BmK AEP, an Anti-Epileptic Peptide Distinctly Affects the Gating of Brain Subtypes of Voltage-Gated Sodium Channels


Abstract
:1. Introduction
2. Results
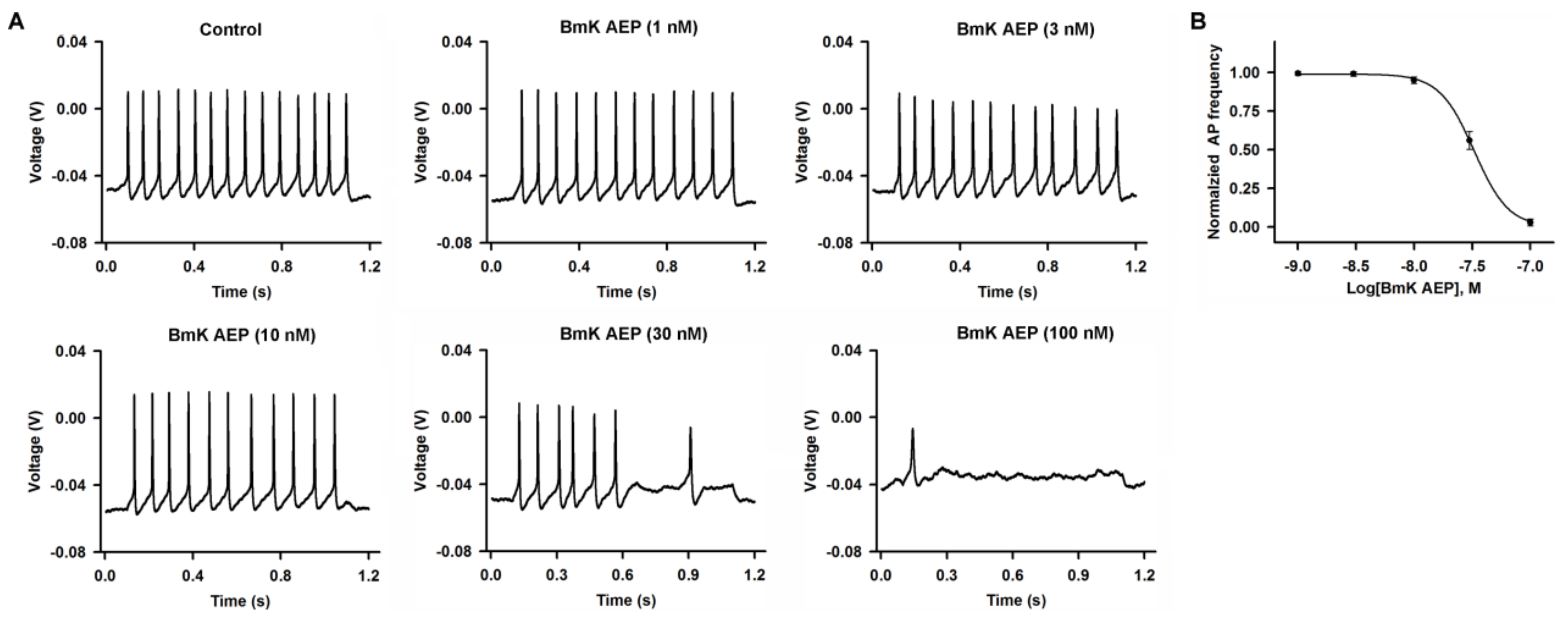
2.1. Influence of BmK AEP on Action Potential Firing in Cerebral Cortical Neurons
2.2. Influence of BmK AEP on VGSCs in Primary Cultured Cortical Neurons
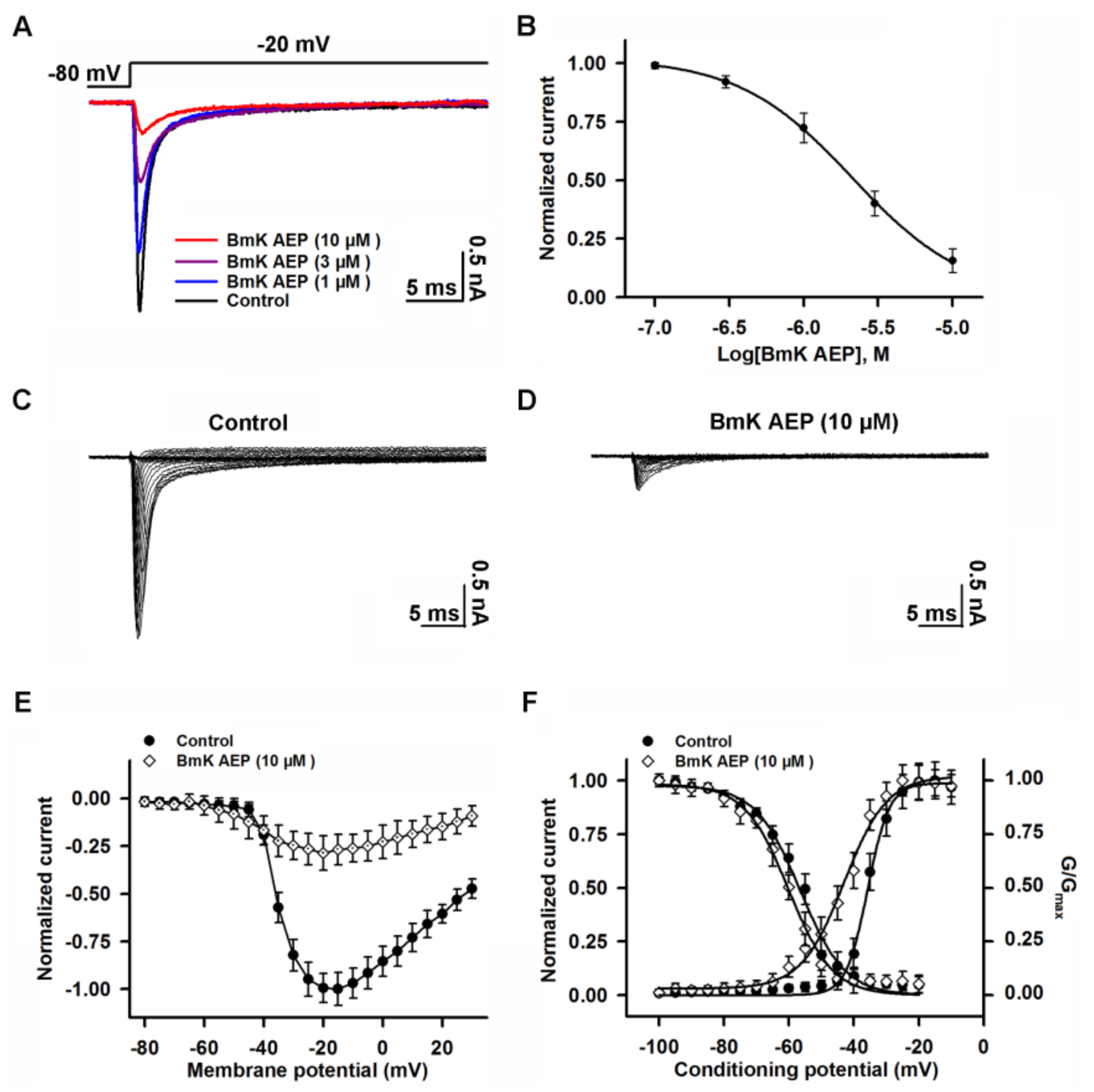
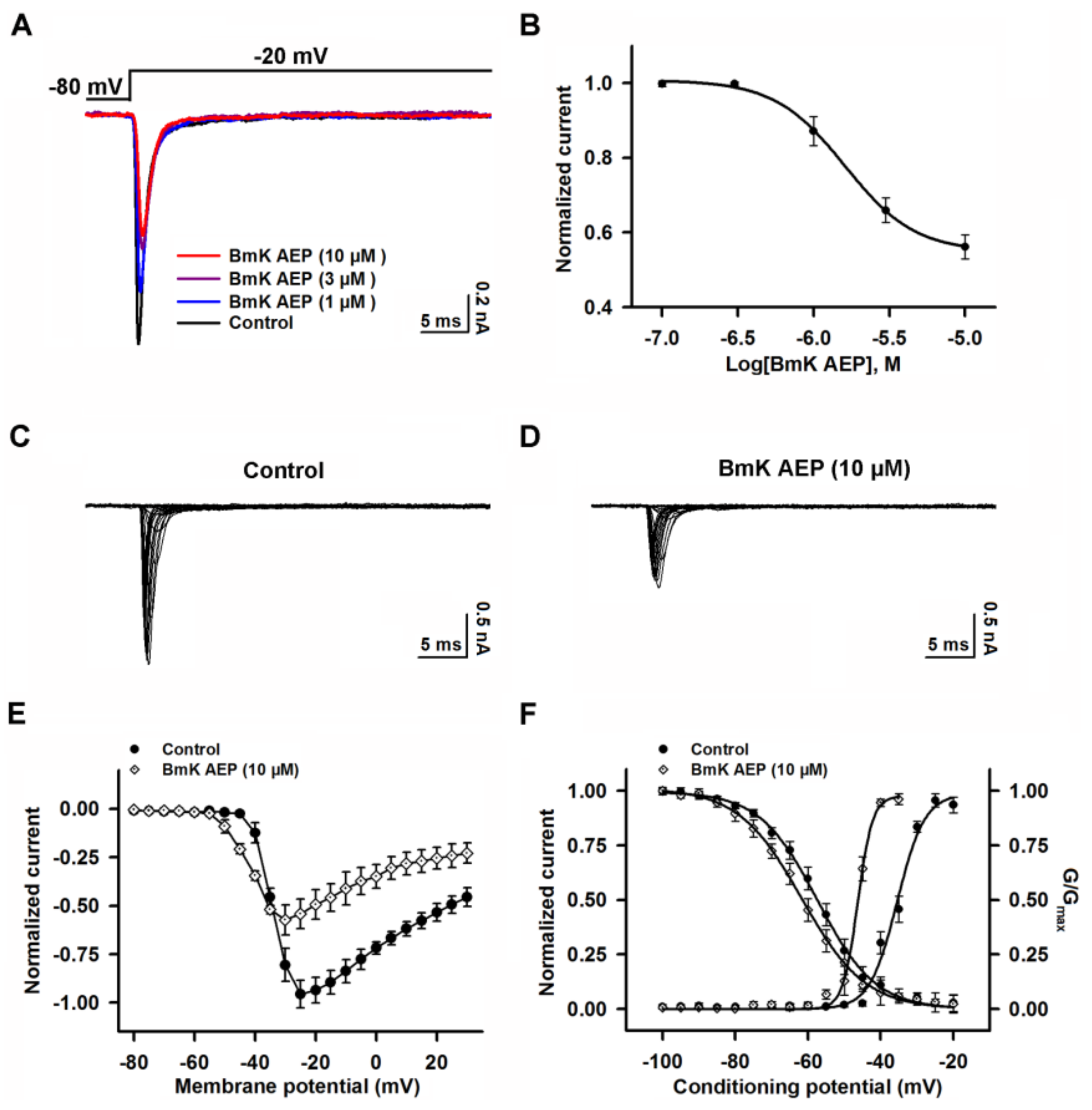
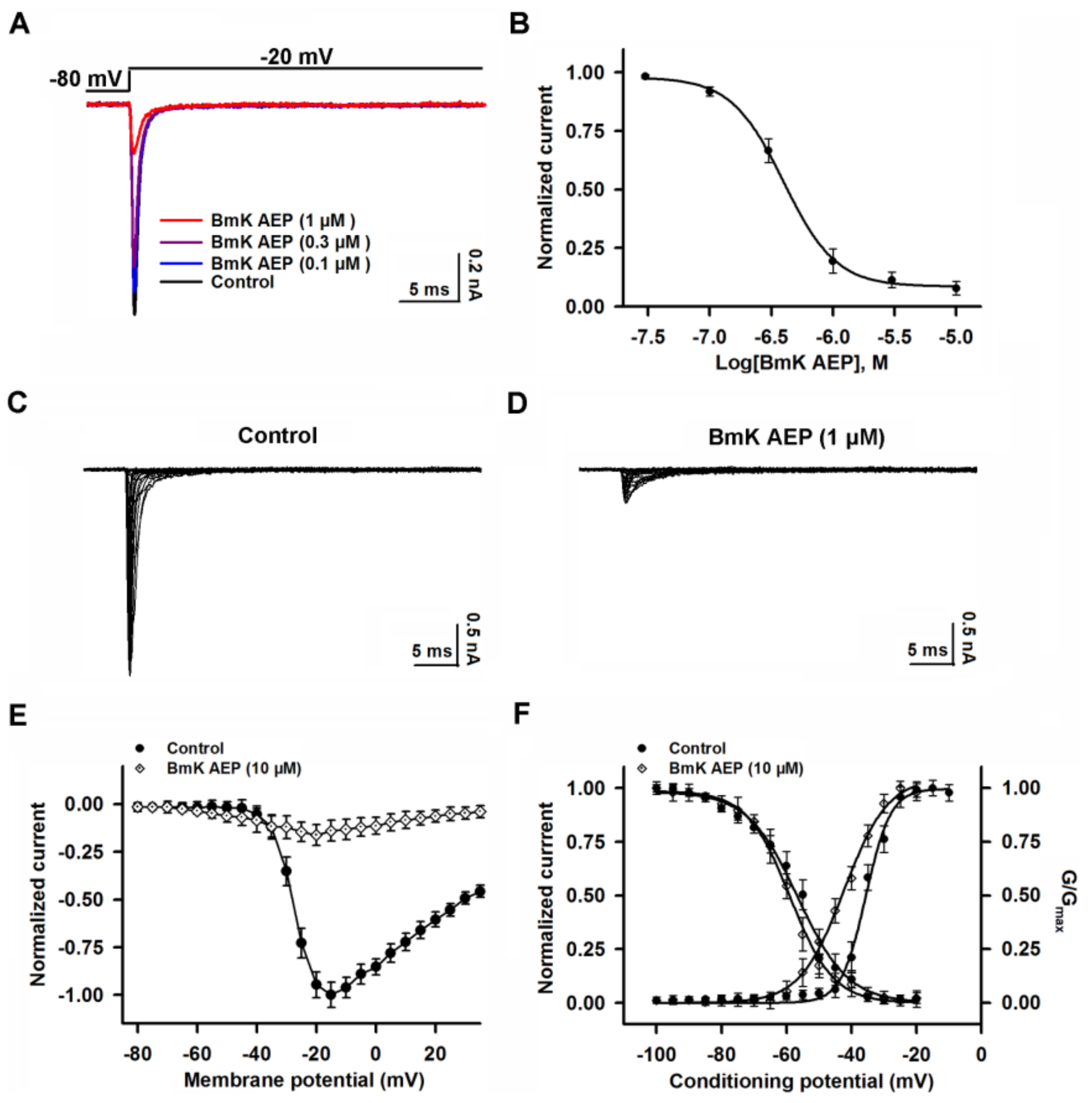
2.3. Influence of BmK AEP on hNav1.1 Expressed in HEK-293 Cells
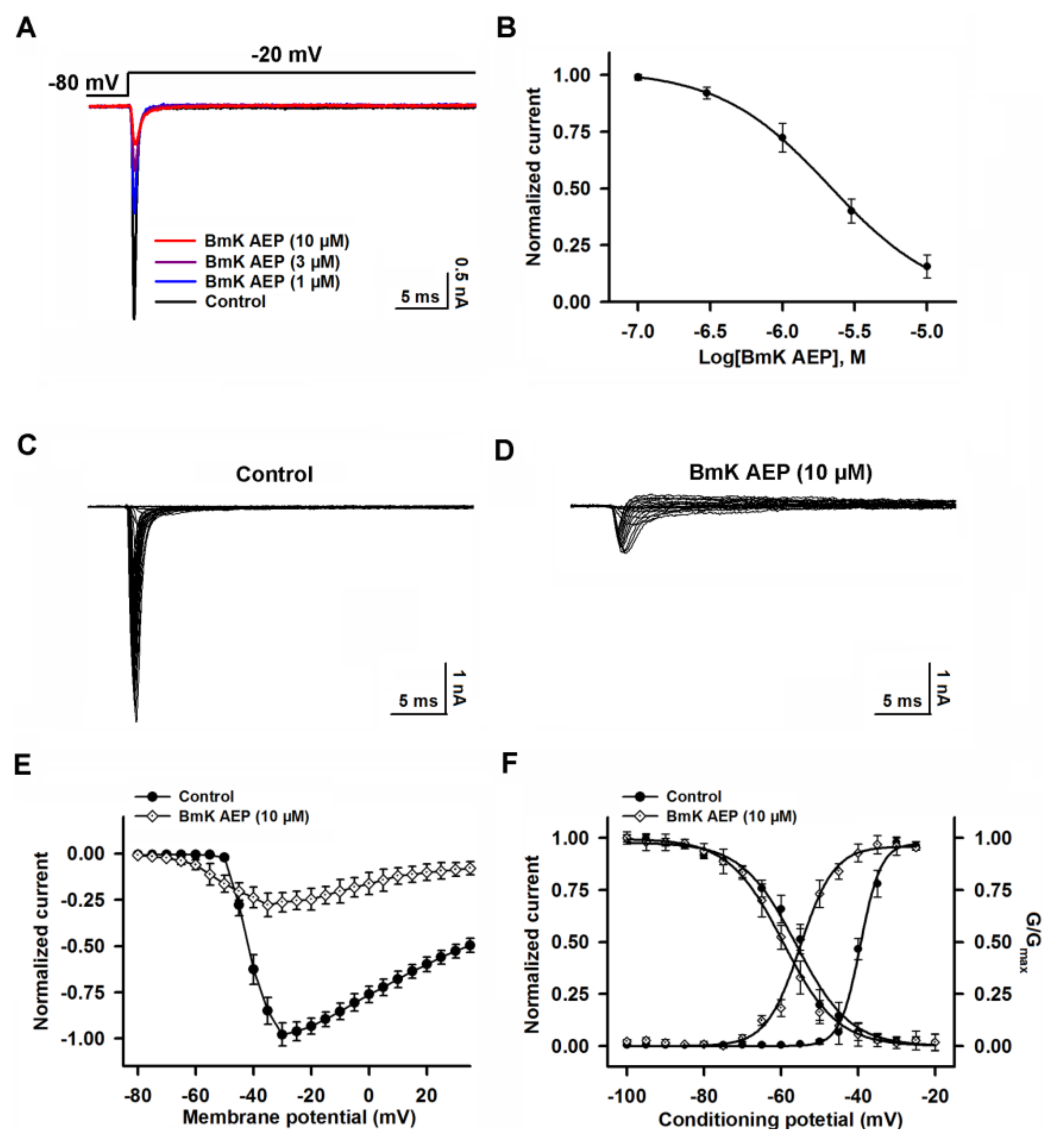
2.4. Influence of BmK AEP on hNav1.2 Expressed in HEK-293 Cells
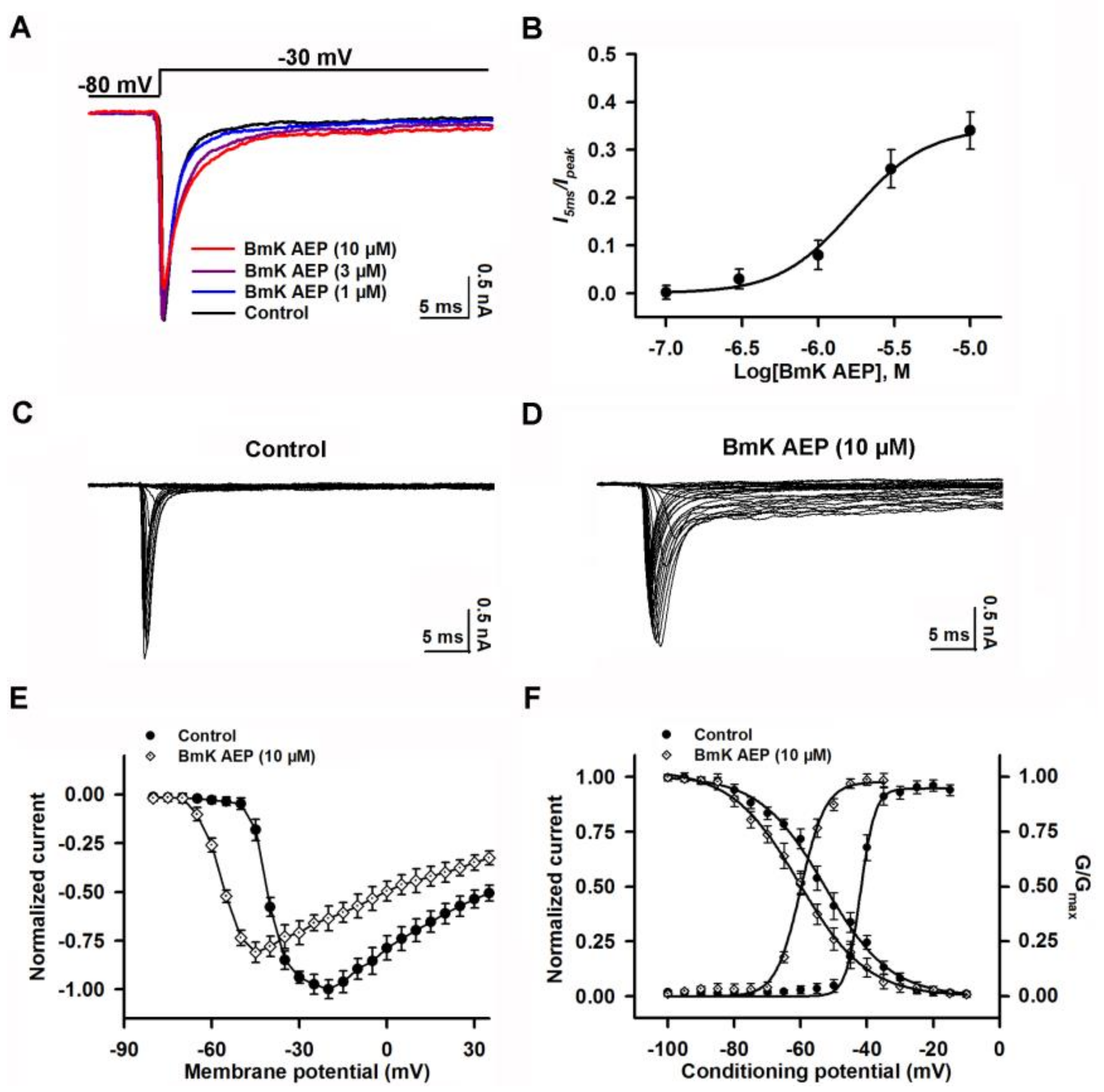
2.5. Influence of BmK AEP on hNav1.3 Stably Expressed in HEK-293 Cells
2.6. Influence of BmK AEP on hNav1.6 Expressed in HEK-293 Cells
3. Discussion
4. Materials and Methods
4.1. Materials and Animals
4.2. BmK AEP Purification
4.3. Primary Cultures of Cortical Neurons
4.4. Culture of HEK-293 Cells Heterologously Expressing VGSC Subtypes
4.5. Voltage-Clamp and Current-Clamp Electrophysiology
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APs | Action potentials |
| BmK | Buthus martensii Karsch |
