Biosynthesis and Signal Transduction of ABA, JA, and BRs in Response to Drought Stress of Kentucky Bluegrass

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Sequence Analysis and Transcript Assembly
2.2. Sequence Annotation
2.3. DEGs under Drought Stress
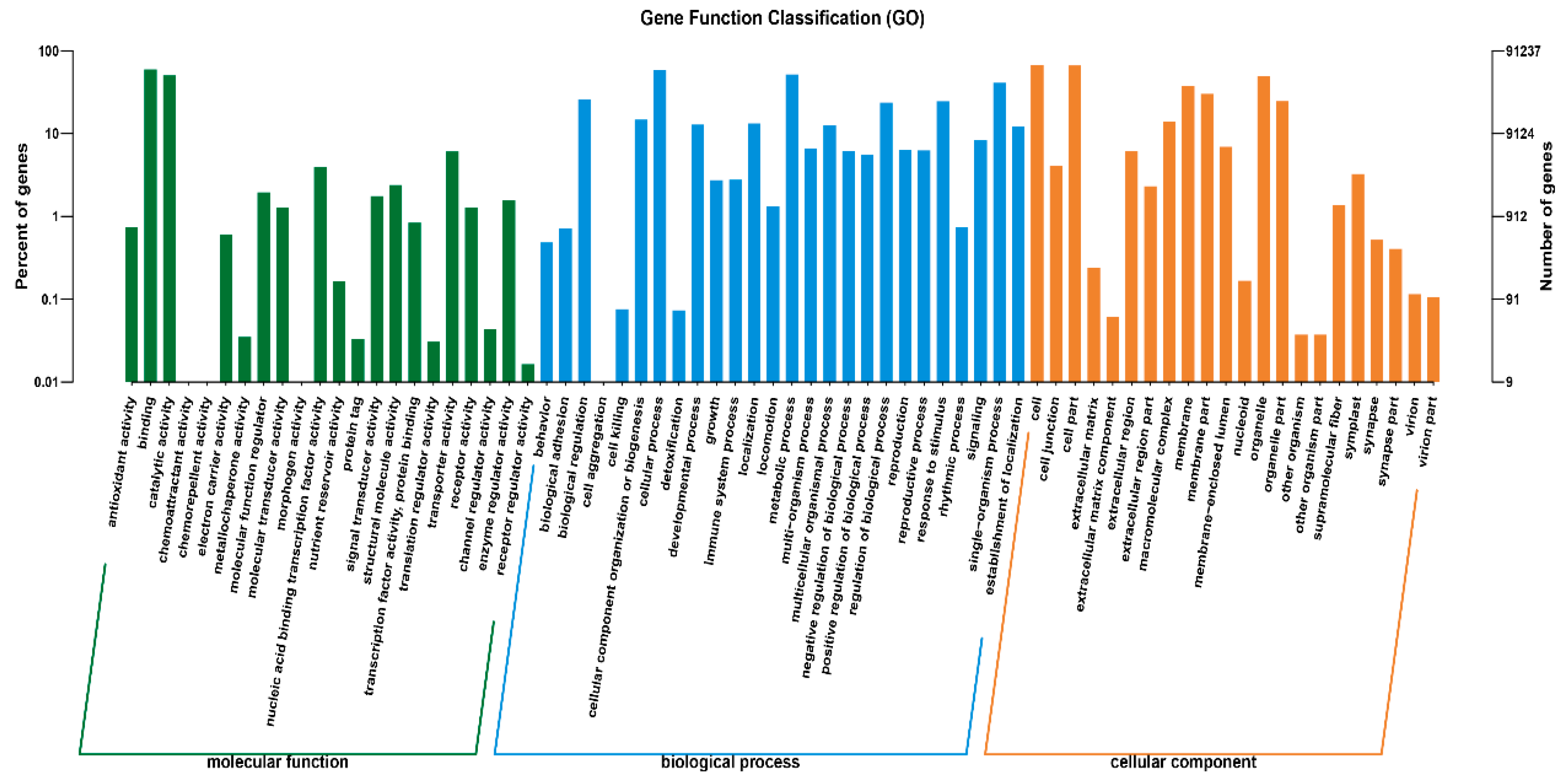
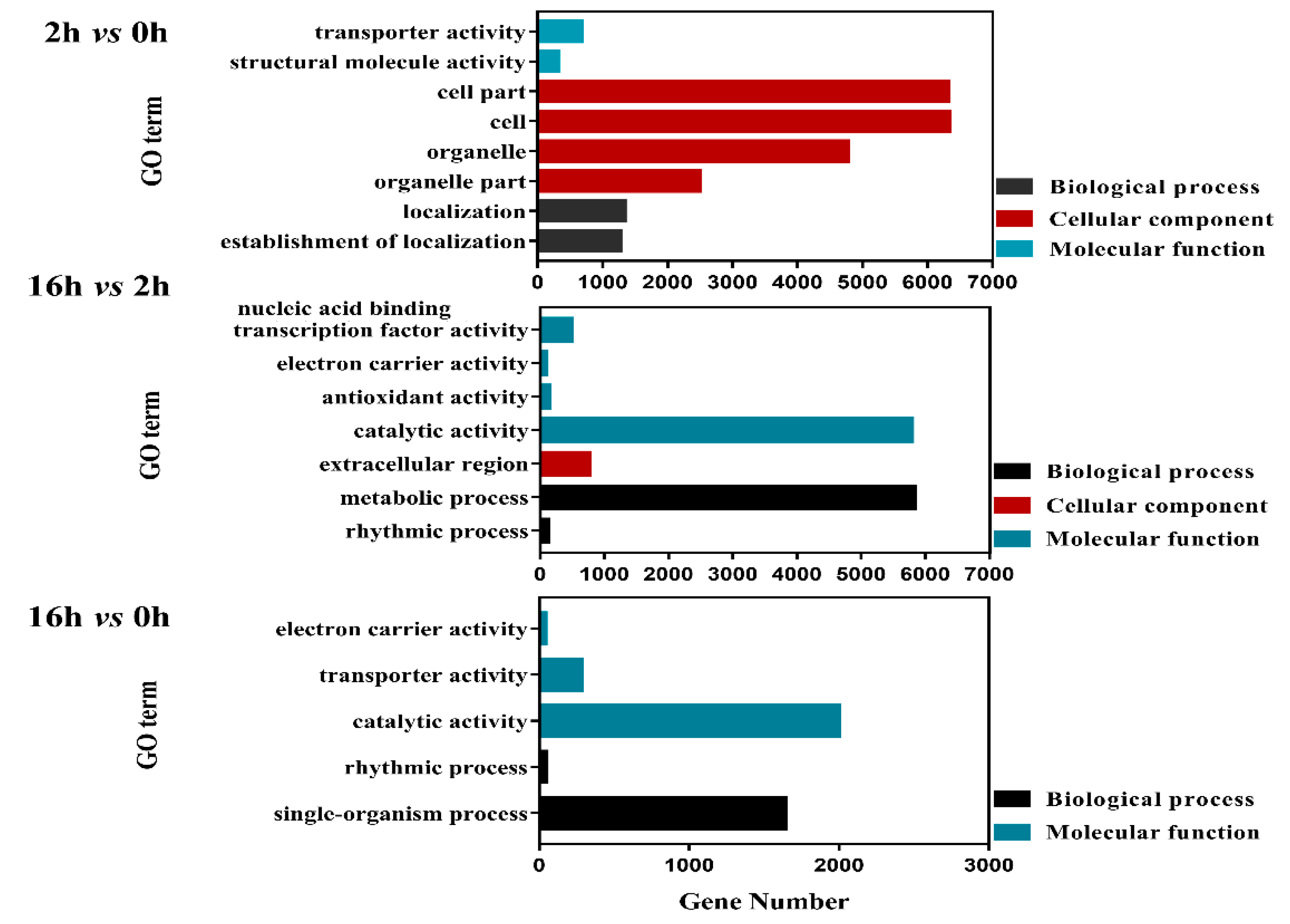
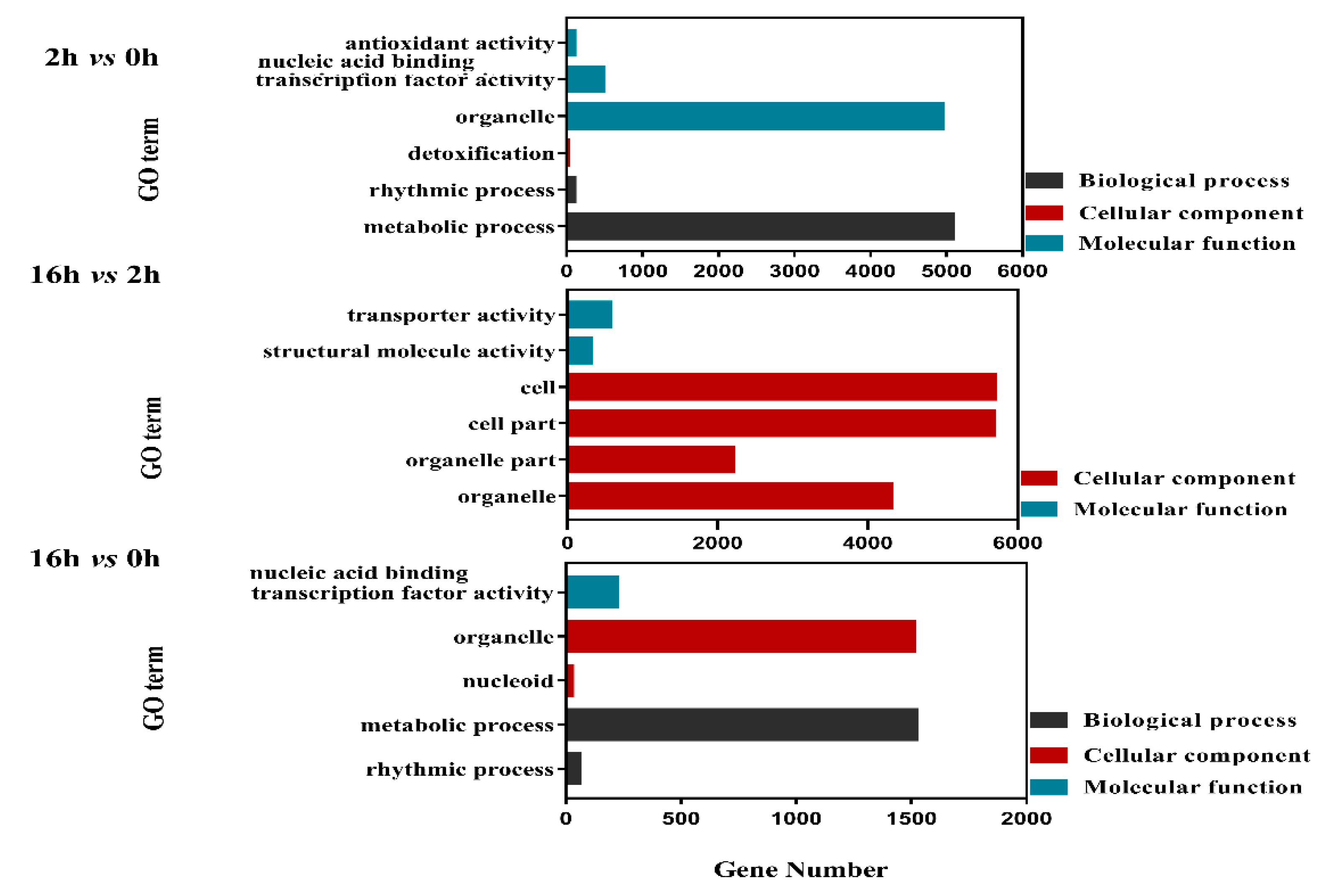
2.4. GO Analysis of DEGs
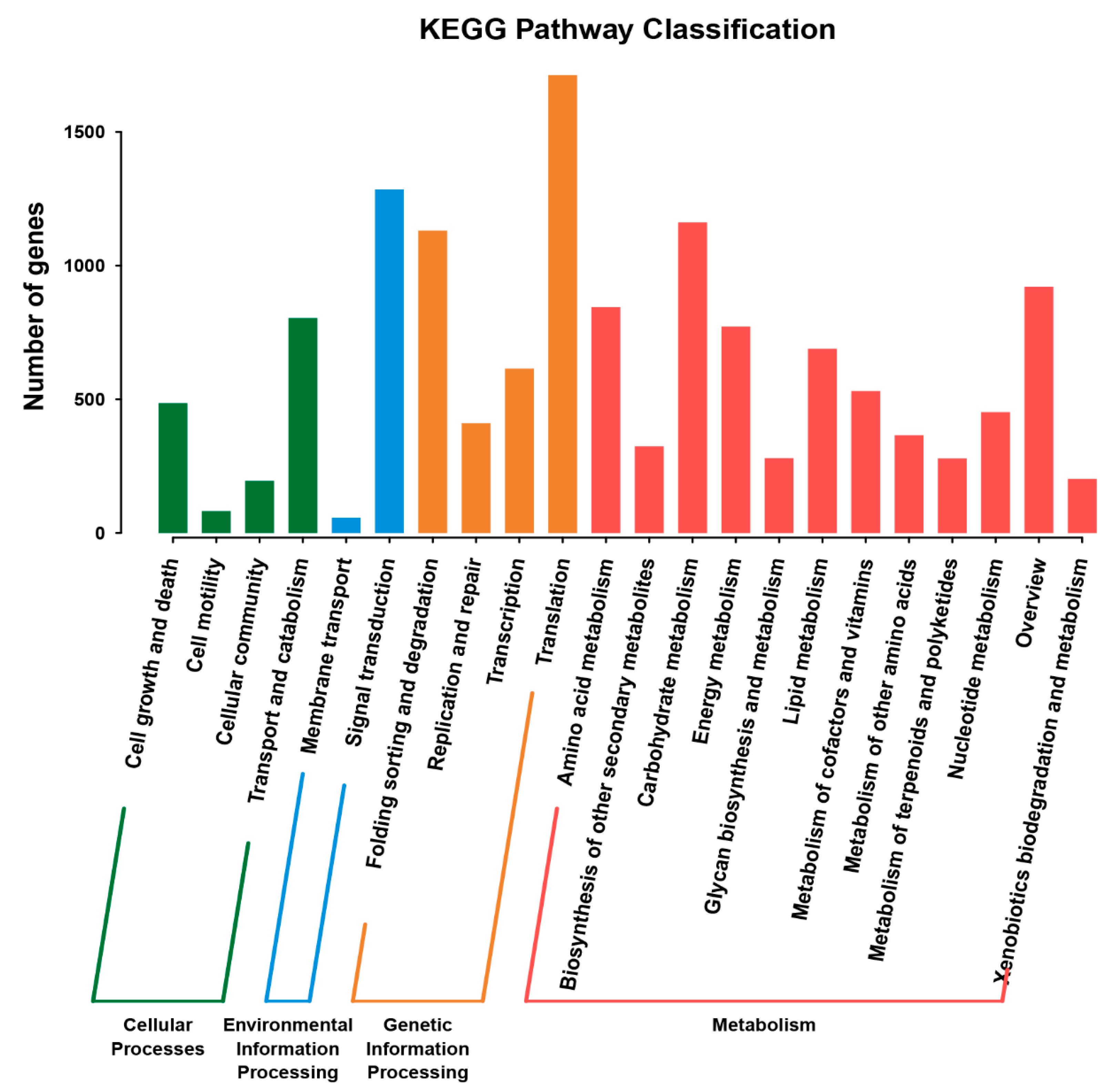
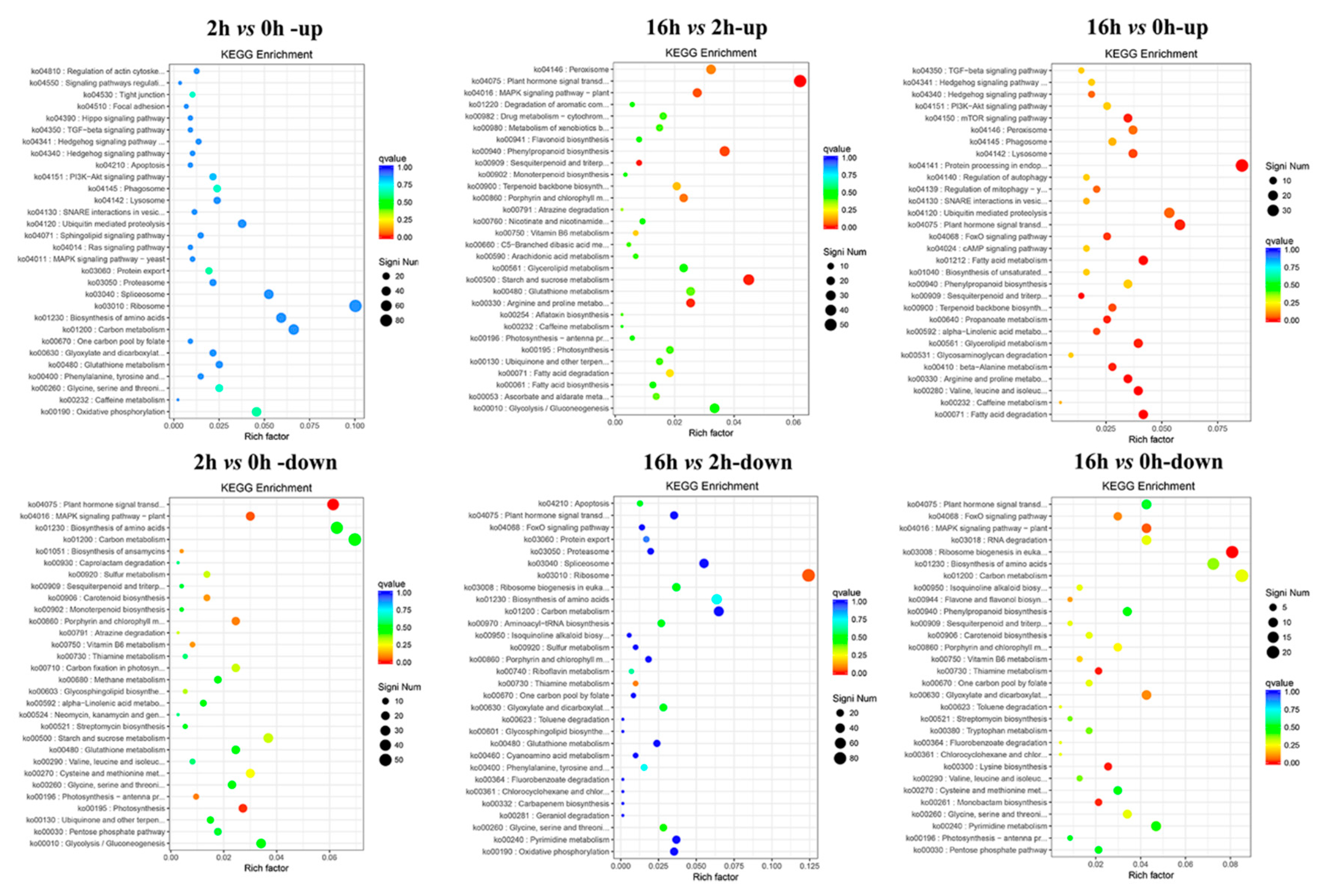
2.5. KEGG Pathway Analysis of DEGs
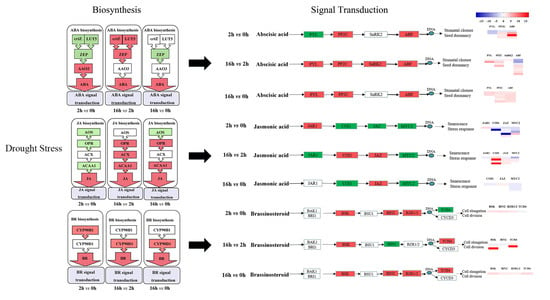
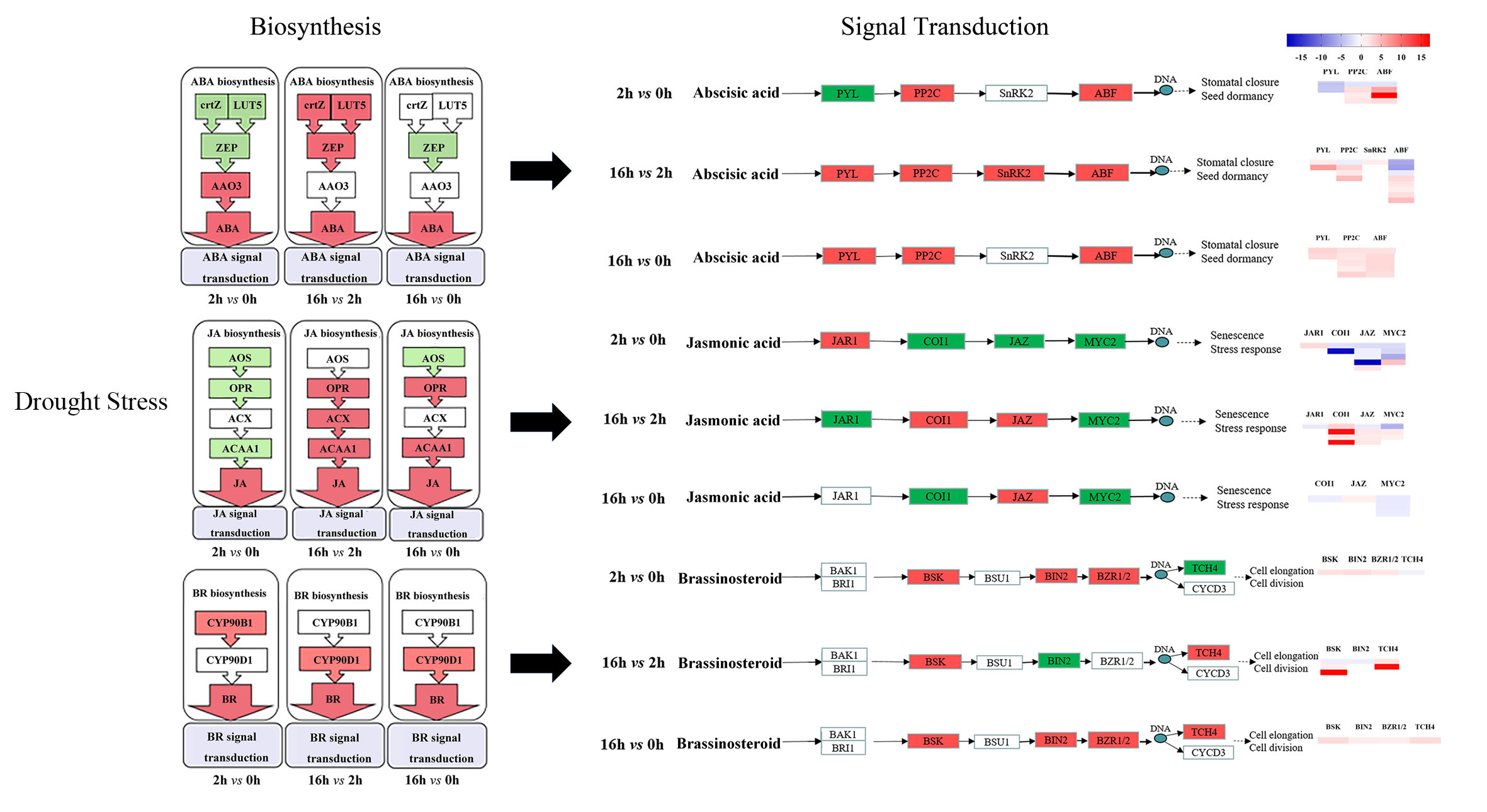
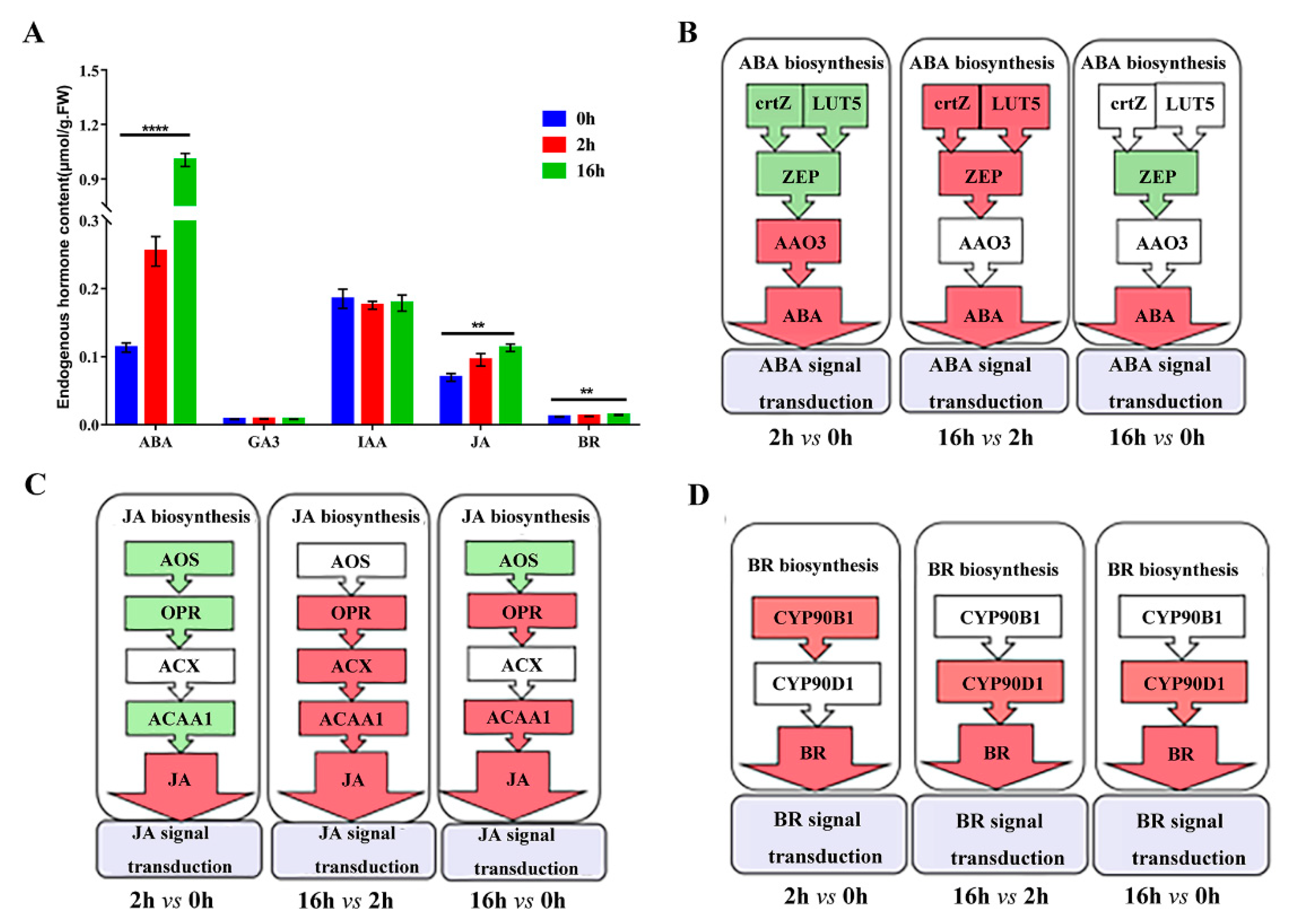
2.6. DEGs Related to Plant Hormone Biosynthesis and Signal Transduction
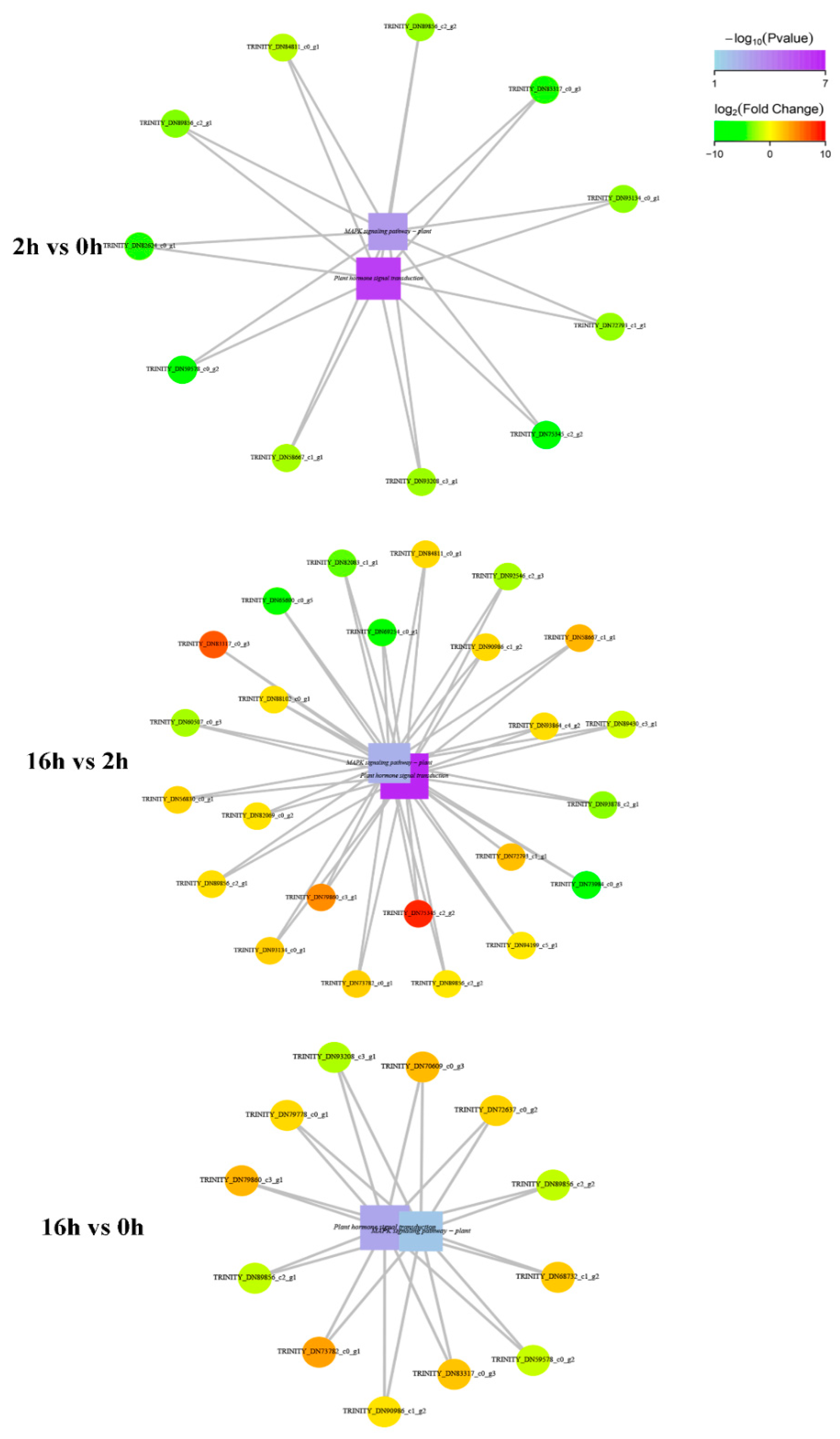
2.7. MAPK Signaling Transduction and Interaction with Hormonal Signaling Transduction
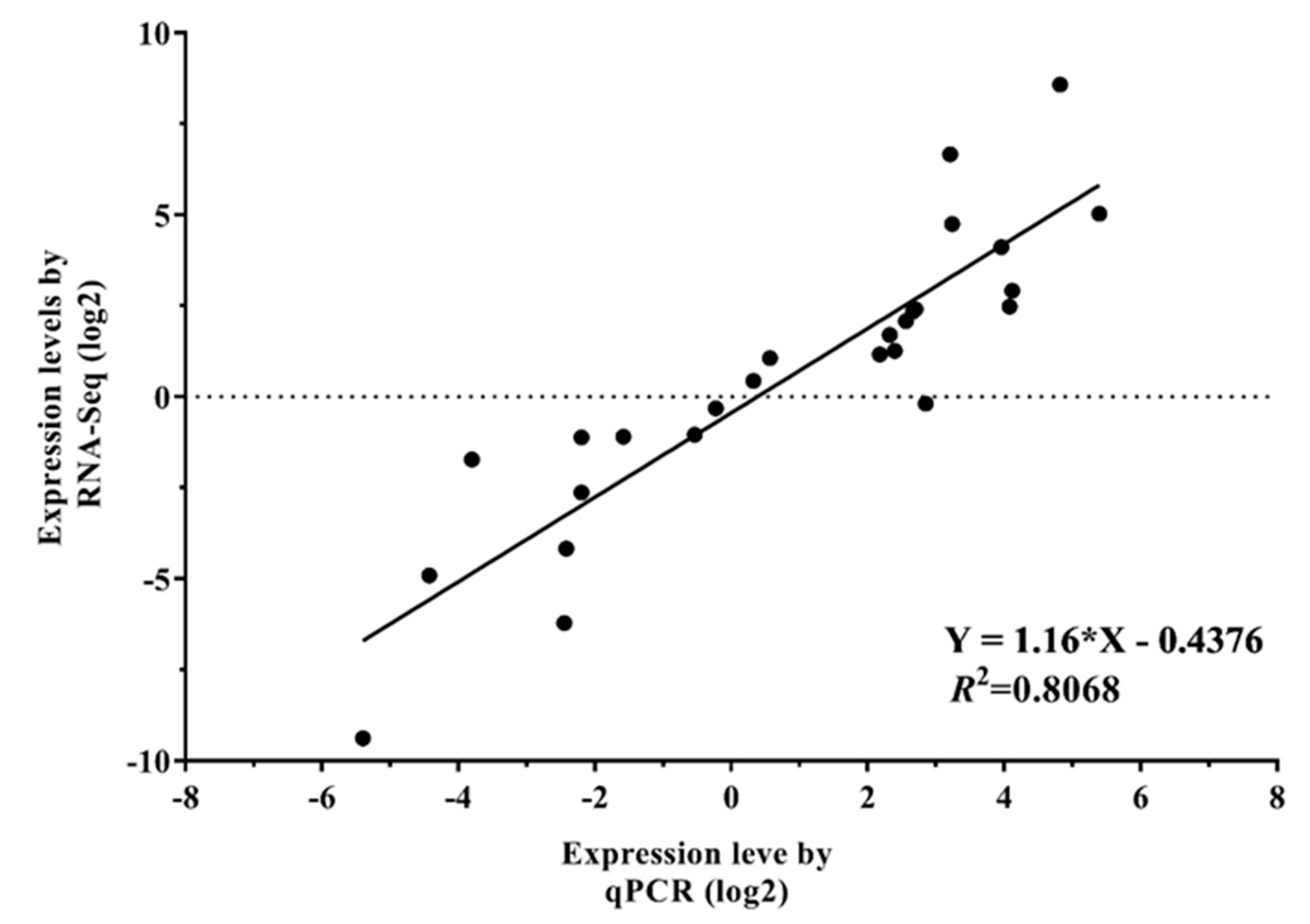
2.8. Validation of RNA-Seq Data by qRT-PCR
2.9. Effects of Drought Stress on Endogenous Hormones
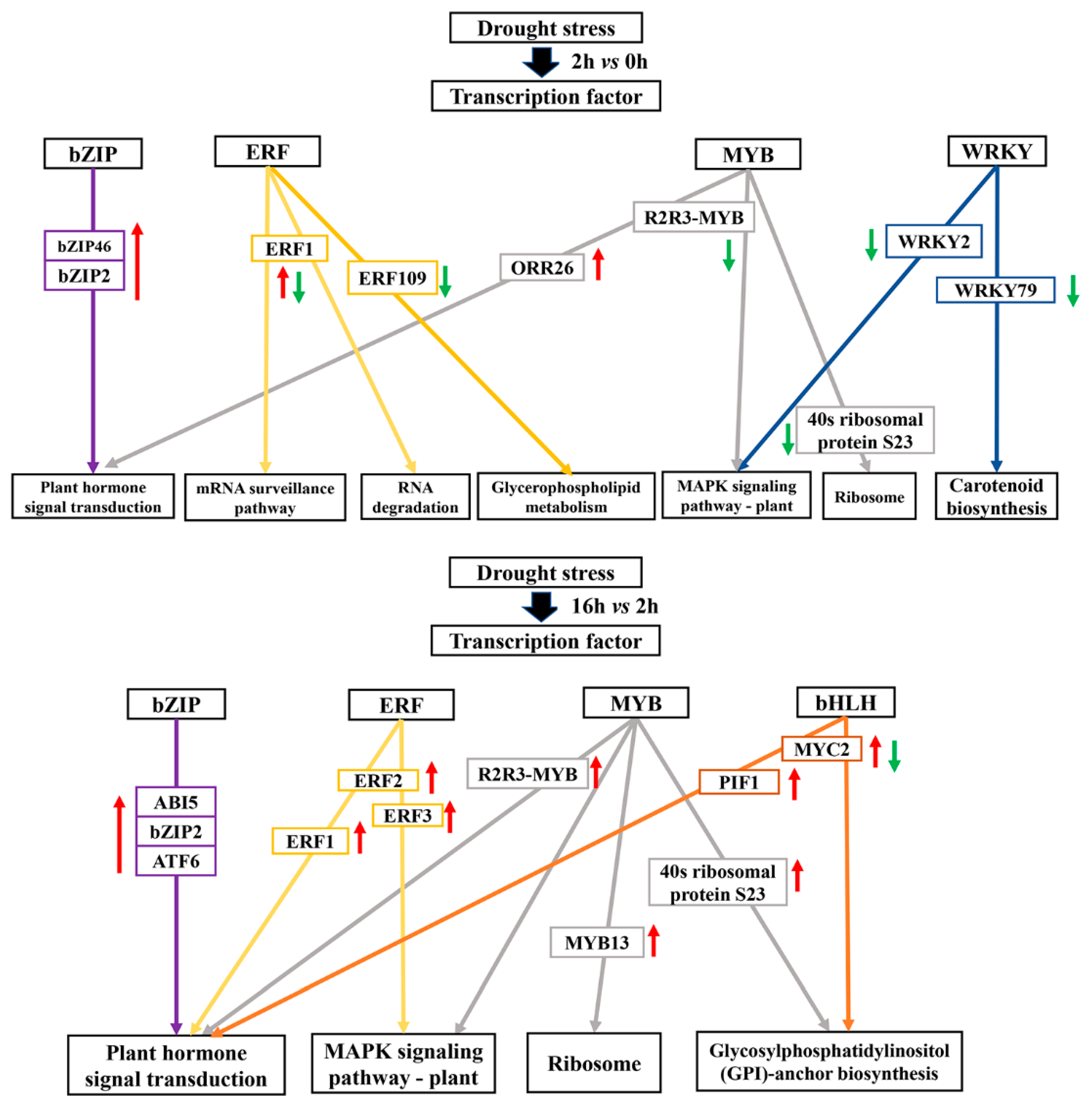
2.10. Transcription Factors Associated with Drought Tolerance
3. Discussion
3.1. Hormone Biosynthesis
3.2. Hormone Signaling Transduction
3.3. MAPK Signaling Transduction and Interaction with Hormones
3.4. Transcription Factors Associated with Drought Tolerance
4. Materials and Methods
4.1. Plant Materials and Treatment
4.2. RNA Extraction, cDNA Library Construction and Sequencing
4.3. De novo Assembly
4.4. Gene Function Annotation
4.5. Quantification of Gene Expression Levels
4.6. Gene Ontology and KEGG Pathway Enrichment Analysis
4.7. Validation of Transcripts by Quantitative Real-Time PCR (qRT-PCR)
4.8. Measurement of Endogenous Hormones
5. Conclusions
Supplementary Materials
Data Availability Statement
Author Contributions
Funding
Conflicts of Interest
References
- Xu, L.; Han, L.; Huang, B. Antioxidant enzyme activities and gene expression patterns in leaves of Kentucky bluegrass in response to drought and post-drought recovery. J. Am. Soc. Hortic. Sci. 2011, 136, 247–255. [Google Scholar] [CrossRef]
- Matzk, F.; Prodanovic, S.; Bäumlein, H.; Schubert, I. The inheritance of apomixis in Poa pratensis confirms a five locus model with differences in gene expressivity and penetrance. Plant Cell 2005, 17, 13–24. [Google Scholar] [CrossRef]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought stress responses in crops. Funct. Integr. Genom. 2014, 14, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, H.; Cline, V. Correlations of leaf relative water content, canopy temperature, and spectral reflectance in perennial ryegrass under water deficit conditions. Hortscience 2009, 44, 459–462. [Google Scholar] [CrossRef]
