Insights into Body Size Evolution: A Comparative Transcriptome Study on Three Species of Asian Sisoridae Catfish


Abstract
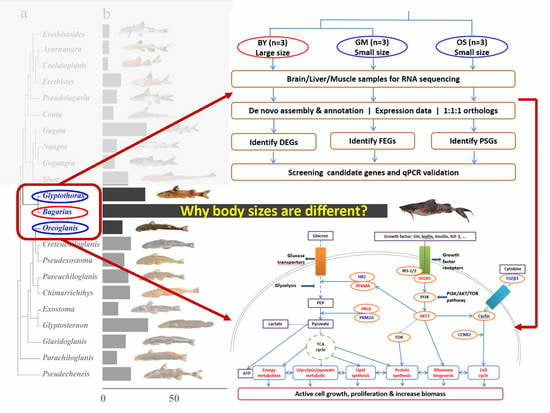
:
1. Introduction
2. Results
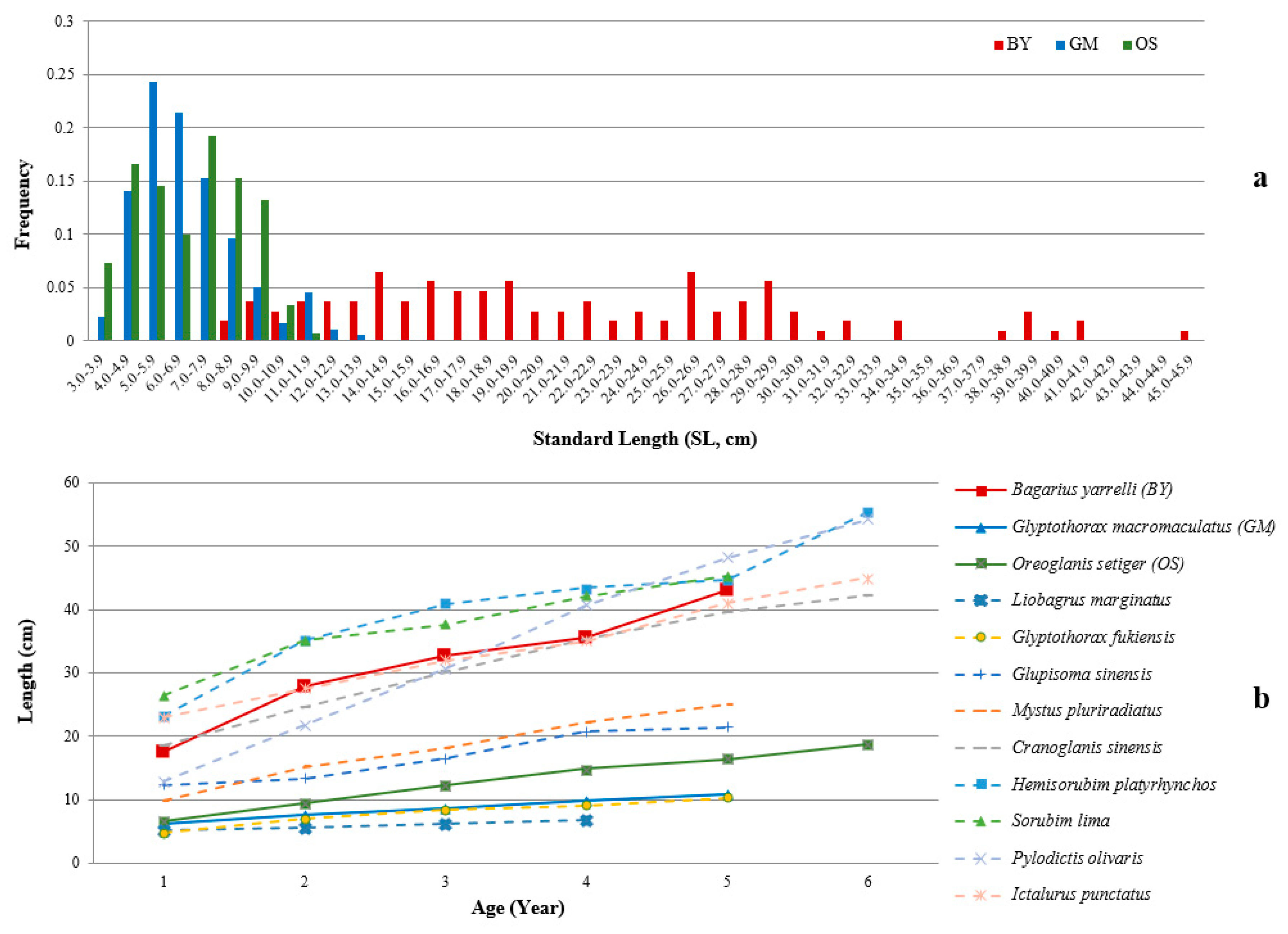
2.1. Length Frequency and Preliminary Age Estimations of BY, GM, and OS
2.2. External Morphology Comparison of BY, GM, and OS
2.3. De Novo Assembly and Annotation of the Three Transcriptomes
2.4. DEGs in BY Relative to GM and OS
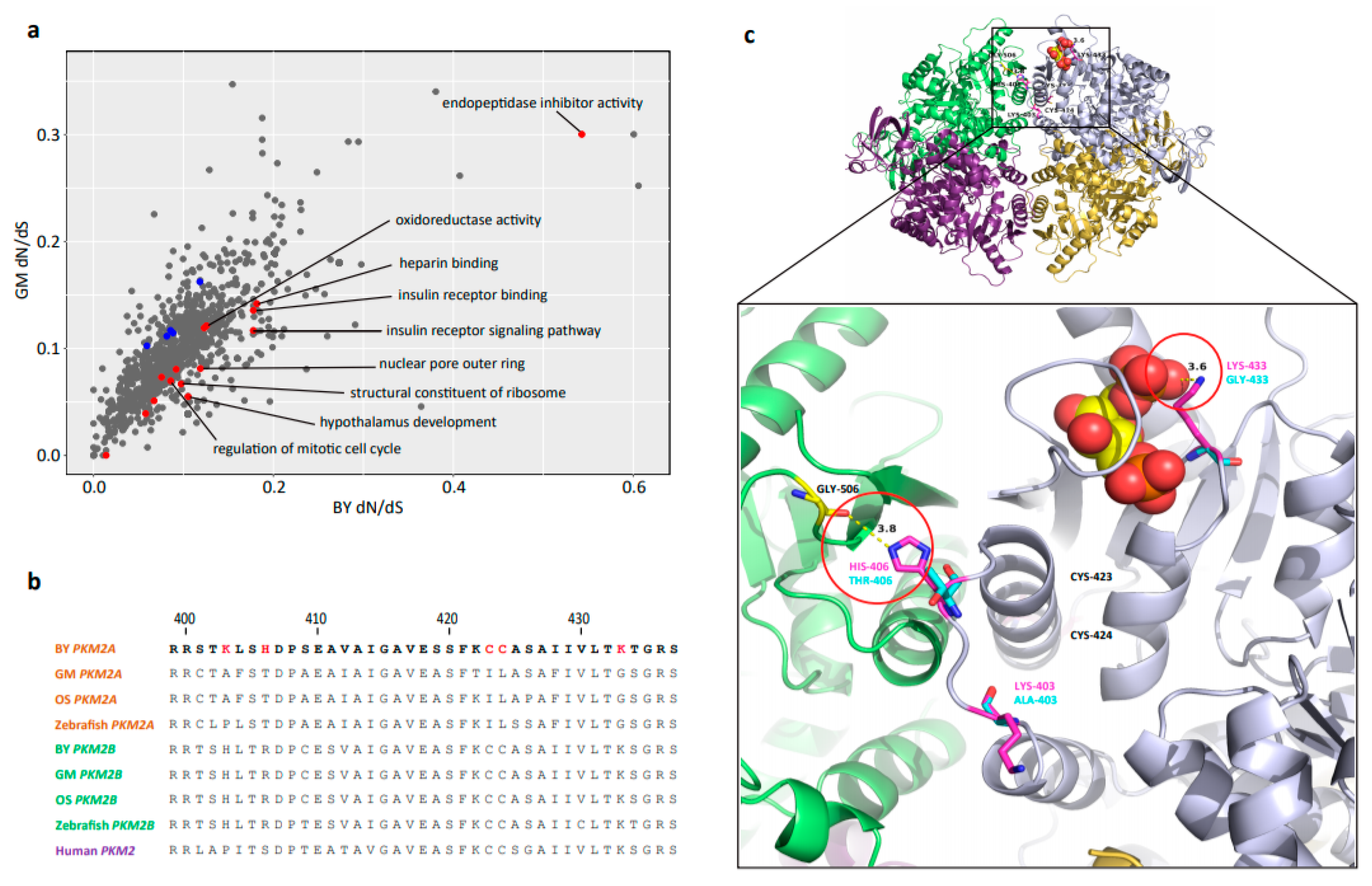
2.5. FEGs and PSGs Involved in BY
2.6. Positive Selection of PKM2A in BY
2.7. Screening for Genes of Interest and Candidate Genes
2.8. qRT-PCR Validation of Candidate Genes
3. Discussion
3.1. Multistep Functional Clusters Converging to Ribosome Biogenesis
3.2. Cell Proliferation Cluster and Metabolism Pathways
3.3. Glycolysis/Pyruvate Metabolic Process
3.4. Other Candidate Genes That May Influence the Large Body Size in BY
3.5. Habitat Preferences May be Implicated in Body Size Evolution of BY
4. Materials and Methods
4.1. Laboratory Preparation
4.2. Fish and Tissue Sampling
4.3. RNA Extraction and Sequencing
4.4. Transcriptome Assembly and Annotation
4.5. Identification of DEGs
4.6. GO Annotations and Pathway Enrichment Analysis
4.7. Identification of FEGs and PSGs
4.8. Quantitative Real-Time PCR (qRT-PCR)
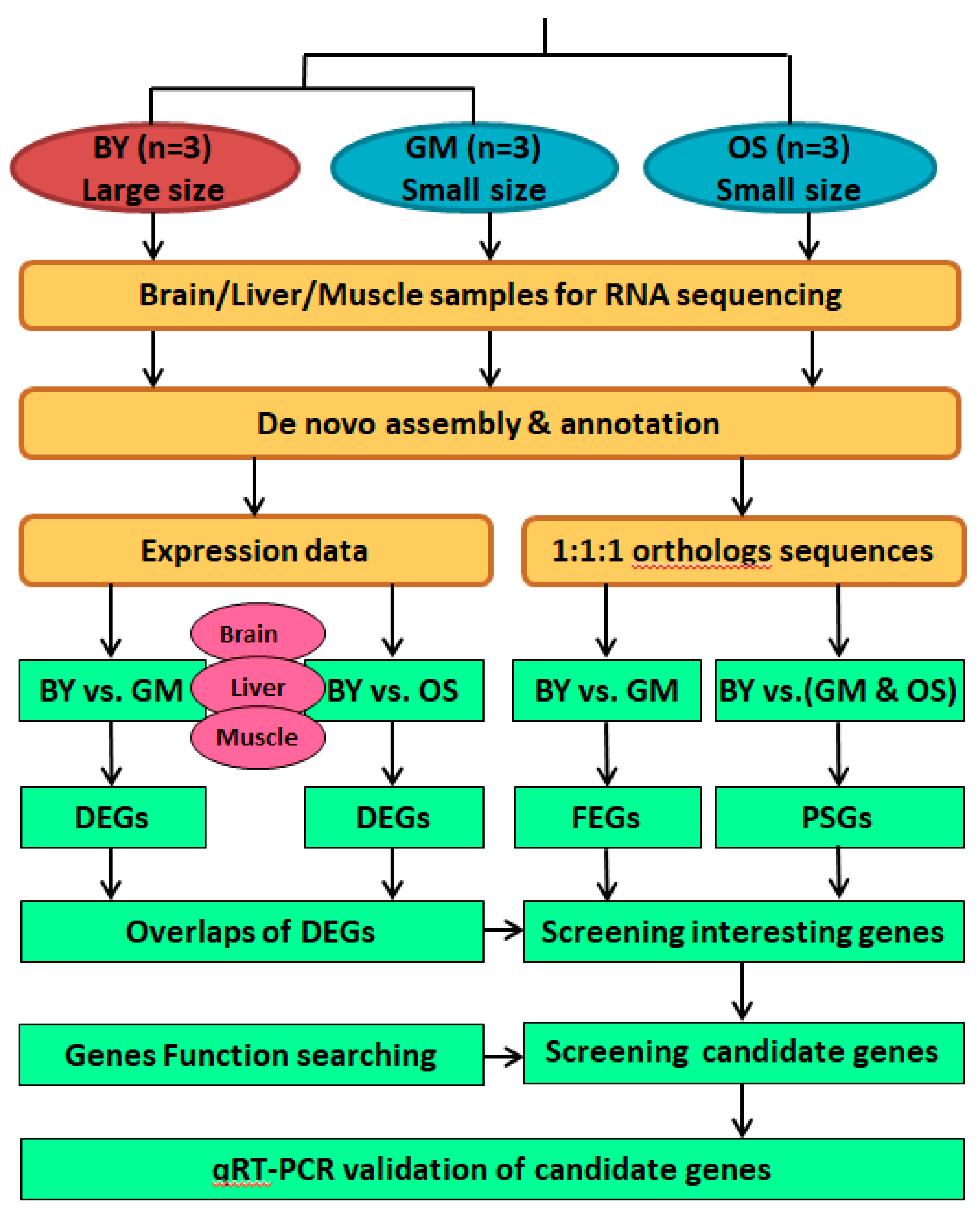
4.9. Overview of Comparative Transcriptome Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haldane, J.B.S. On being the right size. In A Treasury of Science; Shapely, H., Raffort, S., Wright, H., Eds.; Harper: New York, NY, USA, 1928; pp. 321–325. [Google Scholar]
- Feldman, A.; Sabath, N.; Pyron, R.A.; Mayrose, I.; Meiri, S. Body sizes and diversification rates of lizards, snakes, amphisbaenians and the tuatara. Glob. Ecol. Biogeogr. 2016, 25, 187–197. [Google Scholar] [CrossRef]