| CI | Confidence Interval |
| DIV | Days in vitro |
| FBS | Fetal bovine serum |
| I-V | Current-membrane voltage |
| VGSCs | Voltage-gated sodium channels |
References
- Norton, R.S.; Chandy, K.G. Venom-derived peptide inhibitors of voltage-gated potassium channels. Neuropharmacology 2017, 127, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housley, D.M.; Housley, G.D.; Liddell, M.J.; Jennings, E.A. Scorpion toxin peptide action at the ion channel subunit level. Neuropharmacology 2017, 127, 46–78. [Google Scholar] [CrossRef] [PubMed]
- Bourinet, E.; Zamponi, G.W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology 2017, 127, 109–115. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zou, X.; Li, X.; Chen, J.; Jin, L.; Zhang, F.; Yu, B.; Cao, Z. Activation of sodium channels by α-scorpion toxin, BmK NT1, produced neurotoxicity in cerebellar granule cells: An association with intracellular Ca2+ overloading. Arch. Toxicol. 2017, 91, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Sarfo-Poku, C.; Eshun, O.; Lee, K.H. Medical application of scorpion venom to breast cancer: A mini-review. Toxicon 2016, 122, 109–112. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.M.; Goncalves, B.D.C.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef]
- Shen, B.; Cao, Z.; Li, W.; Sabatier, J.M.; Wu, Y. Treating autoimmune disorders with venom-derived peptides. Expert Opin. Biol. Ther. 2017, 17, 1065–1075. [Google Scholar] [CrossRef]
- Chan, Y.S.; Cheung, R.C.; Xia, L.; Wong, J.H.; Ng, T.B.; Chan, W.Y. Snake venom toxins: Toxicity and medicinal applications. Appl. Microbiol. Biotechnol. 2016, 100, 6165–6181. [Google Scholar] [CrossRef]
- De la Vega, R.C.; Possani, L.D. Overview of scorpion toxins specific for Na+ channels and related peptides: Biodiversity, structure-function relationships and evolution. Toxicon 2005, 46, 831–844. [Google Scholar] [CrossRef]
- Bergeron, Z.L.; Bingham, J.P. Scorpion toxins specific for potassium (K+) channels: A historical overview of peptide bioengineering. Toxins 2012, 4, 1082–1119. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Wilson, D. Structural diversity of arthropod venom toxins. Toxicon 2018, 152, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.J.; Wang, C.G.; Wang, M.; Wang, D.C. A depressant insect toxin with a novel analgesic effect from scorpion Buthus martensii Karsch. Biochimica. Biophys. Acta 2001, 1549, 9–18. [Google Scholar] [CrossRef]