- Zhou, Y.; Lambrides, C.J.; Kearns, R.; Ye, C.; Fukai, S. Water use, water use efficiency and drought resistance among warm-season turfgrasses in shallow soil profiles. Funct. Plant Biol. 2012, 39, 116–125. [Google Scholar] [CrossRef]
- Hu, L.; Wang, Z.; Huang, B. Effects of cytokinin and potassium on stomatal and photosynthetic recovery of Kentucky bluegrass from drought stress. Crop Sci. 2013, 53, 221–231. [Google Scholar] [CrossRef]
- Bazargani, M.M.; Sarhadi, E.; Bushehri, A.A.S.; Matros, A.; Mock, H.P.; Naghavi, M.R.; Hajihoseini, V.; Mardia, M.; Hajirezaei, M.R.; Moradid, F.; et al. A proteomics view on the role of drought-induced senescence and oxidative stress defense in enhanced stem reserves remobilization in wheat. J. Proteom. 2011, 74, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Chen, Y.J.; Shah, F.; Saddam, H.; Li, N.; Li, X.; Safa, A.A.F.E.A. Silicate application increases the photosynthesis and its associated metabolic activities in Kentucky bluegrass under drought stress and post-drought recovery. Environ. Sci. Pollut. Res. 2016, 23, 17647–17655. [Google Scholar]
- Shah, S.; Fahad, S.; Chen, Y.; Ihsan, M.Z.; Hammad, H.M.; Nasim, W.; Amanullah, J.; Muhammad, A.; Hesham, A. Effects of nitrogen supply on water stress and recovery mechanisms in Kentucky bluegrass plants. Front. Plant Sci. 2017, 8, 983. [Google Scholar]
- Krishnan, S.; Merewitz, E.B. Drought stress and trinexapac-ethyl modify phytohormone content within Kentucky bluegrass leaves. J. Plant Growth Regul. 2015, 34, 1–12. [Google Scholar] [CrossRef]
- Teng, K.; Li, J.; Liu, L.; Han, Y.; Du, Y.; Zhang, J. Exogenous ABA induces drought tolerance in upland rice: The role of chloroplast and ABA biosynthesis-related gene expression on photosystem II during PEG stress. Acta Physiol. Plant. 2014, 36, 2219–2227. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Yin, Z.; Jiang, J.; Zhang, M.; Guo, X.; Ye, Z.; Zhao, Y.; Xiong, H.; Zhang, Z.; et al. OsASR5 enhances drought tolerance through a stomatal closure pathway associated with ABA and H2O2 signaling in rice. Plant Biotechnol. J. 2017, 15, 183–196. [Google Scholar] [CrossRef]
- Bian, S.; Jiang, Y. Reactive oxygen species, antioxidant enzyme activities and gene expression patterns in leaves and roots of Kentucky bluegrass in response to drought stress and recovery. Sci. Hortic. 2009, 120, 264–270. [Google Scholar] [CrossRef]
- Qin, D.D.; Wu, H.Y.; Peng, H.R.; Yao, Y.Y.; Ni, Z.F.; Li, Z.X.; Zhou, C.L.; Sun, Q.X. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genom. 2008, 9, 432. [Google Scholar] [CrossRef]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2015, 9, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R. Identification of drought-tolerant mechanisms in Maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef]
