ytiB and ythA Genes Reduce the Uranium Removal Capacity of Bacillus atrophaeus

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
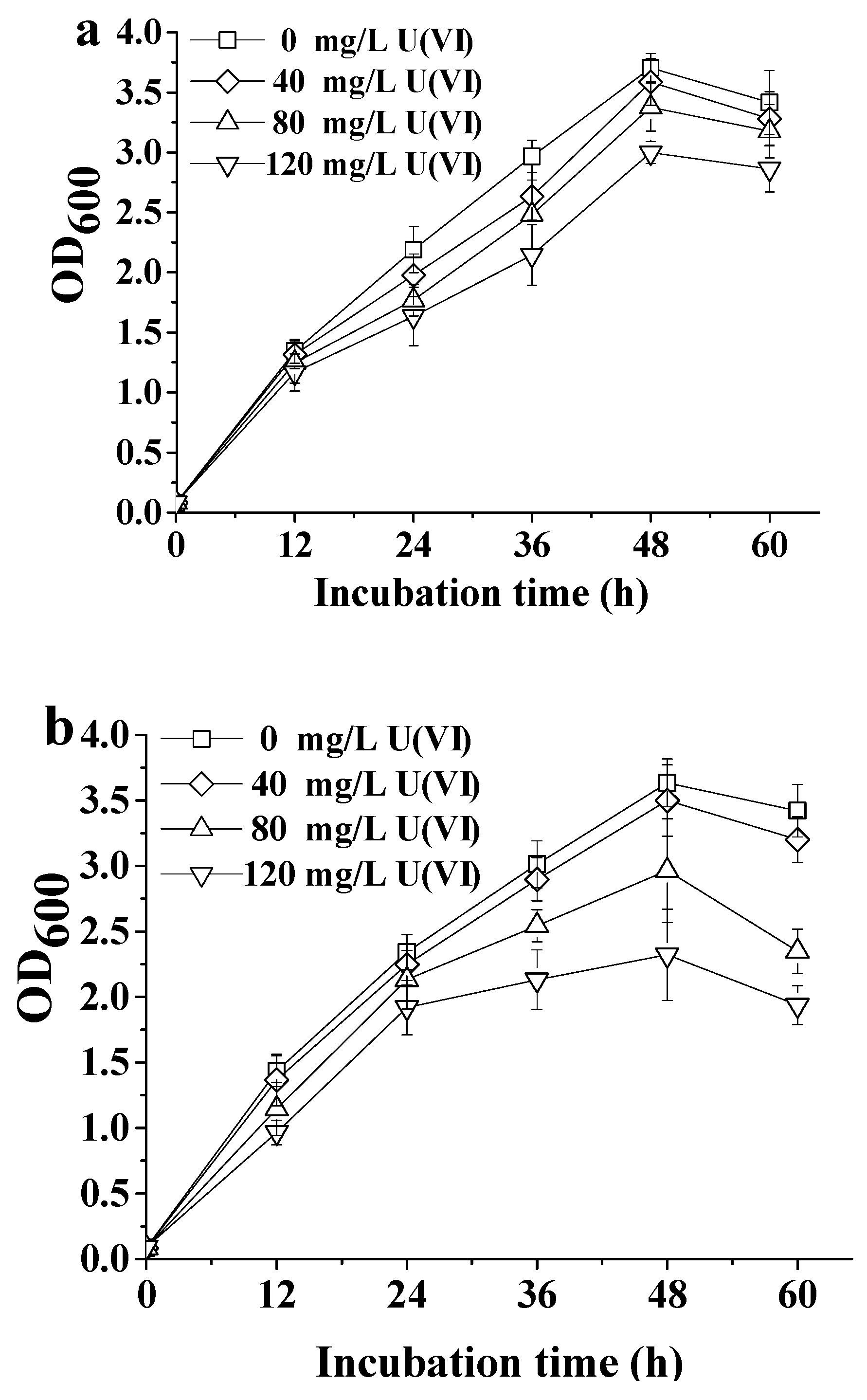
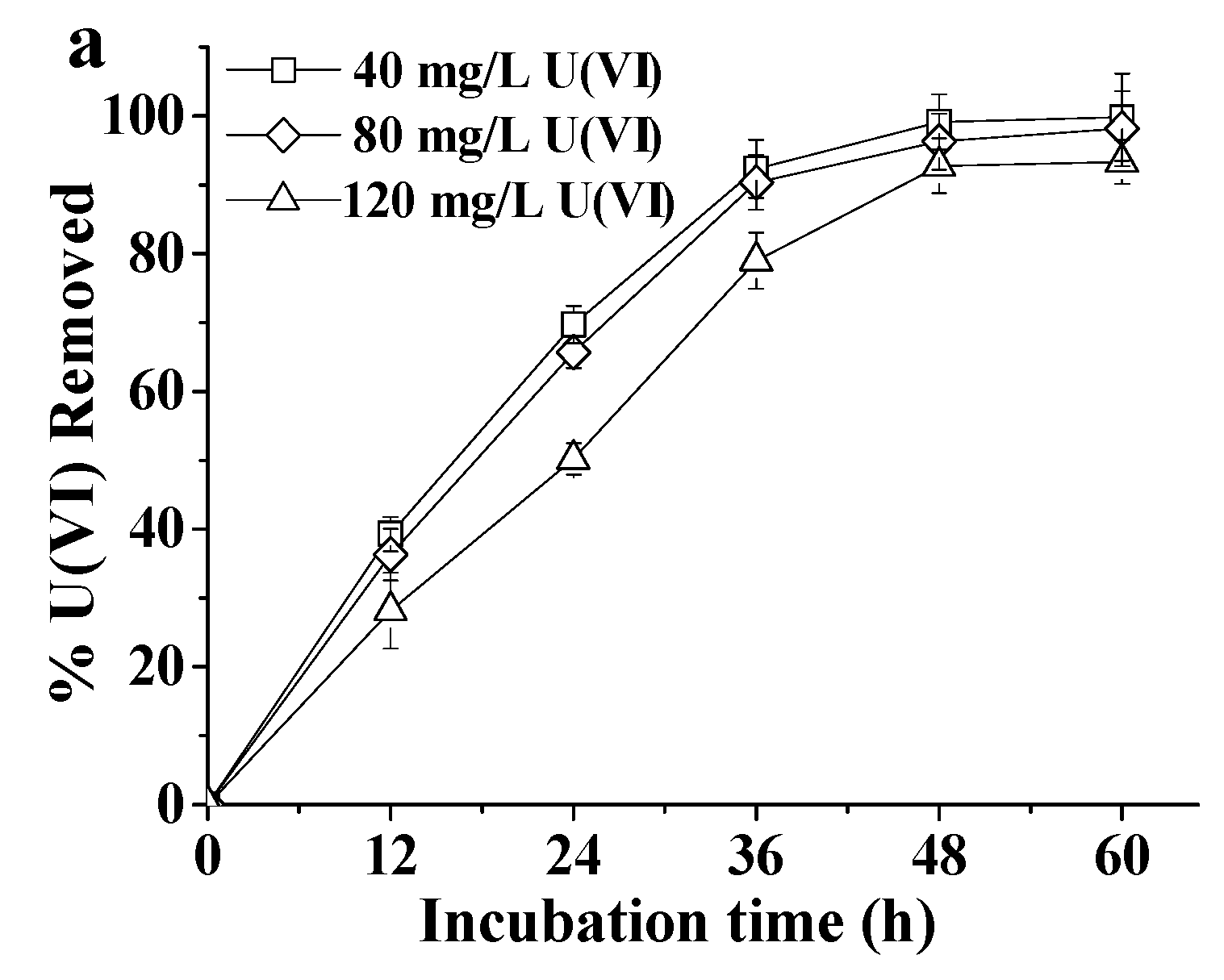
2.1. Tolerance of ATCC 9372 and Ua Strains to Different Concentrations of U(VI)
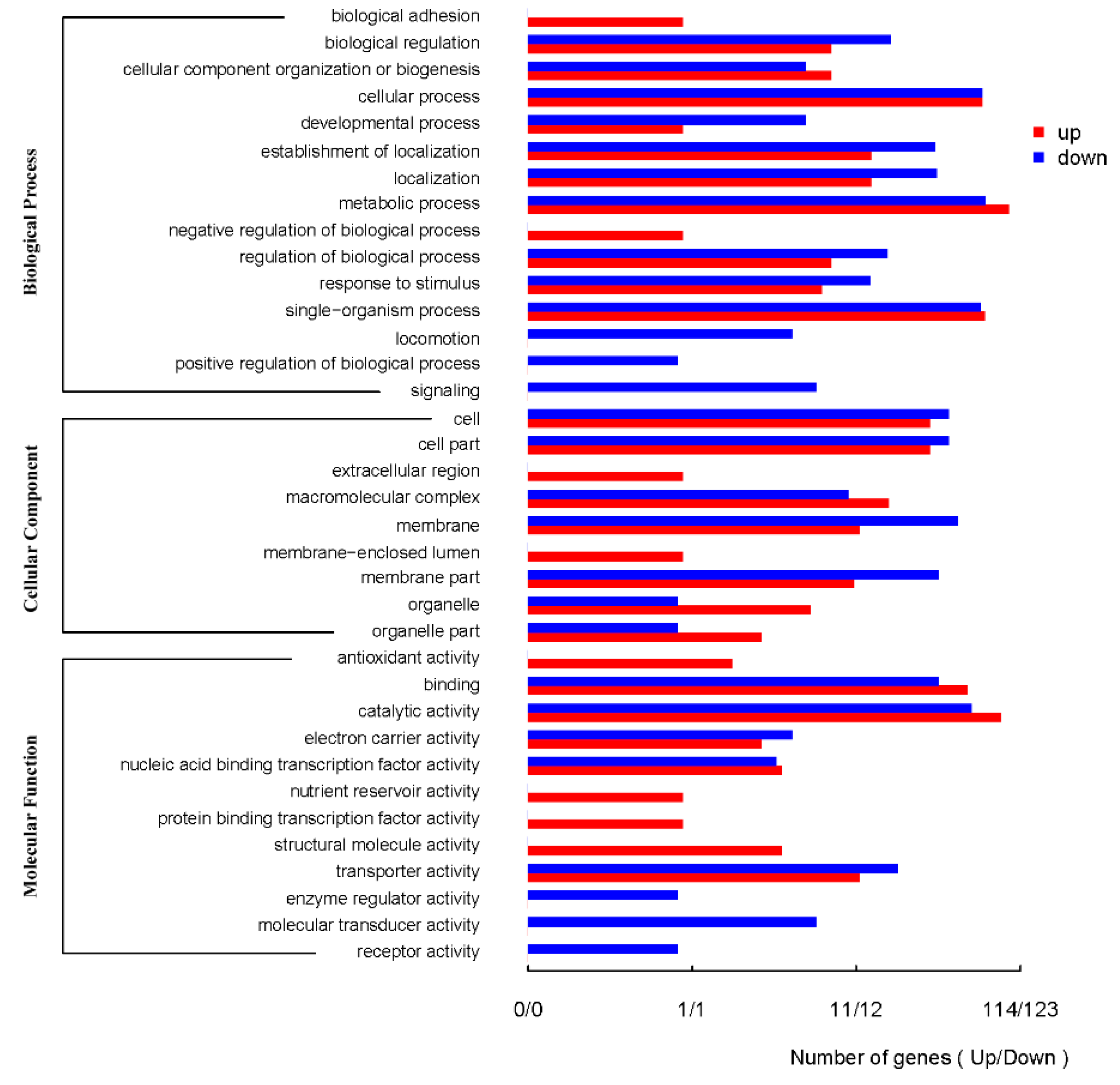
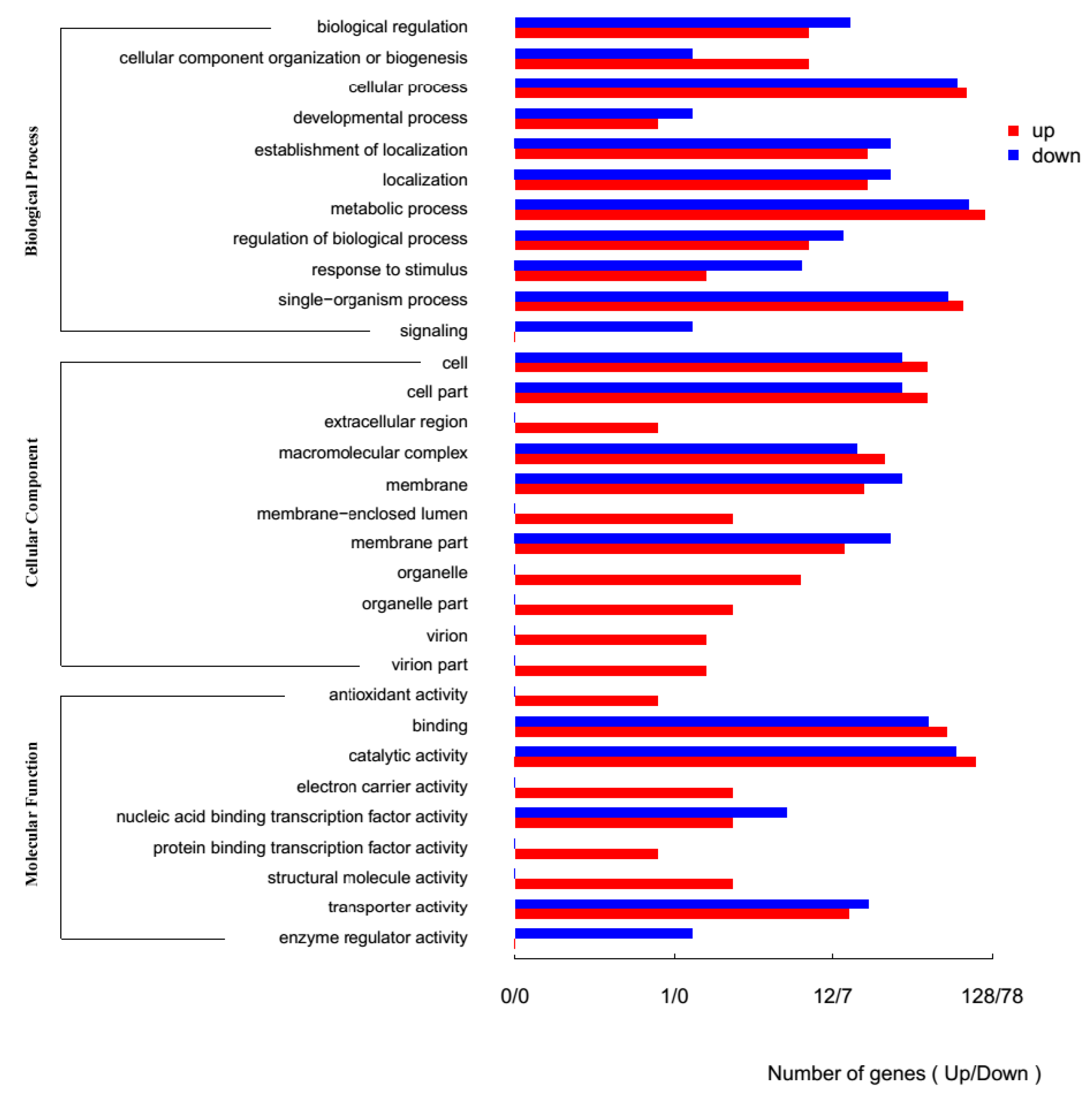
2.2. Comparison of Bacillus atrophaeus Transcriptomes under Control Conditions and under Exposure to Uranium
2.3. Isolation and Sequence Analysis of ytiB and ythA Genes
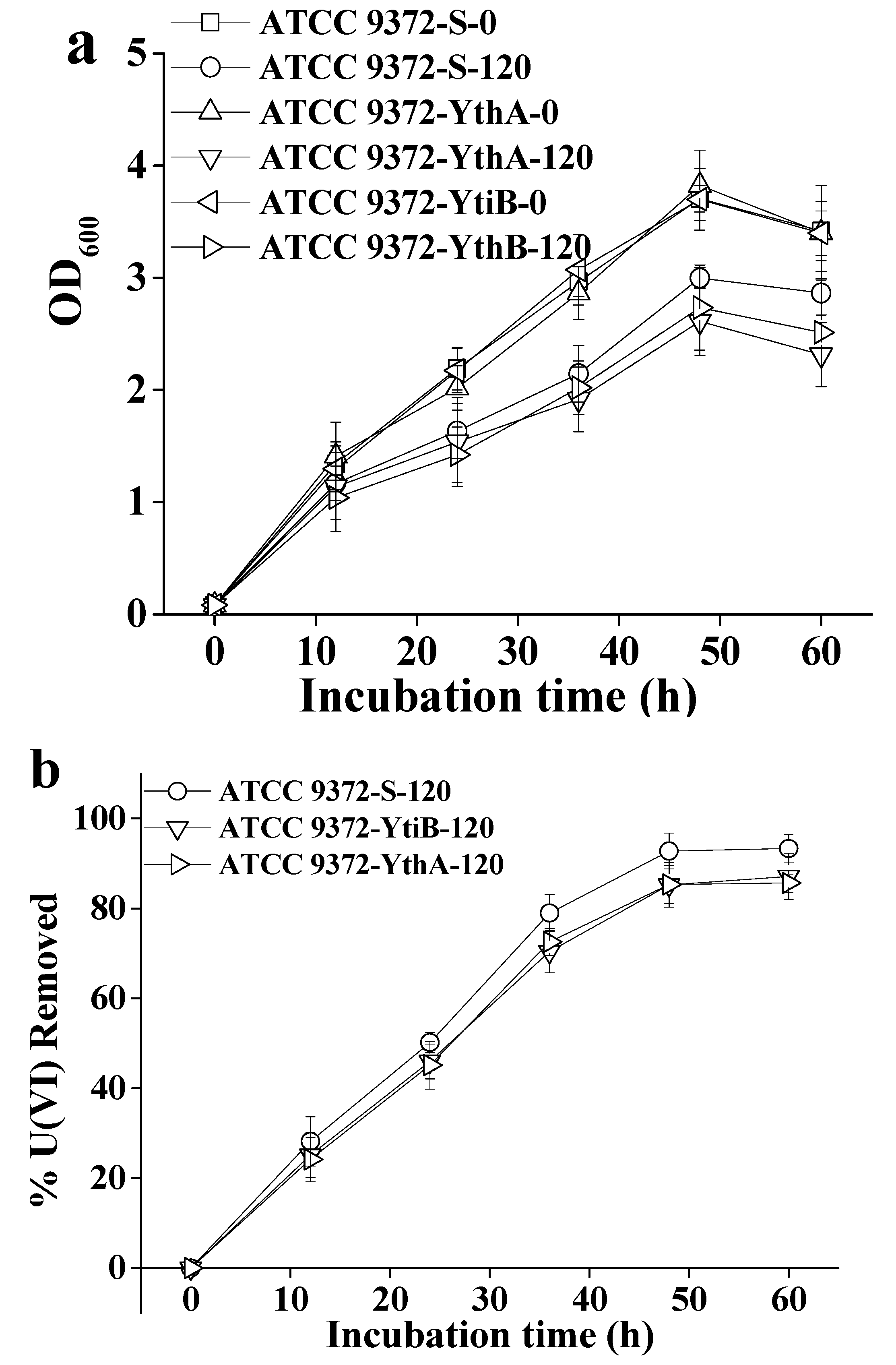
2.4. Influence of ytiB and ythA on the U(VI) Removal Capacity of Bacteria in Transgenics
3. Materials and Methods
3.1. Microorganisms
3.2. Analysis of U(VI) Tolerance and Removal Capacities of Bacterial Strains
3.3. Total RNA Extraction and Transcriptome Sequencing
3.4. Bioinformatic Analysis of ytiB and ythA Genes
3.5. Analysis of the Roles of ytiB and ythA in U(VI) Removal Using a Transgenic Approach
Author Contributions
Funding
Conflicts of Interest
References
- Nair, R.N.; Faby, S.; Manikandan, S.T. Modelling of decay chain transport in groundwater from uranium tailings ponds. Appl. Math. Model. 2010, 34, 2300–2311. [Google Scholar] [CrossRef]