- Woodward, G.; Ebenman, B.; Emmerson, M.; Montoya, J.M.; Olesen, J.M.; Valido, A.; Warren, P.H. Body size in ecological networks. Trends Ecol. Evol. 2005, 20, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Nakano, Y.; Nagamatsu, Y.; Misumi, T.; Ohta, H.; Ohshima, Y. Cyclic GMP-dependent protein kinase EGL-4 controls body size and lifespan in C. elegans. Development 2003, 130, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, T. Molecular mechanism of size control in development and human diseases. Cell Res. 2011, 21, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Conlon, I.; Raff, M. Size Control in Animal Development. Cell 1999, 96, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.F.; Gumienny, T.L.; Gleason, R.J.; Wang, H.; Padgett, R.W. Regulation of genes affecting body size and innate immunity by the DBL-1/BMP-like pathway in Caenorhabditis elegans. BMC Dev. Biol. 2010, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Stocker, H.; Hafen, E. Genetic control of cell size. Curr. Opin. Genet. Dev. 2000, 10, 529–535. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Diniz-Filho, J.A.F.; Tôrres, N.M. Phylogenetic comparative methods and the geographic range size–body size relationship in new world terrestrial carnivora. Evol. Ecol. 2002, 16, 351–367. [Google Scholar] [CrossRef]
- Ng, H.H. Phylogenetic systematics of the Asian catfish family Sisoridae (Actinopterygii: Siluriformes). Ichthyol. Explor. Freshw. 2015, 26, 97–157. [Google Scholar]
- Thomson, A.W.; Page, L.M. Genera of the Asian catfish families Sisoridae and Erethistidae (Teleostei: Siluriformes). Zootaxa 2006, 1345, 1–96. [Google Scholar]
- Ng, H.H.; Jiang, W. Intrafamilial relationships of the Asian hillstream catfish family Sisoridae (Teleostei: Siluriformes) inferred from nuclear and mitochondrial DNA sequences. Ichthyol. Explor. Freshw. 2015, 26, 229–240. [Google Scholar]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, Y.; Hu, G.; Zhang, X.; Ruan, Z.; Zhao, X.; Guo, C.; Tang, Z.; Li, X.; You, X. Comparative transcriptomic study of muscle provides new insights into the growth superiority of a novel grouper hybrid. PLoS ONE 2016, 11, e0168802. [Google Scholar] [CrossRef] [PubMed]
- Penha, J.; Mateus, L.; Barbieri, G. Age and growth of the porthole shovelnose catfish (Hemisorubim platyrhynchos) in the Pantanal. Braz. J. Biol. 2004, 64, 833–840. [Google Scholar] [CrossRef]