- Surget-Groba, Y.; Montoya-Burgos, J. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Kakimoto, M.; Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Tran, L.S. Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 2010, 50, 54–69. [Google Scholar] [CrossRef]
- Bechtold, U.; Albihlal, W.S.; Lawson, T.; Fryer, M.J.; Sparrow, P.A.C.; Richard, F. Arabidopsis Heat shock transcription factor 1b overexpression enhances water productivity, resistance to drought, and infection. J. Exp. Bot. 2013, 64, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef]
- Liu, W.C.; Han, T.T.; Yuan, H.M.; Yu, Z.D.; Zhang, L.Y.; Zhang, B.L.; Zhai, S.; Zheng, S.Q.; Lu, Y.T. CATALASE2 functions for seedling post-germinative growth by scavenging H2O2 and stimulating ACX2/3 activity in Arabidopsis. Plant Cell Environ. 2017, 40, 2720–2728. [Google Scholar] [CrossRef]
- Zhao, W.; Cheng, Y.H.; Zhang, C.; Shen, X.J.; You, Q.B.; Guo, W. Genome-wide identification and characterization of the GmSnRK2 family in soybean. Int. J. Mol. Sci. 2017, 18, 1834. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zheng, X.; Song, Y.; Zhu, L.; Yu, Z.; Gan, L. NtLTP4, a lipid transfer protein that enhances salt and drought stresses tolerance in Nicotiana tabacum. Sci. Rep. 2018, 8, 8873. [Google Scholar] [CrossRef]
- Hu, L.X.; Li, H.Y.; Chen, L.; Lou, Y.H.; Amombo, E.; Fu, J.M. RNA-seq for gene identification and transcript profiling in relation to root growth of bermudagrass (Cynodon dactylon) under salinity stress. BMC Genom. 2015, 16, 575. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, B.R. Transcriptomic analysis reveals unique molecular factors for lipid hydrolysis, secondary cell-walls and oxidative protection associated with thermotolerance in perennial grass. BMC Genom. 2018, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Bushman, B.S.; Amundsen, K.L.; Warnke, S.E.; Robins, J.G.; Johnson, P.G. Transcriptome profiling of Kentucky bluegrass (Poa pratensis L.) accessions in response to salt stress. BMC Genom. 2016, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kuang, T.; He, Y. Conservation between higher plants and the moss Physcomitrella patens in response to the phytohormone abscisic acid: A proteomics analysis. BMC Plant Biol. 2010, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef]