- Teixeira, S.; Vieira, M.N.; Marques, J.E.; Pereira, R. Bioremediation of an iron-rich mine effluent by Lemna minor. Int. J. Phytorem. 2014, 16, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Tang, L.; Zeng, G.M.; Chen, J.; Cai, Y.; Zhang, Y.; Yang, G.D.; Liu, Y.Y.; Zhang, C.; Tang, W.W. Mesoporous carbon nitride based biosensor for highly sensitive and selective analysis of phenol and catechol in compost bioremediation. Biosens. Bioelectron. 2014, 61, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Dong, H.L.; Reguera, G.; Beyenal, H.; Lu, A.H.; Liu, J.; Yu, H.Q.; Fredrickson, J.K. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 2016, 14, 651–662. [Google Scholar] [CrossRef]
- Zammit, C.M.; Shuster, J.P.; Gagen, E.J.; Southam, G. The geomicrobiology of supergene metal deposits. Elements 2015, 11, 337–342. [Google Scholar] [CrossRef]
- Ortiz-Bernad, I.; Anderson, R.T.; Vrionis, H.A.; Lovley, D.R. Resistance of Solid-Phase U(VI) to Microbial Reduction during In Situ Bioremediation of Uranium-Contaminated Groundwater. Appl. Environ. Microbiol. 2004, 70, 7558–7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skennerton, C.T.; Barr, J.J.; Slater, F.R.; Bond, P.L.; Tyson, G.W. Expanding our view of genomic diversity in Candidatus Accumulibacter clades. Environ. Microbiol. 