- Wang, C.G.; He, X.L.; Shao, F.; Liu, W.; Ling, M.H.; Wang, D.C.; Chi, C.W. Molecular characterization of an anti-epilepsy peptide from the scorpion Buthus martensi Karsch. Eur. J. Biochem. 2001, 268, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.G.; Ling, M.H.; Chi, C.W.; Wang, D.C.; Pelhate, M. Purification of two depressant insect neurotoxins and their gene cloning from the scorpion Buthus martensi Karsch. J. Pept. Res. 2003, 61, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zeng, X.C.; He, X.H.; Pu, J.; Li, W.X.; Zhu, Z.H.; Liu, H. Molecular cloning and functional expression of a gene encoding an antiarrhythmia peptide derived from the scorpion toxin. Eur. J. Biochem. 2002, 269, 4468–4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosmans, F.; Martin-Eauclaire, M.F.; Tytgat, J. The depressant scorpion neurotoxin LqqIT2 selectively modulates the insect voltage-gated sodium channel. Toxicon 2005, 45, 501–507. [Google Scholar] [CrossRef]
- He, X.; Peng, F.; Zhang, J.; Li, W.; Zeng, X.; Liu, H. Inhibitory effects of recombinant neurotoxin BmK IM on seizures induced by pentylenetetrazol in Rats. Chin. Med. J. 2003, 116, 1898–1903. [Google Scholar]