- Haider, M.S.; Zhang, C.; Kurjogi, M.M.; Pervaiz, T.; Zheng, T.; Zhang, C.B.; Lide, C.; Shangguan, L.; Fang, F. Insights into grapevine defense response against drought as revealed by biochemical, physiological and RNA-Seq analysis. Sci. Rep. 2017, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Tůmová, L.; Tarkowská, D.; Řehořová, K.; Marková, H.; Kočová, M.; Rothová, O. Drought-tolerant and drought-sensitive genotypes of maize (Zea mays L.) differ in contents of endogenous brassinosteroids and their drought-induced changes. Front. Plant Sci. 2018, 13, e0197870. [Google Scholar] [CrossRef]
- Man, D.; Bao, Y.X.; Han, L.B.; Zhang, X. Drought tolerance associated with proline and hormone metabolism in two tall fescue cultivars. Hortscience 2011, 46, 1027–1032. [Google Scholar] [CrossRef]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D”Angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Xue, S.; Luo, Y.; Tian, B.; Fang, H.; Li, H. Hydrogen sulfide interacting with abscisic acid in stomatal regulation responses to drought stress in Arabidopsis. Plant Physiol. Biochem. 2013, 62, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Seok, H.Y.; Park, B.K.; Kim, S.H.; Goh, C.H.; Lee, B.H. Overexpression of Arabidopsis ZEP enhances tolerance to osmotic stress. Biochem. Biophys. Res. Commun. 2008, 375, 80–85. [Google Scholar] [CrossRef] [PubMed]
- De, S.D.; Bonsegna, S.; Horres, R.; Pastor, V.; Taurino, M.; Poltronieri, P. Transcriptomic analysis of oxylipin biosynthesis genes and chemical profiling reveal an early induction of jasmonates in chickpea roots under drought stress. Plant Physiol. Biochem. 2012, 61, 115–122. [Google Scholar] [Green Version]
- Fariduddin, Q.; Khanam, S.; Hasan, S.A.; Ali, B.; Hayat, S.; Ahmad, A. Effect of 28-homobrassinolide on the drought stress-induced changes in photosynthesis and antioxidant system of Brassica juncea L. Acta Physiol. Plant. 2009, 31, 889–897. [Google Scholar] [CrossRef]
- Yuan, G.F.; Jia, C.G.; Li, Z.; Sun, B.; Zhang, L.P.; Liu, N. Effect of brassinosteroids on drought resistance and abscisic acid concentration in tomato under water stress. Sci. Hortic. 2010, 126, 103–108. [Google Scholar] [CrossRef]
- Ohnishi, T.; Szatmari, A.M.; Watanabe, B.; Fujita, S.; Bancos, S.; Koncz, C.; Lafos, M.; Shibata, K.; Yokota, T.; Sakata, K.; et al. C-23 hydroxylation by Arabidopsis CYP90C1 and CYP90D1 reveals a novel shortcut in brassinosteroid biosynthesis. Plant Cell 2006, 18, 3275–3288. [Google Scholar] [CrossRef]
- Sahni, S.; Prasad, B.D.; Liu, Q.; Grbic, V.; Sharpe, A.; Singh, S.P.; Krishna, P. Overexpression of the brassinosteroid biosynthetic gene DWF4 in Brassica napus simultaneously increases seed yield and stress tolerance. Sci. Rep. 2016, 6, 28298. [Google Scholar] [CrossRef]
- Coello, P.; Hey, S.J.; Halford, N.G. The sucrose non-fermenting-1-related (SnRK) family of protein kinases: Potential for manipulation to improve stress tolerance and increase yield. J. Exp. Bot. 2011, 62, 883–893. [Google Scholar] [CrossRef]