2015, 17, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.Y.; Tian, Z.G.; Xu, Z.; Zhang, L.; Wang, Y.X.; Chen, J. Construction and application of a micro-array for profiling micro-RNA expression. Prog. Biochem. Biophys. 2007, 34, 31–41. [Google Scholar]
- Wright, E.S.; Yilmaz, L.S.; Corcoran, A.M.; Okten, H.E.; Noguera, D.R. Automated design of probes for rRNA-targeted fluorescence in situ hybridization reveals the advantages of using dual probes for accurate identification. Appl. Environ. Microbiol. 2014, 80, 5124–5133. [Google Scholar] [CrossRef]
- Murai, J.; Marchand, C.; Shahane, S.A.; Sun, H.M.; Huang, R.L.; Zhang, Y.P.; Chergui, A.; Ji, J.P.; Doroshow, J.H.; Jadhav, A. Identification of novel PARP inhibitors using a cell-based TDP1 inhibitory assay in a quantitative high-throughput screening platform. DNA Repair 2014, 21, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.L.; Shao, H.H.; Liu, Y.C.; Liu, G.; Xie, C.Y.; Cheng, X.J.; Wang, H.Y.; Tan, X.M.; Feng, H. Transcriptome profiling analysis reveals metabolic changes across various growth phases in Bacillus pumilus BA06. BMC Microbiol. 2017, 17, 156. [Google Scholar] [CrossRef]
