Inhibition of Heme Oxygenase-1 Activity Enhances Wilms Tumor-1-Specific T-Cell Responses in Cancer Immunotherapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
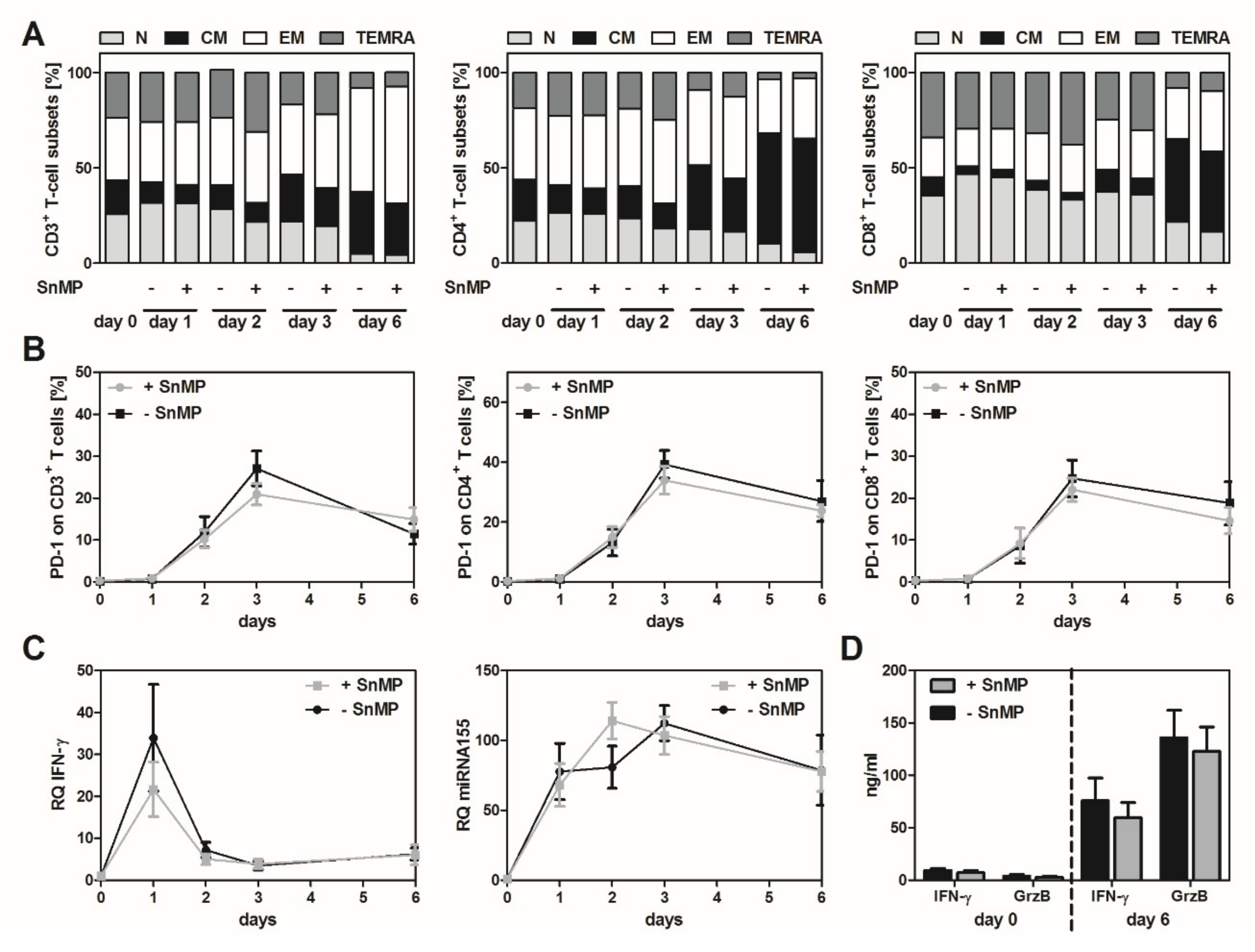
2.1. HO-1 Inhibition Had No Significant Effect on T-Cell Activation and Subsets in an Antigen-Independent Setting
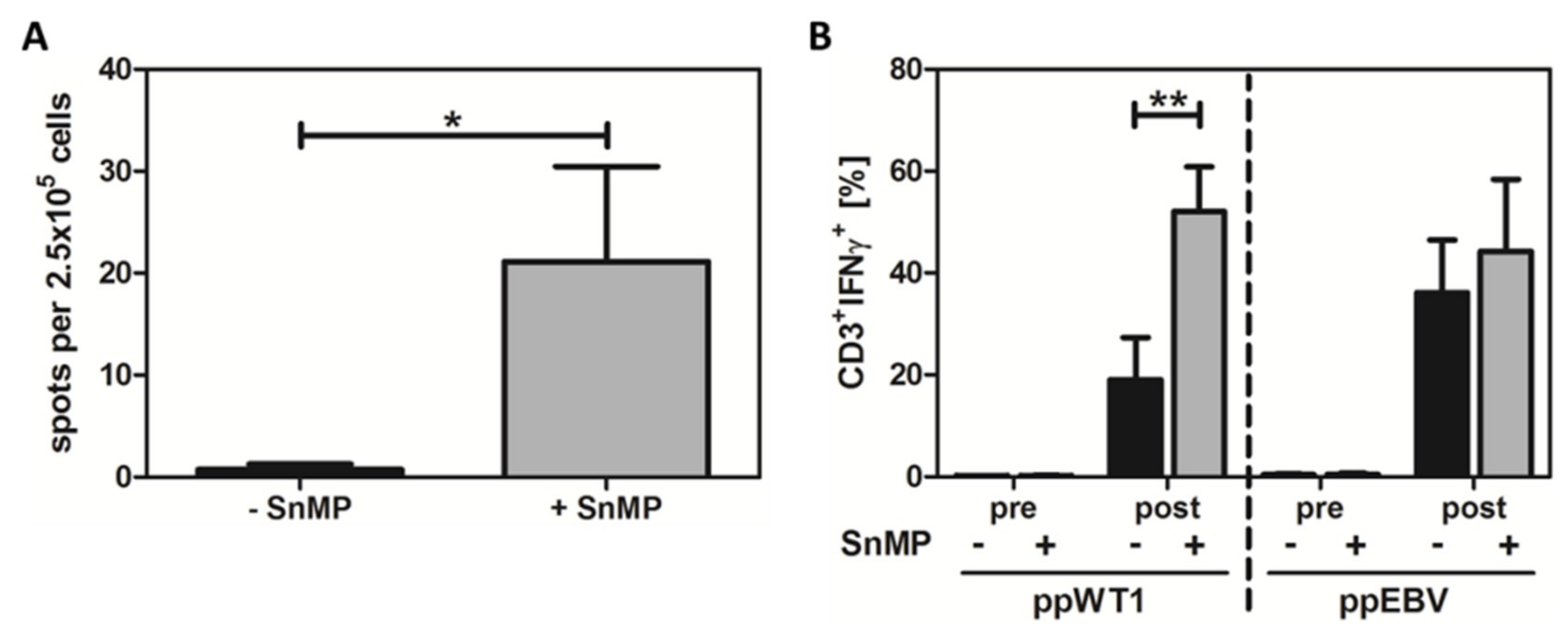
2.2. SnMP Resulted in Higher T-Cell Response to WT1 in Healthy Donors
2.3. Combined Stimulation with ppWT1 + SnMP Enhanced the Enrichment of IFN-γ-Secreting T Cells in a Preclinical Setting
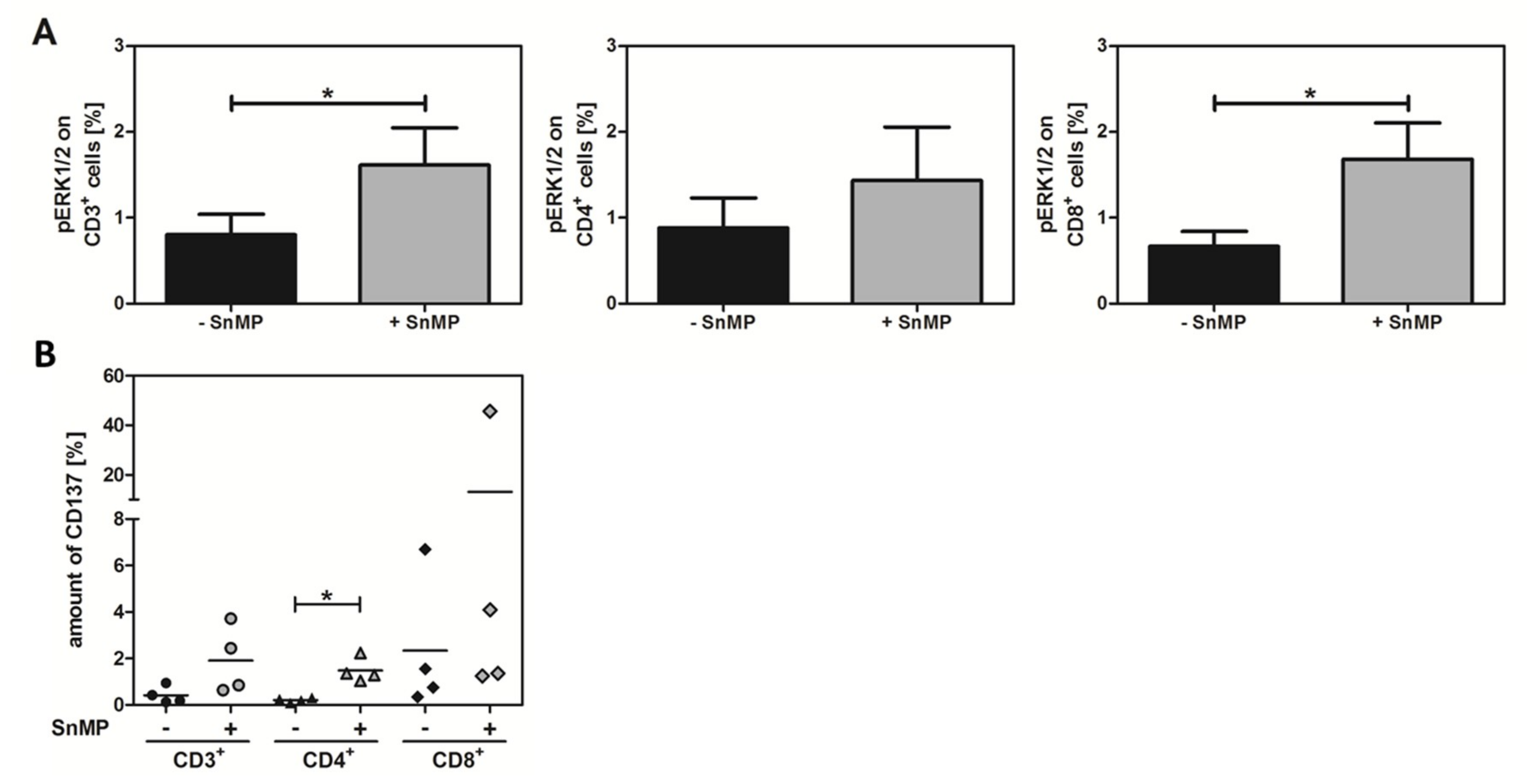
2.4. SnMP Increases Phosphorylated ERK Expression
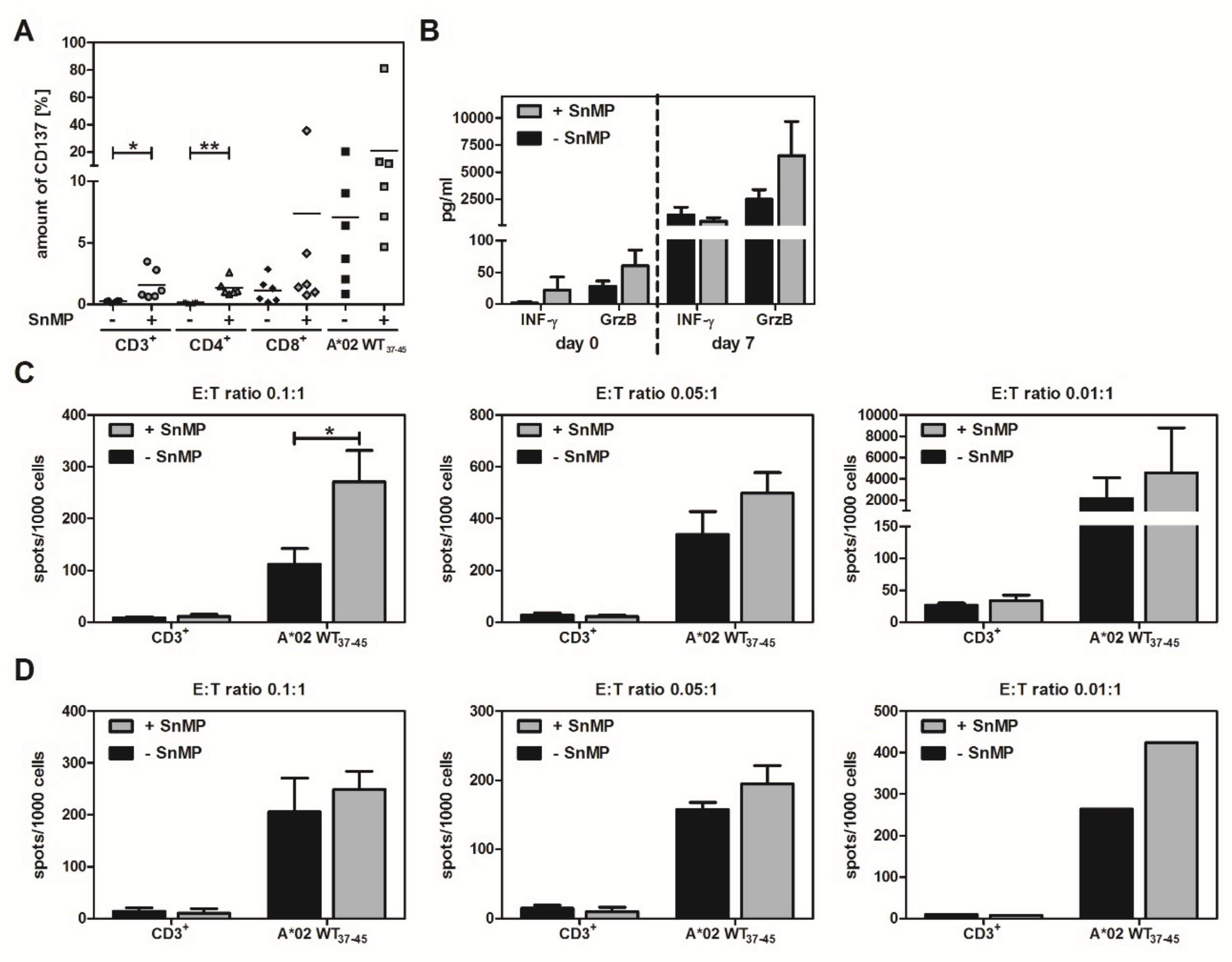
2.5. CD137 Expression Increases in Response to SnMP
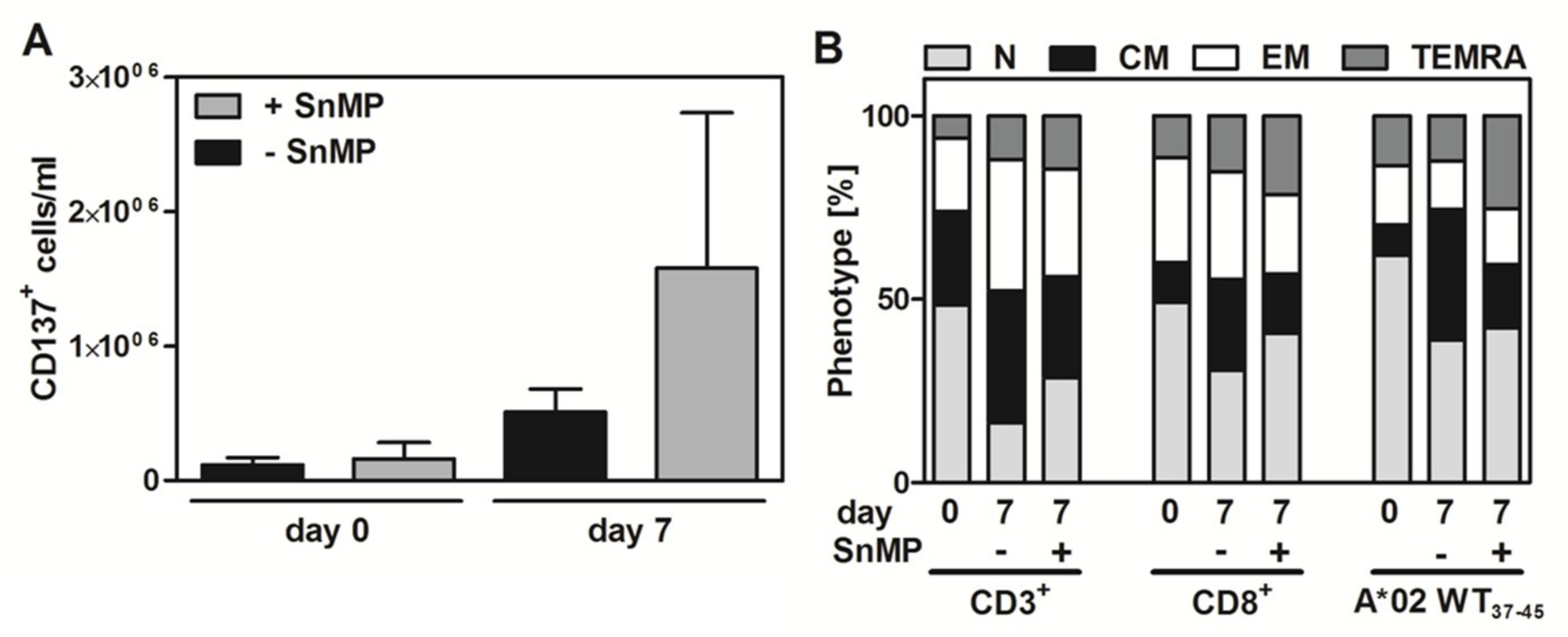
2.6. Effect of SnMP on WT1 Peptide-Specific T-Cell Expansion and T-cell Functionality
3. Discussion
3.1. HO-1 Inhibition Improves Clinical Enrichment Strategies for WT1-Specific T Cells
3.2. HO-1 Inhibition in the Absence of Antigenic Stimulation Did Not Alter T-Cell-Functionality
3.3. HO-1 Inhibition Increases pWT137–45 Peptide-Specific T-Cell Expansion and Functionality Without Altering the Composition of T-Cell Subsets
4. Material and Methods
4.1. Study Population
4.2. Synthetic Peptide Pools and Peptides
4.3. Influence of SnMP in an Antigen-Independent Setting
4.4. Evaluation of Changes the T-Cell Frequencies and Phenotypes
4.5. Gene and microRNA Expression Analysis
4.6. WT1-Specific T-Cell Stimulation Using HLA-Ig-Based aAPCs
4.7. Analyzing Cell Culture Supernatants via IFN-γ and Granzyme B ELISA
4.8. Cell Activation After ppWT Stimulation, as Determined via IFN-γ ELISpot
4.9. Flow Cytometric Assessment of Phosphorylated ERK
4.10. Use of SnMP to Enhance the Isolation of WT1-Specific T Cells in a Near-Clinical Setting
4.11. Effect of SnMP on the Expression of CD137
4.12. ELISpot for Detection of Granzyme B Release After Expansion with aAPCs
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aAPC | artificial antigen-presenting cell |
| APC | allophycocyanin |
| CO | Carbon monoxide |
| CTL | Cytotoxic T lymphocyte |
| EBV | Epstein-Barr virus |
| GvHD | graft-versus-host disease |
| IFN-γ | interferon–gamma |