- Nishimura, N.; Sarkeshik, A.; Nito, K.; Park, S.Y.; Wang, A.; Carvalho, P.C. PYR/PYL/RCAR family members are major in-vivo ABI1 protein phosphatase 2C-interacting proteins in Arabidopsis. Plant J. Cell Mol. Biol. 2010, 61, 290–299. [Google Scholar] [CrossRef]
- Han, Y.L.; Jang, G.; Um, T.; Kim, J.K.; Lee, J.S.; Yang, D.C. The soluble ABA receptor PYL8 regulates drought resistance by controlling ABA signaling in Arabidopsis. Plant Biotechnol. Rep. 2015, 9, 319–330. [Google Scholar]
- Yoshida, T.; Fujita, Y.; Maruyama, K.; Mogami, J.; Todaka, D.; Shinozaki, K. Four Arabidopsis AREB/ABF transcription factors function predominantly in gene expression downstream of SnRK2 kinases in abscisic acid signaling in response to osmotic stress. Plant Cell Environ. 2015, 38, 35–49. [Google Scholar] [CrossRef]
- Cao, M.J.; Zhang, Y.L.; Liu, X.; Huang, H.; Zhou, X.E.; Wang, W.L.; Zeng, A.; Zhao, C.Z.; Si, T.; Du, J.M. Combining chemical and genetic approaches to increase drought resistance in plants. Nat. Commun. 2017, 8, 12. [Google Scholar] [CrossRef]
- Shen, X.; Guo, X.; Zhao, D.; Zhang, Q.; Jiang, Y.; Wang, Y. Cloning and expression profiling of the PacSnRK2 and PacPP2C gene families during fruit development, ABA treatment, and dehydration stress in sweet cherry. Plant Physiol. Biochem. 2017, 119, 275–285. [Google Scholar] [CrossRef]
- Fan, W.; Zhao, M.; Li, S.; Bai, X. Contrasting transcriptional responses of PYR1/PYL/RCAR ABA receptors to ABA or dehydration stress between maize seedling leaves and roots. BMC Plant Biol. 2016, 16, 99. [Google Scholar] [CrossRef]
- Dalal, M.; Inupakutika, M. Transcriptional regulation of ABA core signaling component genes in sorghum (Sorghum bicolor L. Moench). Mol. Breed. 2014, 34, 1517–1525. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Min, M.K.; Choi, E.H.; Kim, N.; Moon, S.J.; Yoon, I. The protein phosphatase 2C clade A protein OsPP2C51 positively regulates seed germination by directly inactivating OsbZIP10. Plant Mol. Biol. 2017, 93, 389–401. [Google Scholar] [CrossRef]
- Yoshida, R.; Hobo, T.; Ichimura, K.; Mizoguchi, T.; Takahashi, F.; Aronso, J.; Ecker, J.R.; Shinozaki, K. ABA-activated SnRK2 protein kinase is required for dehydration stress signaling in Arabidopsis. Plant Cell Physiol. 2002, 43, 1473–1483. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.P.; Chen, P.; Ren, J.; Ji, K.; Li, Q. Transcriptional regulation of SlPYL, SlPP2C, and SlSnRK2 gene families encoding ABA signal core components during tomato fruit development and drought stress. J. Exp. Bot. 2011, 62, 5659–5669. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Yoshida, T.; Yamaguchi-Shinozaki, K. Pivotal role of the AREB/ABF-SnRK2 pathway in ABRE-mediated transcription in response to osmotic stress in plants. Physiol. Plant. 2013, 147, 15–27. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.H.; Yin, H.; Wang, W.X.; Zhao, X.M.; Du, Y.G. Alginate oligosaccharides enhanced Triticum aestivum L. tolerance to drought stress. Plant Physiol. Biochem. 2013, 62, 33–40. [Google Scholar] [CrossRef]