- Liu, S.; Hao, H.; Lu, X.; Zhao, X.; Wang, Y.; Zhang, Y.; Xie, Z.; Wang, R. Transcriptome profiling of genes involved in induced systemic salt tolerance conferred by Bacillus amyloliquefaciens FZB42 in Arabidopsis thaliana. Sci. Rep. 2017, 7, 10795. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Wei, C.L.; Liu, E.T.; Ruan, Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009, 19, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Han, Y.J.; Kim, H.J.; Kwak, D.W.; Park, S.Y.; Chun, S.H.; Ryu, H.M. Genome-wide gene expression analysis in the placenta from fetus with trisomy 21. BMC Genom. 2017, 18, 720. [Google Scholar] [CrossRef] [Green Version]
- Leimena, M.M.; Wels, M.; Bongers, R.S.; Smid, E.J.; Zoetendal, E.G.; Kleerebezem, M. Comparative Analysis of Lactobacillus plantarum WCFS1 Transcriptomes by Using DNA Microarray and Next-Generation Sequencing Technologies. Appl. Environ. Microbiol. 2012, 78, 4141–4148. [Google Scholar] [CrossRef]
- McKenzie, A.T.; Pomerantsev, A.P.; Sastalla, I.; Martens, C.; Ricklefs, S.M.; Virtaneva, K.; Anzick, S.; Porcella, S.F.; Leppla, S.H. Transcriptome analysis identifies Bacillus anthracis genes that respond to CO2 through an AtxA-dependent mechanism. BMC Genom. 2014, 15, 229. [Google Scholar] [CrossRef]