- Penha, J.; Mateus, L.; Barbieri, G. Age and growth of the duckbill catfish (Sorubim cf. lima) in the Pantanal. Braz. J. Biol. 2004, 64, 125–134. [Google Scholar] [CrossRef]
- Kwak, T.J.; Waters, D.S.; Pine, W.E., III. Age, growth, and mortality of introduced flathead catfish in Atlantic rivers and a review of other populations. N. Am. J. Fish Manag. 2006, 26, 73–87. [Google Scholar] [CrossRef]
- Colombo, R.E.; Phelps, Q.E.; Miller, C.M.; Garvey, J.E.; Heidinger, R.C.; Richards, N.S. Comparison of channel catfish age estimates and resulting population demographics using two common structures. N. Am. J. Fish Manag. 2010, 30, 305–308. [Google Scholar] [CrossRef]
- Tian, Z.F.; Zhang, J.B.; Zhu, B.K.; Du, X.T. Comparison on age material of Mystus pluriradiatus in Yuanjiang River. Freshw. Fish. 2010, 40, 16–19. [Google Scholar]
- Yin, S.; Liu, M.; Tian, H.; Deng, H.; Jin, J.; Liu, S. Age Discrimination and Growth Characteristics of Clupisoma sinensis in Lancang River. Chin. J. Zool. 2011, 46, 12–21. [Google Scholar]
- Wang, H.-L. Studies on the Age and Growth of Liobagrus marginatus (Günther) in the Downstream of Jialing River. J. Anhui Agric. Sci. 2011, 15, 174. [Google Scholar]
- Shen, Y.; Pu, D.; Gao, M.; Wang, H.L.; Wang, Z.J. Studies on the relationship between the age and growth of Glyptothorax fukiensis in the Jialing River. J. Southwest Agric. Univ. 2005, 27, 106–110. [Google Scholar]
- Liu, Z.; Liu, S.; Yao, J.; Bao, L.; Zhang, J.; Li, Y.; Jiang, C.; Sun, L.; Wang, R.; Zhang, Y. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 2016, 7, 11757. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucl. Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Romero, I.G.; Ruvinsky, I.; Gilad, Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012, 13, 505. [Google Scholar] [CrossRef]
- Alvarez, M.; Schrey, A.W.; Richards, C.L. Ten years of transcriptomics in wild populations: What have we learned about their ecology and evolution? Mol. Ecol. 2015, 24, 710–725. [Google Scholar] [CrossRef]
