscape of chronic lymphocytic leukaemia: Clinical implications. Clin. Lymphoma Myeloma Leuk. 2018, 18, S112–S115. [Google Scholar] [CrossRef]
- Kumar, K.; Kaur, J.; Walia, S.; Pathak, T.; Aggarwal, D. L-asparaginase: An effective agent in the treatment of acute lymphoblastic leukemia. Leuk. Lymphoma 2014, 55, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Pui, C.H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef] [Green Version]
- Dyczynski, M.; Vesterlund, M.; Bjorklund, A.C.; Zachariadis, V.; Janssen, J.; Gallart-Ayala, H.; Daskalaki, E.; Wheelock, C.E.; Lehtio, J.; Grander, D.; et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018, 9, 846. [Google Scholar] [CrossRef]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered expression of autophagy-related genes might contribute to glucocorticoid resistance in precursor b-cell-type acute lymphoblastic leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under l-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef]
- Polak, R.; Bierings, M.B.; van der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for etv6-runx1 driven b-cell precursor acute lymphoblastic leukemia. Haematologica 2018. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. Mir-15 and mir-16 induce apoptosis by targeting bcl2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bologna, C.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Audrito, V.; Brusa, D.; Pagnani, A.; Coscia, M.; D’Arena, G.; Mereu, E.; et al. Slamf1 regulation of chemotaxis and autophagy determines cll patient response. J. Clin. Investig. 2016, 126, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevated expression of il-3ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined mtor and autophagy inhibition: Phase i trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase i/ii trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and hdac inhibition: A phase i safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the hdac inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel atg4b antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Bosc, D.; Vezenkov, L.; Bortnik, S.; An, J.; Xu, J.; Choutka, C.; Hannigan, A.M.; Kovacic, S.; Loo, S.; Clark, P.G.K.; et al. A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease atg4b. Sci. Rep. 2018, 8, 11653. [Google Scholar] [CrossRef]
- Sathiyaseelan, P.; Rothe, K.; Yang, K.C.; Xu, J.; Chow, N.S.; Bortnik, S.; Choutka, C.; Ho, C.; Jiang, X.; Gorski, S.M. Diverse mechanisms of autophagy dysregulation and their therapeutic implications: Does the shoe fit? Autophagy 2019, 15, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Watanabe, A.; Forrest, D.L.; Gorski, S.; Young, R.; Jiang, X. Inhibiting the core autophagy enzyme atg4b with novel drugs sensitizes resistant leukemic stem/progenitor cells to standard targeted therapy. Blood 2018, 132, 933. [Google Scholar]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 43. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting bcr-abl-independent tki resistance in chronic myeloid leukemia by mtor and autophagy inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting hedgehog signaling pathway and autophagy overcomes drug resistance of bcr-abl-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef]
- Bosnjak, M.; Ristic, B.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Perovic, V.; Bogdanovic, A.; Paunovic, V.; Markovic, I.; Bumbasirevic, V.; et al. Inhibition of mtor-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS ONE 2014, 9, e94374. [Google Scholar] [CrossRef]
- Palmeira dos Santos, C.; Pereira, G.J.; Barbosa, C.M.; Jurkiewicz, A.; Smaili, S.S.; Bincoletto, C. Comparative study of autophagy inhibition by 3ma and cq on cytarabineinduced death of leukaemia cells. J. Cancer Res. Clin. Oncol. 2014, 140, 909–920. [Google Scholar] [CrossRef]