| mAb | monoclonal antibody |
| pERK | phosphorylated extracellular signal-regulated kinase |
| ppEBV | pool of peptides derived from Epstein-Barr virus |
| ppWT1 | overlapping pool of peptides derived from WT1 |
| PCR | polymerase chain reaction |
| RT | room temperature |
| SnMP | tin mesoporphyrin |
| TCR | T-cell receptor |
| WT1 | Wilms tumor protein-1 |
References
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kersey, J.; Kolb, H.J.; Rimm, A.A.; Ringden, O.; Rozman, C.; Speck, B.; et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar]
- Ohminami, H.; Yasukawa, M.; Fujita, S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood 2000, 95, 286–293. [Google Scholar]
- Oka, Y.; Elisseeva, O.A.; Tsuboi, A.; Ogawa, H.; Tamaki, H.; Li, H.; Oji, Y.; Kim, E.H.; Soma, T.; Asada, M.; et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms’ tumor gene (WT1) product. Immunogenetics 2000, 51, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Tyler, E.M.; Jungbluth, A.A.; O’Reilly, R.J.; Koehne, G. WT1-specific T-cell responses in high-risk multiple myeloma patients undergoing allogeneic T cell-depleted hematopoietic stem cell transplantation and donor lymphocyte infusions. Blood 2013, 121, 308–317. [Google Scholar] [CrossRef]
- Koesters, R.; Linnebacher, M.; Coy, J.F.; Germann, A.; Schwitalle, Y.; Findeisen, P.; von Knebel Doeberitz, M. WT1 is a tumor-associated antigen in colon cancer that can be recognized by in vitro stimulated cytotoxic T cells. Int. J. Cancer 2004, 109, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Loeb, D.M.; Evron, E.; Patel, C.B.; Sharma, P.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Korz, D.; Sukumar, S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001, 61, 921–925. [Google Scholar] [PubMed]
- Oji, Y.; Miyoshi, S.; Maeda, H.; Hayashi, S.; Tamaki, H.; Nakatsuka, S.; Yao, M.; Takahashi, E.; Nakano, Y.; Hirabayashi, H.; et al. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int. J. Cancer 2002, 100, 297–303. [Google Scholar] [CrossRef]