- Maksup, S.; Roytrakul, S.; Supaibulwatana, K. Physiological and comparative proteomic analyses of Thai jasmine rice and two check cultivars in response to drought stress. J. Plant Interact. 2014, 9, 43–55. [Google Scholar] [CrossRef]
- Lima, J.M.; Nath, M.; Dokku, P.; Raman, K.V.; Kulkarni, K.P.; Vishwakarma, C.; Sahoo, S.P.; Mohapatra, U.B.; AmithaMithra, S.V.; Chinnusamy, V.; Robin, S.; et al. Physiological, anatomical and transcriptional alterations in a rice mutant leading to enhanced water stress tolerance. AoB Plants 2015, 7. [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Fernandez, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; Garcia-Casado, G.; Lopez-Vidriero, I.; Lozano, F.; Ponce, M. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Liu, N.; Staswick, P.E.; Avramova, Z. Memory responses of jasmonic acid-associated Arabidopsis genes to a repeated dehydration stress. Plant Cell Environ. 2016, 39, 2515–2529. [Google Scholar] [CrossRef]
- Li, Z.Y.; Xu, Z.S.; He, G.Y.; Yang, G.X.; Chen, M.; Li, L.C.; Ma, Y.Z. A mutation in Arabidopsis BSK5 encoding a brassinosteroid-signaling kinase protein affects responses to salinity and abscisic acid. Biochem. Biophys. Res. Commun. 2012, 426, 522–527. [Google Scholar] [CrossRef]
- Fariduddin, Q.; Yusuf, M.; Ahmad, I.; Ahmad, A. Brassinosteroids and their role in response of plants to abiotic stresses. Biol. Plant. 2014, 58, 9–17. [Google Scholar] [CrossRef]
- Zhang, S.; Cai, Z.; Wang, X. The primary signaling outputs of brassinosteroids are regulated by abscisic acid signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 4543–4548. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Burgess, P.; Huang, B. Transcriptional regulation of hormone-synthesis and signaling pathways by overexpressing cytokinin-synthesis contributes to improved drought tolerance in creeping bentgrass. Physiol. Plant. 2017, 161, 235–256. [Google Scholar] [CrossRef]
- Saha, G.; Park, J.I.; Jung, H.J.; Ahmed, N.U.; Kayum, M.A.; Kang, J.G. Molecular characterization of BZR transcription factor family and abiotic stress-induced expression profiling in Brassica rapa. Plant Physiol. Biochem. 2015, 92, 92–104. [Google Scholar] [CrossRef]