- Bonilha, L.; Cendes, F.; Ghizoni, E.; Vieira, R.J.; Li, L.M. Epilepsy due to a destructive brain lesion caused by a scorpion sting. Arch. Neurol. 2004, 61, 1294–1296. [Google Scholar] [CrossRef]
- Bahloul, M.; Souissi, B.; Turki, O.; Dlela, M.; Ben Mahfoudh, K.; Bouaziz, M. Evidence of direct toxicological effects of scorpion venom on central nervous system in Tunisian children. Case Rep. Crit. Care 2018, 2018, 8304375. [Google Scholar] [CrossRef]
- Isbister, G.K.; Bawaskar, H.S. Scorpion envenomation. N. Engl. J. Med. 2014, 371, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, H.; Zhang, F.; Zhang, C.; Zou, X.; Cao, Z. Selective voltage-gated sodium channel peptide toxins from animal venom: Pharmacological probes and analgesic drug development. ACS Chem. Neurosci. 2018, 9, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.I.; Isom, L.L.; Petrou, S. Role of Sodium Channels in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, S.P.; Van Poppel, K.; McGregor, A.L.; Mudigoudar, B.; Wheless, J.W. Vagus nerve stimulation in intractable epilepsy associated with SCN1A gene abnormalities. J. Child Neurol. 