- Ueda, T.; Oji, Y.; Naka, N.; Nakano, Y.; Takahashi, E.; Koga, S.; Asada, M.; Ikeba, A.; Nakatsuka, S.; Abeno, S.; et al. Overexpression of the Wilms’ tumor gene WT1 in human bone and soft-tissue sarcomas. Cancer Sci. 2003, 94, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kramarzova, K.; Boublikova, L.; Stary, J.; Trka, J. Evaluation of WT1 expression in bone marrow vs peripheral blood samples of children with acute myeloid leukemia-impact on minimal residual disease detection. Leukemia 2013, 27, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Yong, A.S.; Savani, B.N.; Mielke, S.; Keyvanfar, K.; Gostick, E.; Price, D.A.; Douek, D.C.; Barrett, A.J. Graft-versus-leukemia effects associated with detectable Wilms tumor-1 specific T lymphocytes after allogeneic stem-cell transplantation for acute lymphoblastic leukemia. Blood 2007, 110, 1924–1932. [Google Scholar] [CrossRef]
- Weber, G.; Gerdemann, U.; Caruana, I.; Savoldo, B.; Hensel, N.F.; Rabin, K.R.; Shpall, E.J.; Melenhorst, J.J.; Leen, A.M.; Barrett, A.J.; et al. Generation of multi-leukemia antigen-specific T cells to enhance the graft-versus-leukemia effect after allogeneic stem cell transplant. Leukemia 2013, 27, 1538–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannage, M.; Abel, M.; Michallet, A.S.; Delluc, S.; Lambert, M.; Giraudier, S.; Kratzer, R.; Niedermann, G.; Saveanu, L.; Guilhot, F.; et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: Implications for vaccine development and adoptive cellular immunotherapy. J. Immunol. 2005, 174, 8210–8218. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Caruana, I.; Rouce, R.H.; Barrett, A.J.; Gerdemann, U.; Leen, A.M.; Rabin, K.R.; Bollard, C.M. Generation of tumor antigen-specific T cell lines from pediatric patients with acute lymphoblastic leukemia--implications for immunotherapy. Clin. Cancer Res. 2013, 19, 5079–5091. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Niiya, H.; Azuma, T.; Uchida, N.; Yakushijin, Y.; Sakai, I.; Hato, T.; Takahashi, M.; Senju, S.; Nishimura, Y.; et al. Direct recognition and lysis of leukemia cells by WT1-specific CD4+ T lymphocytes in an HLA class II-restricted manner. Blood 2005, 106, 1415–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslak, P.G.; Dao, T.; Krug, L.M.; Chanel, S.; Korontsvit, T.; Zakhaleva, V.; Zhang, R.; Wolchok, J.D.; Yuan, J.; Pinilla-Ibarz, J.; et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood 2010, 116, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Pittet, M.J.; Valmori, D.; Dunbar, P.R.; Speiser, D.E.; Lienard, D.; Lejeune, F.; Fleischhauer, K.; Cerundolo, V.; Cerottini, J.C.; Romero, P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999, 190, 705–715. [Google Scholar] [CrossRef]
- Schmied, S.; Gostick, E.; Price, D.A.; Abken, H.; Assenmacher, M.; Richter, A. Analysis of the functional WT1-specific T-cell repertoire in healthy donors reveals a discrepancy between CD4(+) and CD8(+) memory formation. Immunology 2015, 145, 558–569. [Google Scholar] [CrossRef]
- Burt, T.D.; Seu, L.; Mold, J.E.; Kappas, A.; McCune, J.M. Naive human T cells are activated and proliferate in response to the heme oxygenase-1 inhibitor tin mesoporphyrin. J. Immunol. 2010, 185, 5279–5288. [Google Scholar] [CrossRef]
- Lundvig, D.M.; Immenschuh, S.; Wagener, F.A. Heme oxygenase, inflammation, and fibrosis: The good, the bad, and the ugly? Front. Pharmacol. 2012, 3, 81. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Mahidhara, R.S.; Zhou, Z.; Hoffman, R.A.; Seol, D.W.; Flavell, R.A.; Billiar, T.R.; Otterbein, L.E.; Choi, A.M. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J. Immunol. 2004, 172, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.O.; Oh, G.S.; Choi, B.M.; Chae, S.C.; Kim, Y.M.; Chung, K.R.; Chung, H.T. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J. Immunol. 2004, 172, 4744–4751. [Google Scholar] [CrossRef] [PubMed]
- Bunse, C.E.; Fortmeier, V.; Tischer, S.; Zilian, E.; Figueiredo, C.; Witte, T.; Blasczyk, R.; Immenschuh, S.; Eiz-Vesper, B. Modulation of heme oxygenase-1 by metalloporphyrins increases anti-viral T cell responses. Clin. Exp. Immunol. 2015, 179, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bellantuono, I.; Elsasser, A.; Marley, S.B.; Gordon, M.Y.; Goldman, J.M.; Stauss, H.J. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood 2000, 95, 2198–2203. [Google Scholar]