- Iliev, E.A.; Xu, W.; Polisensky, D.H.; Oh, M.H.; Torisky, R.S.; Clouse, S.D.; Braam, J. Transcriptional and posttranscriptional regulation of Arabidopsis TCH4 expression by diverse stimuli. roles of cis regions and brassinosteroids. Plant Physiol. 2002, 130, 770–783. [Google Scholar] [CrossRef]
- Ning, J.; Li, X.; Hicks, L.M.; Xiong, L. A Raf-like MAPKKK gene DSM1 mediates drought resistance through reactive oxygen species scavenging in rice. Plant Physiol. 2010, 152, 876–890. [Google Scholar] [CrossRef]
- Li, Y.; Cai, H.; Liu, P.; Wang, C.; Gao, H.; Wu, C. Arabidopsis MAPKKK18 positively regulates drought stress resistance via downstream MAPKK3. Biochem. Biophys. Res. Commun. 2017, 484, 292–297. [Google Scholar] [CrossRef]
- Yoo, S.D.; Cho, Y.H.; Tena, G.; Xiong, Y.; Sheen, J. Dual control of nuclear EIN3 by bifurcate MAPK cascades in C2H4 signalling. Nature 2008, 451, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Arraes, F.B.M.; Beneventi, M.A.; Lisei de Sa, M.E.; Paixao, J.F.; Albuquerque, E.V.S.; Marin, S.R.R.M. Implications of ethylene biosynthesis and signaling in soybean drought stress tolerance. BMC Plant Biol. 2015, 15, 213. [Google Scholar] [CrossRef]
- Takahashi, F.; Yoshida, R.; Ichimura, K.; Mizoguchi, T.; Seo, S.; Yonezawa, M.; Maruyama, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Mitogen-activated protein kinase cascade MKK3-MPK6 is an important part of the jasmonate signal transduction pathway in Arabidopsis. Plant Cell 2007, 19, 805–818. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Lu, W.; Meng, F.; Wu, C.A.; Guo, X. Cotton Ghmkk5 affects disease resistance, induces HR-like cell death, and reduces the tolerance to salt and drought stress in transgenic Nicotiana benthamiana. J. Exp. Bot. 2012, 63, 3935–3951. [Google Scholar] [CrossRef]
- Yao, P.F.; Li, C.L.; Zhao, X.R.; Li, M.F.; Zhao, H.X.; Guo, J.Y.; Cai, Y.; Chen, H.; Wu, Q. Overexpression of a tartary buckwheat gene, FtbHLH3, enhances drought/oxidative stress tolerance in transgenic Arabidopsis. Front. Plant Sci. 2017, 8, 625. [Google Scholar] [CrossRef]
- Zhang, L.L.; Cheng, J.; Sun, X.M.; Zhao, T.T.; Li, M.J.; Wang, Q.F.; Li, S.H.; Xin, H.P. Overexpression of VaWRKY14 increases drought tolerance in Arabidopsis by modulating the expression of stress-related genes. Plant Cell Rep. 2018, 37, 1159–1172. [Google Scholar] [CrossRef]
- Tang, N.; Ma, S.; Zong, W.; Yang, N.; Lv, Y.; Yan, C.; Guo, Z.; Li, J.; Li, X.; Xiang, Y.; et al. MODD mediates deactivation and degradation of OsbZIP46 to negatively regulate ABA signaling and drought resistance in rice. Plant Cell 2016, 28, 2161–2177. [Google Scholar] [CrossRef]
- Chen, N.; Yang, Q.L.; Su, M.W.; Pan, L.J.; Chi, X.Y.; Chen, M.N.; He, Y.N.; Yang, Z.; Wang, T.; Wang, M.; et al. Cloning of six ERF family transcription factor genes from peanut and analysis of their expression during abiotic stress. Plant Mol. Biol. Rep. 2012, 30, 1415–1425. [Google Scholar] [CrossRef]