- Xie, S.; Wu, H.; Chen, L.; Zang, H.; Xie, Y.; Gao, X. Transcriptome profiling of Bacillus subtilis OKB105 in response to rice seedlings. BMC Microbiol. 2015, 15, 21. [Google Scholar] [CrossRef]
- Liu, X.; Yang, H.; Zheng, J.; Ye, Y.; Pan, L. Identification of strong promoters based on the transcriptome of Bacillus licheniformis. Biotechnol Lett. 2017, 39, 873–881. [Google Scholar] [CrossRef]
- Nakamura, L.K. Taxonomic Relationship of Black-Pigmented Bacillus subtilis Strains and a Proposal for Bacillus atrophaeus sp. nov. Int. J. Syst. Bacteriol. 1989, 39, 295–300. [Google Scholar] [CrossRef]
- Wang, C.; Chen, X.M.; Xu, Y.; Ruan, C.; Liu, X.L.; Hao, X.C.; Song, S.; Luo, X.G.; Chen, C.X.; Luo, Y.X. Analyzation on Genetic Variations Associated With Cr(VI) Removal Between two Bacillus atrophaeus Strains Applied PFGE Technology. J. Nucl. Agricul. Sci. 2015, 29, 1052–1060. [Google Scholar]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Chiou, C.S.; Torpdahlb, M.; Liao, Y.S.; Liao, C.H.; Tsao, C.S.; Liang, S.Y.; Wang, Y.W.; Kuo, J.C.; Liu, Y.Y. Usefulness of pulsed-field gel electrophoresis profiles for the determination of Salmonella serovars. Int. J. Food Microbiol. 2015, 214, 1–3. [Google Scholar] [CrossRef]