- Li, Y.; Wang, J.; Li, X.; Jia, Y.; Huai, L.; He, K.; Yu, P.; Wang, M.; Xing, H.; Rao, Q.; et al. Role of the Wilms’ tumor 1 gene in the aberrant biological behavior of leukemic cells and the related mechanisms. Oncol. Rep. 2014, 32, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, A.; Oka, Y.; Ogawa, H.; Elisseeva, O.A.; Li, H.; Kawasaki, K.; Aozasa, K.; Kishimoto, T.; Udaka, K.; Sugiyama, H. Cytotoxic T-lymphocyte responses elicited to Wilms’ tumor gene WT1 product by DNA vaccination. J. Clin. Immunol. 2000, 20, 195–202. [Google Scholar] [CrossRef]
- Inoue, K.; Tamaki, H.; Ogawa, H.; Oka, Y.; Soma, T.; Tatekawa, T.; Oji, Y.; Tsuboi, A.; Kim, E.H.; Kawakami, M.; et al. Wilms’ tumor gene (WT1) competes with differentiation-inducing signal in hematopoietic progenitor cells. Blood 1998, 91, 2969–2976. [Google Scholar]
- Priesner, C.; Esser, R.; Tischer, S.; Marburger, M.; Aleksandrova, K.; Maecker-Kolhoff, B.; Heuft, H.G.; Goudeva, L.; Blasczyk, R.; Arseniev, L.; et al. Comparative Analysis of Clinical-Scale IFN-gamma-Positive T-Cell Enrichment Using Partially and Fully Integrated Platforms. Front. Immunol. 2016, 7, 393. [Google Scholar] [CrossRef]
- Tischer, S.; Priesner, C.; Heuft, H.G.; Goudeva, L.; Mende, W.; Barthold, M.; Kloess, S.; Arseniev, L.; Aleksandrova, K.; Maecker-Kolhoff, B.; et al. Rapid generation of clinical-grade antiviral T cells: Selection of suitable T-cell donors and GMP-compliant manufacturing of antiviral T cells. J. Transl. Med. 2014, 12, 336. [Google Scholar] [CrossRef]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvani, K.; Brenchley, J.M.; Price, D.A.; Kilical, Y.; Gostick, E.; Sewell, A.K.; Li, J.; Mielke, S.; Douek, D.C.; Barrett, A.J. T-cell responses directed against multiple HLA-A*0201-restricted epitopes derived from Wilms’ tumor 1 protein in patients with leukemia and healthy donors: Identification, quantification, and characterization. Clin. Cancer Res. 2005, 11, 8799–8807. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Sommermeyer, D.; Michel, C.; Wilde, S.; Schendel, D.; Uckert, W.; Blankenstein, T.; Kyewski, B. Misinitiation of intrathymic MART-1 transcription and biased TCR usage explain the high frequency of MART-1-specific T cells. Eur. J. Immunol. 