2017, 32, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Willow, M.; Gonoi, T.; Catterall, W.A. Voltage clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in neuroblastoma cells. Mol. Pharmacol. 1985, 27, 549–558. [Google Scholar] [PubMed]
- Mattson, R.H.; Cramer, J.A.; Collins, J.F. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic-clonic seizures in adults. The department of veterans affairs epilepsy cooperative study No. 264 group. N. Engl. J. Med. 1992, 327, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; O’Brien, J.E.; Conravey, A.; Hammer, M.F.; Waxman, S.G.; Dib-Hajj, S.D.; Meisler, M.H. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol. Dis. 2014, 69, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, E.; Possani, L.D. Scorpion toxins to unravel the conundrum of ion channel structure and functioning. Toxicon 2018, 150, 17–27. [Google Scholar] [CrossRef]
- Dutertre, S.; Lewis, R.J. Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 2010, 285, 13315–13320. [Google Scholar] [CrossRef]
- Wang, J.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Catterall, W.A. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar] [CrossRef]
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Rogers, J.C.; Qu, Y.; Tanada, T.N.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 1996, 271, 15950–15962. [Google Scholar] [CrossRef]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef]
- Zou, X.; Wu, Y.; Chen, J.; Zhao, F.; Zhang, F.; Yu, B.; Cao, Z. Activation of sodium channel by a novel α-scorpion toxin, BmK NT2, stimulates ERK1/2 and CERB phosphorylation through a Ca2+ dependent pathway in neocortical neurons. Int. J. Biol. Macromol. 2017, 104, 70–77. [Google Scholar] [CrossRef]
- Coleman, N.; Nguyen, H.M.; Cao, Z.; Brown, B.M.; Jenkins, D.P.; Zolkowska, D.; Chen, Y.J.; Tanaka, B.S.; Goldin, A.L.; Rogawski, M.A.; et al. The riluzole derivative 2-amino-6-trifluoromethylthio-benzothiazole (SKA-19), a mixed KCa2 activator and Nav blocker, is a potent novel anticonvulsant. Neurotherapeutics 2015, 12, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, F.H.; Sharp, E.M.; Beacham, D.; Scheuer, T.; Catterall, W.A. Functional properties and differential neuromodulation of Nav1.6 channels. Mol. Cell Neurosci. 2008, 38, 607–615. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VGSC Subtypes | Influence | Maximum Inhibition/Delay (%) | IC50 (μM)a 95% CI | Selectivity for Nav1.6 |
|---|---|---|---|---|