- Elmer, K.R.; Fan, S.; Gunter, H.; Jones, J.; Boekhoff, S.; Kuraku, S.; Meyer, A. Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol. Ecol. 2010, 19, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Li, M.; Ding, F.; Ding, Y.; Ao, J.; Hu, S.; Chen, X. De novo characterization of the spleen transcriptome of the large yellow croaker (Pseudosciaena crocea) and analysis of the immune relevant genes and pathways involved in the antiviral response. PLoS ONE 2014, 9, e97471. [Google Scholar] [CrossRef]
- Coppe, A.; Pujolar, J.M.; Maes, G.E.; Larsen, P.F.; Hansen, M.M.; Bernatchez, L.; Zane, L.; Bortoluzzi, S. Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered European eel. BMC Genom. 2010, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Li, Q.; Zhou, B.; Song, G.; Li, T.; Cui, Z. De novo assembly of mud loach (Misgurnus anguillicaudatus) skin transcriptome to identify putative genes involved in immunity and epidermal mucus secretion. PLoS ONE 2013, 8, e56998. [Google Scholar] [CrossRef]
- Popovici, C.; Hiver, S.; Birnbaum, D.; Roubin, R. LET-756/FGF is Implicated in the Control of C. elegans Body Size. Open Cell Dev. Biol. J. 2008, 1, 24–32. [Google Scholar] [CrossRef]
- Sutter, N.B.; Bustamante, C.D.; Chase, K.; Gray, M.M.; Zhao, K.; Zhu, L.; Padhukasahasram, B.; Karlins, E.; Davis, S.; Jones, P.G. A single IGF1 allele is a major determinant of small size in dogs. Science 2007, 316, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Lempiäinen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Montanaro, L.; Derenzini, M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Cancer Res. 2012, 72, 1602–1607. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, M.S.; Kim, H.D.; Kim, J. Ribosomal protein S3 (rpS3) secreted from various cancer cells is N-linked glycosylated. Oncotarget 2016, 7, 80350. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Brandeis, M.; Rosewell, I.; Carrington, M.; Crompton, T.; Jacobs, M.A.; Kirk, J.; Gannon, J.; Hunt, T. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad. Sci. USA 1998, 95, 4344–4349. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, N.; Shum, L.; Fukumoto, S.; Nomura, T.; Ohishi, M.; Nonaka, K. Transforming growth factor-β3 promotes mesenchymal cell proliferation and angiogenesis mediated by the enhancement of cyclin D1, Flk-1, and CD31 gene expression during CL/Fr mouse lip fusion. Birth Defects Res. A 2005, 73, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.A.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Ito, M.; Sato, K.; Kuester, C.; Veit, K.; Tremp, G.; Liao, R.; Colucci, W.S.; Ivashchenko, Y.; Walsh, K. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J. Mol. Cell. Cardiol. 2005, 38, 375–385. [Google Scholar] [CrossRef]
- Faridi, J.; Fawcett, J.; Wang, L.; Roth, R.A. Akt promotes increased mammalian cell size by stimulating protein synthesis and inhibiting protein degradation. Am. J. Physiol.-Endoc. Matab. 2003, 285, E964–E972. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 336–342. [Google Scholar] [CrossRef]
- Chan, J.C.; Hannan, K.M.; Riddell, K.; Ng, P.Y.; Peck, A.; Lee, R.S.; Hung, S.; Astle, M.V.; Bywater, M.; Wall, M. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci. Signal. 2011, 4, ra56. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. C.S.H. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef]
- Rui, L. SH2B1 regulation of energy balance, body weight, and glucose metabolism. World J. Diabetes 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Cho, K.W.; Rui, L. Critical role of the Src homology 2 (SH2) domain of neuronal SH2B1 in the regulation of body weight and glucose homeostasis in mice. Endocrinology 2010, 151, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Desbuquois, B.; Carré, N.; Burnol, A.F. Regulation of insulin and type 1 insulin-like growth factor signaling and action by the Grb10/14 and SH2B1/B2 adaptor proteins. FEBS J. 2013, 280, 794–816. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, N.; Kose, S. Heat-shock stress activates a novel nuclear import pathway mediated by Hikeshi. Nucleus 2012, 3, 422–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kose, S.; Furuta, M.; Imamoto, N. Hikeshi, a nuclear import carrier for Hsp70s, protects cells from heat shock-induced nuclear damage. Cell 2012, 149, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Zheng, B.; Dai, D. Fauna Sinica (Osteichthyes, Siluriformes); Science Press: Beijing, China, 1999; p. 230. [Google Scholar]