2014, 44, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Steger, B.; Milosevic, S.; Doessinger, G.; Reuther, S.; Liepert, A.; Braeu, M.; Schick, J.; Vogt, V.; Schuster, F.; Kroell, T.; et al. CD4(+)and CD8(+)T-cell reactions against leukemia-associated- or minor-histocompatibility-antigens in AML-patients after allogeneic SCT. Immunobiology 2014, 219, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Grube, M.; Brenchley, J.M.; Sconocchia, G.; Fujiwara, H.; Price, D.A.; Gostick, E.; Yamada, K.; Melenhorst, J.; Childs, R.; et al. Functional leukemia-associated antigen-specific memory CD8+ T cells exist in healthy individuals and in patients with chronic myelogenous leukemia before and after stem cell transplantation. Blood 2003, 102, 2892–2900. [Google Scholar] [CrossRef] [Green Version]
- Whitehurst, C.E.; Geppert, T.D. MEK1 and the extracellular signal-regulated kinases are required for the stimulation of IL-2 gene transcription in T cells. J. Immunol. 1996, 156, 1020–1029. [Google Scholar]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Vibhakar, R.; Juan, G.; Traganos, F.; Darzynkiewicz, Z.; Finger, L.R. Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp. Cell Res. 1997, 232, 25–28. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Yong, A.S.; Mielke, S.; Savani, B.N.; Musse, L.; Superata, J.; Jafarpour, B.; Boss, C.; Barrett, A.J. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood 2008, 111, 236–242. [Google Scholar] [CrossRef]
- Uttenthal, B.; Martinez-Davila, I.; Ivey, A.; Craddock, C.; Chen, F.; Virchis, A.; Kottaridis, P.; Grimwade, D.; Khwaja, A.; Stauss, H.; et al. Wilms’ Tumour 1 (WT1) peptide vaccination in patients with acute myeloid leukaemia induces short-lived WT1-specific immune responses. Br. J. Haematol. 2014, 164, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Holmich, L.R.; Hendel, H.W.; Met, O.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutani, V.K.; Poland, R.; Meloy, L.D.; Hegyi, T.; Fanaroff, A.A.; Maisels, M.J. Clinical trial of tin mesoporphyrin to prevent neonatal hyperbilirubinemia. J. Perinatol. 2016, 36, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Kappas, A.; Drummond, G.S.; Valaes, T. A single dose of Sn-mesoporphyrin prevents development of severe hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient newborns. Pediatrics 2001, 108, 25–30. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Nagler, A.; Niederwieser, D.; Castagna, L.; Tabrizi, R.; Stadler, M.; Kuball, J.; Cornelissen, J.; Vorlicek, J.; et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood 2012, 119, 1599–1606. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Nagler, A.; Bornhauser, M.; Finke, J.; Fassas, A.; Volin, L.; Gurman, G.; Maertens, J.; Bordigoni, P.; et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: A retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J. Clin. Oncol. 2007, 25, 4938–4945. [Google Scholar] [CrossRef]
- Nikiforow, S.; Alyea, E.P. Maximizing GVL in allogeneic transplantation: Role of donor lymphocyte infusions. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 570–575. [Google Scholar] [CrossRef]
- Ishikawa, T.; Fujii, N.; Imada, M.; Aoe, M.; Meguri, Y.; Inomata, T.; Nakashima, H.; Fujii, K.; Yoshida, S.; Nishimori, H.; et al. Graft-versus-leukemia effect with a WT1-specific T-cell response induced by azacitidine and donor lymphocyte infusions after allogeneic hematopoietic stem cell transplantation. Cytotherapy 2017, 19, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Ishikawa, T.; Egawa, S.; Koido, S.; Yanagimoto, H.; Ishii, J.; Kanno, Y.; Kokura, S.; Yasuda, H.; Oba, M.S.; et al. Combination Gemcitabine and WT1 Peptide Vaccination Improves Progression-Free Survival in Advanced Pancreatic Ductal Adenocarcinoma: A Phase II Randomized Study. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef]
- Di Stasi, A.; Jimenez, A.M.; Minagawa, K.; Al-Obaidi, M.; Rezvani, K. Review of the Results of WT1 Peptide Vaccination Strategies for Myelodysplastic Syndromes and Acute Myeloid Leukemia from Nine Different Studies. Front. Immunol. 2015, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.A.; Gao, L.; Hart, D.; Gillmore, R.; Qasim, W.; Thrasher, A.; Apperley, J.; Engels, B.; Uckert, W.; Morris, E.; et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood 2005, 106, 3062–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Verboom, M.; Hallensleben, M.; Horn, P.A.; Blasczyk, R. Bioinformatic integration of biomechanics makes HLA sequencing universally applicable. Tissue Antigens 2007, 70, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Bunse, C.E.; Tischer, S.; Lahrberg, J.; Oelke, M.; Figueiredo, C.; Blasczyk, R.; Eiz-Vesper, B. Granulocyte colony-stimulating factor impairs CD8(+) T cell functionality by interfering with central activation elements. Clin. Exp. Immunol. 