- Azimi, L.; Talebi, M.; Khodaei, F.; Najafi, M.; Lari, A.R. Comparison of multiple-locus variable-number tandem-repeat analysis with pulsed-field gel electrophoresis typing of carbapenemases producing Acinetobacter baumannii isolated from burn patients. Burns 2015, 42, 441–445. [Google Scholar] [CrossRef]
- Bopp, D.J.; Baker, D.J.; Thompson, L.; Saylors, A.; Root, T.P.; Armstrong, L.; Mitchell, K.; Dumas, N.B.; Musser, K.A. Implementation of Salmonella serotype determination using pulsed-field gel electrophoresis in a state public health laboratory. Diagn. Micr. Infec. Dis. 2016, 85, 416–418. [Google Scholar] [CrossRef]
- Bassi, D.; Colla, F.; Gazzola, S.; Puglisi, E.; Delledonne, M.; Cocconcelli, P.S. Transcriptome analysis of Bacillus thuringiensis spore life, germination and cell outgrowth in a vegetable-based food model. Food Microbiol. 2016, 55, 73–85. [Google Scholar] [CrossRef]
- Misture, S.T.; Naylor, M.O.; Jin, T.; Shelby, J.E. Galliosilicate glasses for viscous sealants in solid oxide fuel cell stacks: Part II: Interactions with yttria stabilized zirconia and stainless steel coated with alumina. Int. J. Hydrogen. Energ. 2013, 38, 16328–16337. [Google Scholar] [CrossRef]