- Leopold, L.B.; Wolman, M.G.; Miller, J.P. Fluvial Processes in Geomorphology; Dover Publications: New York, NY, USA, 1964. [Google Scholar]
- Benda, L.; Poff, N.L.; Miller, D.; Dunne, T.; Reeves, G.; Pess, G.; Pollock, M. The network dynamics hypothesis: How channel networks structure riverine habitats. AIBS Bull. 2004, 54, 413–427. [Google Scholar] [CrossRef]
- Lu, B.; Shen, F.; Tong, X.; Wang, D.; Yang, L. The correlation between fish diversity and hydrological parameters in Langcang-Mekong river and its upper tributaries. J. Kunming Univ. 2010, 32, 58–61. [Google Scholar]
- Schermerhorn, T. Normal glucose metabolism in carnivores overlaps with diabetes pathology in non-carnivores. Front. Endocrinol. 2013, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-J.; Xie, C.-X. Age and growth of the Tibetan catfish Glyptosternum maculatum in the Brahmaputra River, China. Zool. Stud. 2008, 47, 555–563. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Prot. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Prot. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Gene Name † | Main Functional Keywords | BY vs. GM | BY vs. OS | ||||
|---|---|---|---|---|---|---|---|---|
| Brain | Liver | Muscle | Brain | Liver | Muscle | |||
| 1 | AKT3 | multi-functional protein kinases | 2.71 | 2.46 | 3.6 | 2.07 § | 4.31 | 3.33 |
| 2 | SH2B1 | multi-functional adapter protein | 10.97 | 2.38 | 2.74 | 10.47 | 2.72 | 10.82 |
| 3 | RPS3 | multi-functional ribosomal protein | 18.09 | 16.3 | 8.01 | 17.59 | 16.07 | 16.84 |
| 4 | HSP70.2 | heat shock protein in wide processes | 1.63 | 2.14 | 4.65 | 0.91 § | 2.41 | 2.55 |
| 5 | HIKESHI | HSP70 carrier for heat-shock stress | 2.69 | 0.97 § | 2.67 | 2.64 | 2.02 | 2.16 |
| 6 | HNRNPD | telomere elongation | 6.64 | 4.94 | 5.07 | 11.02 | 7.39 | 5.92 |
| 7 | RINT1 | telomere length control | 5.92 | 2.19 | 2.9 | 3.64 | 2.29 | 3.82 |