- Vilimanovich, U.; Bosnjak, M.; Bogdanovic, A.; Markovic, I.; Isakovic, A.; Kravic-Stevovic, T.; Mircic, A.; Trajkovic, V.; Bumbasirevic, V. Statin-mediated inhibition of cholesterol synthesis induces cytoprotective autophagy in human leukemic cells. Eur. J. Pharmacol. 2015, 765, 415–428. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, X.; Wang, S.; Fan, J.; Wang, Z.; Song, P.; Mei, X.; Ju, D. Blocking autophagy enhanced leukemia cell death induced by recombinant human arginase. Tumor Biol. 2016, 37, 6627–6635. [Google Scholar] [CrossRef]
- Tanios, R.; Bekdash, A.; Kassab, E.; Stone, E.; Georgiou, G.; Frankel, A.E.; Abi-Habib, R.J. Human recombinant arginase i(co)-peg5000 [huargi(co)-peg5000]-induced arginine depletion is selectively cytotoxic to human acute myeloid leukemia cells. Leuk. Res. 2013, 37, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; Lo, A.; Ng, M.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Booth, S.; Vardon, A.; Cousins, A.; Tubb, V.; Perry, T.; Noyvert, B.; Beggs, A.; Ng, M.; Halsey, C.; et al. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated-recombinant arginase i bct-100. Int. J. Cancer 2018, 142, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Wang, R.Y.; Qiu, Y.H.; Mak, D.H.; Coombes, K.; Yoo, S.Y.; Zhang, Q.; Jessen, K.; Liu, Y.; Rommel, C.; et al. Mln0128, a novel mtor kinase inhibitor, disrupts survival signaling and triggers apoptosis in aml and aml stem/ progenitor cells. Oncotarget 2016, 7, 55083–55097. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Bradstock, K.F.; Bendall, L.J. Rad001 (everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009, 5, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Cisterne, A.; Thien, M.; Hewson, J.; Baraz, R.; Bradstock, K.F.; Bendall, L.J. Potentiating effects of rad001 (everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood 2009, 113, 3297–3306. [Google Scholar] [CrossRef]
- Hartwell, K.A.; Miller, P.G.; Mukherjee, S.; Kahn, A.R.; Stewart, A.L.; Logan, D.J.; Negri, J.M.; Duvet, M.; Jaras, M.; Puram, R.; et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat. Chem. Biol. 2013, 9, 840–848. [Google Scholar] [CrossRef]
- Xie, N.; Zhong, L.; Liu, L.; Fang, Y.; Qi, X.; Cao, J.; Yang, B.; He, Q.; Ying, M. Autophagy contributes to dasatinib-induced myeloid differentiation of human acute myeloid leukemia cells. Biochem. Pharmacol. 2014, 89, 74–85. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Autophagy Modifier | Combination Strategy | Leukemia Types | Context | Ref |
|---|---|---|---|---|
| HCQ, Lys05, PIK-III (VPS34), Spautin-1, shATG4B, LV-320, NSC185058 | +TKIs | CML | In vitro and in vivo | [6,7,83,142,143] |
| CQ | +l-asp | ALL | In vitro and in vivo | [130] |
| CQ, siATG5 | +Tigecycline | CML | In vitro | [144] |
| HCQ | +NVP-BEZ235 | CML | In vitro and in vivo | [145] |
| HCQ, shATG7 | +Vismodegib | CML | In vitro | [146] |
| atg7−/− | +AraC | Murine AML | In vitro and in vivo | [121] |
| CQ, Bafilomycin A1, siMAP1LC3, siSQSTM1 | +Cytarabine | AML | In vitro | [147] |
| CQ, 3-MA, shATG7 | +AraC | AML | In vitro and in vivo | [54,148] |
| Bafilomycin A1, siBECN1 | +Statins | CLL, CML, AML, ALL | In vitro | [149] |
| − | shS100A8+adriamycin or vincristine | CML, AML, ALL | In vitro | [17] |
| CQ, 3-MA | +rhArginase | AML, ALL | In vitro | [150,151] |
| − | rhArginase+AraC or dexamethasone | CML, AML, ALL | In vitro and in vivo | [152,153] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030461
Rothe K, Porter V, Jiang X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. International Journal of Molecular Sciences. 2019; 20(3):461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030461
Chicago/Turabian StyleRothe, Katharina, Vanessa Porter, and Xiaoyan Jiang. 2019. "Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions" International Journal of Molecular Sciences 20, no. 3: 461. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030461