- Draberova, L.; Bugajev, V.; Potuckova, L.; Halova, I.; Bambouskova, M.; Polakovicova, I.; Xavier, R.J.; Seed, B.; Draber, P. Transmembrane adaptor protein PAG/CBP is involved in both positive and negative regulation of mast cell signaling. Mol. Cell Biol. 2014, 34, 4285–4300. [Google Scholar] [CrossRef]
- Barbe, V.; Cruveiller, S.; Kunst, F.; Lenoble, P.; Meurice, G.; Sekowska, A.; Vallenet, D.; Wang, T.; Moszer, I.; Medigue, C.; Danchin, A. From a consortium sequence to a unified sequence: The Bacillus subtilis 168 reference genome a decade later. Microbiol 2009, 155, 1758–1775. [Google Scholar] [CrossRef]
- Jauberty, L.; Drogat, N.; Decossas, J.L.; Delpech, V.; Vincent, G.; Sol, V. Optimization of the arsenazo-III method for the determination of uranium in water and plant samples. Talanta 2013, 115, 751–754. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Clean Reads | Similarity (%) |
|---|---|---|
| ATCC 9372-0 | 22534064/28206904 | 79.89% |
| ATCC 9372-U | 19228202/24475060 | 78.56% |
| Ua-0 | 30370551/30654342 | 99.10% |
| Ua-U | 39516637/40055216 | 98.70% |