- Yu, Y.W.; Yang, D.X.; Zhou, S.R.; Gu, J.T.; Wang, F.R.; Dong, J.G.; Huang, R.F. The ethylene response factor OsERF109 negatively affects ethylene biosynthesis and drought tolerance in rice. Protoplasma 2016, 254, 401–408. [Google Scholar] [CrossRef]
- Butt, H.I.; Yang, Z.; Gong, Q.; Chen, E.; Wang, X.; Zhao, G.; Ge, X.Y.; Zhang, X.Y.; Li, F.G. GaMYB85, an R2R3 MYB gene, in transgenic Arabidopsis plays an important role in drought tolerance. BMC Plant Biol. 2017, 17, 142. [Google Scholar] [CrossRef]
- Gao, F.; Wang, J.; Wei, S.; Li, Z.; Wang, N.; Li, H.; Feng, J.; Li, H.; Zhou, Y.; Zhang, F. Transcriptomic analysis of drought stress responses in ammopiptanthus mongolicus leaves using the RNA-Seq technique. PLoS ONE 2015, 10, e0124382. [Google Scholar]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.; Terol, J.; Williams, T.; Nagaraj, S.; Nueda, M.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theor. Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.; Wang, Z.; Zhu, Q.; Wang, W. Hormonal changes in the grains of rice subjected to water stress during grain filling. Plant Physiol. 2001, 127, 315–323. [Google Scholar] [CrossRef]
- Zhao, J.; Li, G.; Wang, B.M.; Liu, W.; Nan, T.G.; Zhai, Z.X. Development of a monoclonal antibody-based enzyme-linked immunosorbent assay for the analysis of glycyrrhizic acid. Anal. Bioanal. Chem. 2006, 386, 1735–1740. [Google Scholar] [CrossRef]
- Xu, Y.G.; Liu, R.R.; Sui, N.; Shi, W.W.; Wang, L.; Tian, C.Y. Changes in endogenous hormones and seed coat phenolics during seed storage of two Suaeda salsa populations. Aust. J. Bot. 2016, 64, 325–332. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Chen, Y.; Shi, Z.; Jin, Y.; Sun, H.; Xie, F.; Zhang, L. Biosynthesis and Signal Transduction of ABA, JA, and BRs in Response to Drought Stress of Kentucky Bluegrass. Int. J. Mol. Sci. 2019, 20, 1289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061289
Chen Y, Chen Y, Shi Z, Jin Y, Sun H, Xie F, Zhang L. Biosynthesis and Signal Transduction of ABA, JA, and BRs in Response to Drought Stress of Kentucky Bluegrass. International Journal of Molecular Sciences. 2019; 20(6):1289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061289
Chicago/Turabian StyleChen, Yajun, Yang Chen, Zhenjie Shi, Yifeng Jin, Huashan Sun, Fuchun Xie, and Lu Zhang. 2019. "Biosynthesis and Signal Transduction of ABA, JA, and BRs in Response to Drought Stress of Kentucky Bluegrass" International Journal of Molecular Sciences 20, no. 6: 1289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061289