| 8 | NOX5 | cell growth and apoptosis | 6.69 | 5.85 | 6.44 § | 5.86 | 3.26 | 0.96 § |
| 9 | IMPDH2 | regulation of cell growth | 6.32 | 1.39 § | 3.27 | 5.19 | 3.43 | 2.58 |
| 10 | PCGF1 | cycle progression and cell proliferation | 2.76 | 2.84 § | 3.34 | 7.21 | 1.21 § | 5.05 |
| 11 | NRF1 | cellular growth and development | 1.04 § | 1.46 | 1.99 | 3.25 | 3.43 | 3.5 |
| 12 | TRAF4 | cell survival and apoptosis | 3.07 | 11.79 | 7.95 | 5.54 | 4.1 | 4.44 |
| 13 | USP9X | neuronal cell migration and growth | 3.59 | 3.44 | 3.12 | 1.5 | 1.14 | 0.88 § |
| 14 | TFB2M | mitochondrial transcription factor | 5.47 | 4.98 | 5 | 3.14 | 2.59 | 3.58 |
| 15 | TFDP2 | cell-cycle progression | 9.08 | 2.01 | 1.50 § | 3.17 | 1.78 | 2.21 § |
| 16 | ACHE | growth factor like actions | 2.57 | 0.44 § | 0.40 § | 3.80 | 0.98 § | 1.63 § |
| 17 | PKM2A‡ | glycolytic rate-limiting pyruvate kinase | 2.49 | 0.21 | 1.81 § | −0.65 § | -0.82 § | −0.38 § |
| 18 | TGFβ3‡ | cell growth and differentiation | 0.29 § | −0.11 § | −0.22 § | 1.43 § | -0.07 § | 0.01 § |
| 19 | CCNB2‡ | regulation of cell cycle | −1.57 § | −0.10 § | −4.00 | −2.64 § | 1.28 § | −2.66 § |
| 20 | HK2‡ | hexose metabolism | −3.57 | −2.89 | −4.01 | −0.98 § | −1.44 § | −0.17 § |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Guo, Y.; Yang, K.; Shi, Q.; Yang, J. Insights into Body Size Evolution: A Comparative Transcriptome Study on Three Species of Asian Sisoridae Catfish. Int. J. Mol. Sci. 2019, 20, 944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040944
Jiang W, Guo Y, Yang K, Shi Q, Yang J. Insights into Body Size Evolution: A Comparative Transcriptome Study on Three Species of Asian Sisoridae Catfish. International Journal of Molecular Sciences. 2019; 20(4):944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040944
Chicago/Turabian StyleJiang, Wansheng, Yicheng Guo, Kunfeng Yang, Qiong Shi, and Junxing Yang. 2019. "Insights into Body Size Evolution: A Comparative Transcriptome Study on Three Species of Asian Sisoridae Catfish" International Journal of Molecular Sciences 20, no. 4: 944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040944