| Gene ID | String_Tophit_Description | KEGG_Gene_Name | Length | String_topHSP_%-Simil | NR_tophit_Description | Ua_0 Count | Ua_U Count | Up- or Down-Regulation |
|---|---|---|---|---|---|---|---|---|
| 2626 | YtiB | CynT | 570 | 95.72 | carbonic anhydrase | 294 | 281 | down |
| 2629 | YthA | CydA | 1332 | 97.26 | cytochrome D ubiquinol oxidase subunit I | 350 | 256 | down |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Xiao, S.; Chen, X.; Chen, S.; Wang, S.; Wang, C.; Tang, Y.; Dong, F. ytiB and ythA Genes Reduce the Uranium Removal Capacity of Bacillus atrophaeus. Int. J. Mol. Sci. 2019, 20, 1766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071766
Wang L, Xiao S, Chen X, Chen S, Wang S, Wang C, Tang Y, Dong F. ytiB and ythA Genes Reduce the Uranium Removal Capacity of Bacillus atrophaeus. International Journal of Molecular Sciences. 2019; 20(7):1766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071766
Chicago/Turabian StyleWang, Li, Shiqi Xiao, Xiaoming Chen, Shilin Chen, Shanqiang Wang, Chao Wang, Yunlai Tang, and Faqin Dong. 2019. "ytiB and ythA Genes Reduce the Uranium Removal Capacity of Bacillus atrophaeus" International Journal of Molecular Sciences 20, no. 7: 1766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071766