2016, 185, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bunse, C.E.; Borchers, S.; Varanasi, P.R.; Tischer, S.; Figueiredo, C.; Immenschuh, S.; Kalinke, U.; Kohl, U.; Goudeva, L.; Maecker-Kolhoff, B.; et al. Impaired functionality of antiviral T cells in G-CSF mobilized stem cell donors: Implications for the selection of CTL donor. PLoS ONE 2013, 8, e77925. [Google Scholar] [CrossRef] [PubMed]
- Sukdolak, C.; Tischer, S.; Dieks, D.; Figueiredo, C.; Goudeva, L.; Heuft, H.G.; Verboom, M.; Immenschuh, S.; Heim, A.; Borchers, S.; et al. CMV-, EBV- and ADV-specific T cell immunity: Screening and monitoring of potential third-party donors to improve post-transplantation outcome. Biol. Blood Marrow Transplant. 2013, 19, 1480–1492. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schillingmann, D.A.; Riese, S.B.; Vijayan, V.; Tischer-Zimmermann, S.; Schmetzer, H.; Maecker-Kolhoff, B.; Blasczyk, R.; Immenschuh, S.; Eiz-Vesper, B. Inhibition of Heme Oxygenase-1 Activity Enhances Wilms Tumor-1-Specific T-Cell Responses in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030482
Schillingmann DA, Riese SB, Vijayan V, Tischer-Zimmermann S, Schmetzer H, Maecker-Kolhoff B, Blasczyk R, Immenschuh S, Eiz-Vesper B. Inhibition of Heme Oxygenase-1 Activity Enhances Wilms Tumor-1-Specific T-Cell Responses in Cancer Immunotherapy. International Journal of Molecular Sciences. 2019; 20(3):482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030482
Chicago/Turabian StyleSchillingmann, Damaris A., Sebastian B. Riese, Vijith Vijayan, Sabine Tischer-Zimmermann, Helga Schmetzer, Britta Maecker-Kolhoff, Rainer Blasczyk, Stephan Immenschuh, and Britta Eiz-Vesper. 2019. "Inhibition of Heme Oxygenase-1 Activity Enhances Wilms Tumor-1-Specific T-Cell Responses in Cancer Immunotherapy" International Journal of Molecular Sciences 20, no. 3: 482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030482