| Nav1.1 | Inhibition of peak current | 82% | 3.20 (1.73–6.32) | 9.30 |
| Nav1.2 | Delay of inactivation | 34% | 1.69 (0.93–2.76) | - |
| Nav1.3 | Inhibition of peak current | 56% | 1.46 (0.91–2.56) | 6.21 |
| Nav1.6 | Inhibition of peak current | 93% | 0.39 (0.26–0.57) | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Wu, Y.; Zou, X.; Tang, Q.; Zhao, F.; Cao, Z. BmK AEP, an Anti-Epileptic Peptide Distinctly Affects the Gating of Brain Subtypes of Voltage-Gated Sodium Channels. Int. J. Mol. Sci. 2019, 20, 729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030729
Zhang F, Wu Y, Zou X, Tang Q, Zhao F, Cao Z. BmK AEP, an Anti-Epileptic Peptide Distinctly Affects the Gating of Brain Subtypes of Voltage-Gated Sodium Channels. International Journal of Molecular Sciences. 2019; 20(3):729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030729
Chicago/Turabian StyleZhang, Fan, Ying Wu, Xiaohan Zou, Qinglian Tang, Fang Zhao, and Zhengyu Cao. 2019. "BmK AEP, an Anti-Epileptic Peptide Distinctly Affects the Gating of Brain Subtypes of Voltage-Gated Sodium Channels" International Journal of Molecular Sciences 20, no. 3: 729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030729