Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Bile Acids Biochemistry
2. BAs Physiology
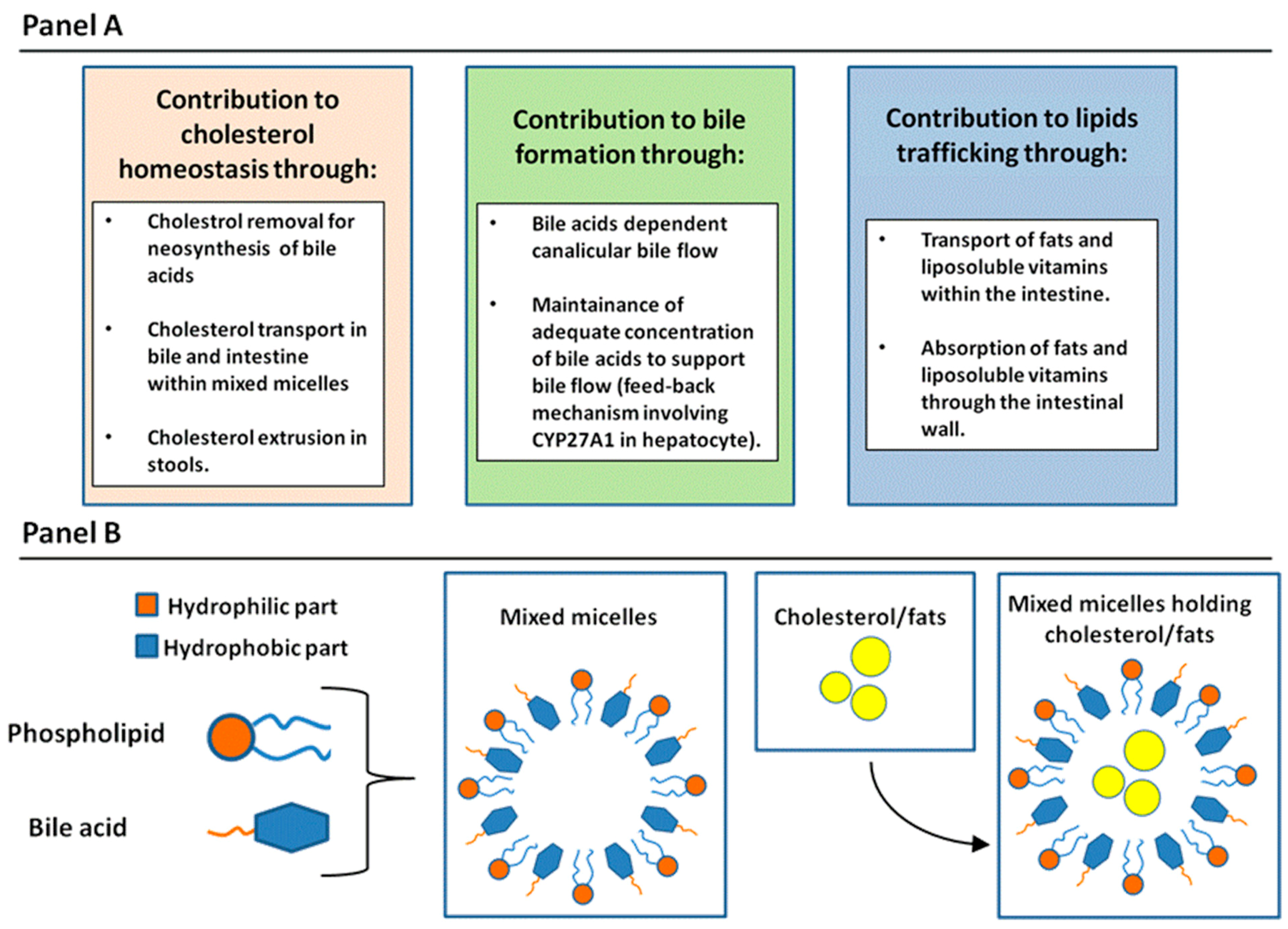
2.1. General Physiologic Functions of BAs
2.2. Enterohepatic Circulation of BAs
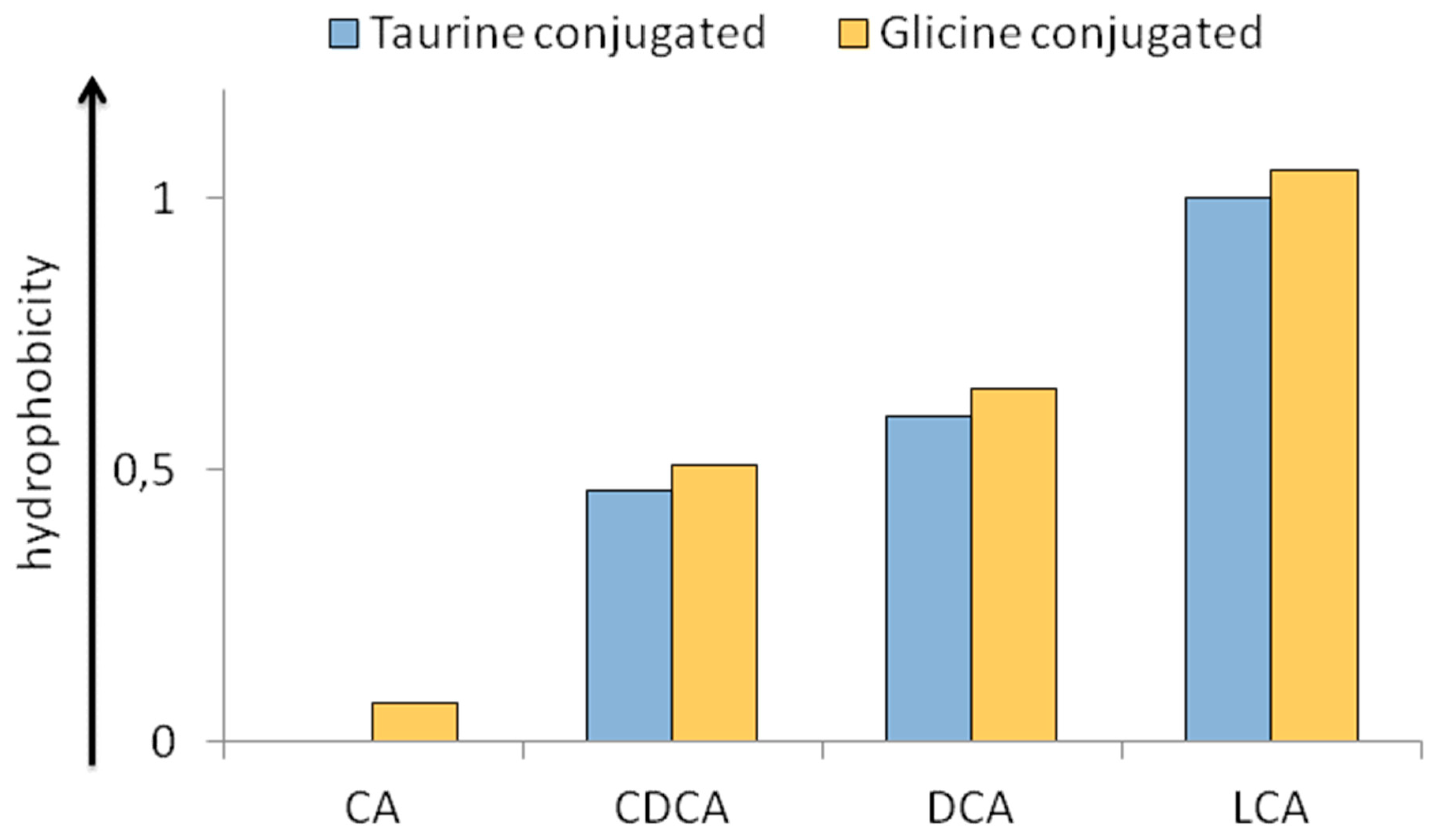
2.3. Physico-Chemical Heterogeneity of BAs with Relevance in Physiopathology
2.4. Physiologic Effects of BAs on Biliary Epithelium
2.5. Protective Effects of BAs on Biliary Epithelium
2.6. Injurious Effects of BAs on Biliary Epithelium
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | Adenosine Tri-Phosphate binding cassette |
| AE2 | apical Cl−/HCO3− anion exchanger 2 |
| ASBT | apical sodium bile acid co-transporter |
| AKR1C4 | 3a-hydroxysteroid dehydrogenase |
| AKR1D1 | D4–3-oxosteroid-5b-reductase |
| BAAT | bile acid CoA-amino acid N-acyltransferase |
| BACS | bile acid CoA synthetase |
| BDL | bile duct ligated, (BSEP |
| cAMP | adenosine 3′,5′-cyclic monophosphate |
| CCA | cholangiocarcinoma |
| CCl4 | carbon tetrachloride |
| CDCA | chenodeoxycholic acid |
| CYP27A1 | Sterol 27-hydroxylase |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CYP7B1 | oxysterol 7a-hydroxylase |
| CYP8B1 | 12a-hydroxylase |
| cSrc | Rous sarcoma oncogene |
| DCA | Deoxycholic Acid |
| DHCA | 3a,7a-dihydroxy-5b-cholestanoic acid |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular regulated protein kinase |
| FXR | Farnesoid receptor X |
| HI | hydrophobicity index |
| HPLC | high pressure liquid chromatography |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL6 | interleukin-6 |
| LCA | Lithocholic Acid |
| MAPK | mitogen-activated protein kinase |
| Mdr | Multi-Drug Resistance |
| MDR/TAP | multi-drug-resistance/transporter associated with antigen processing |
| NTCP | sodium/taurocholate co-transporting polypeptide |
| OST | organic solute transporter |
| PBP | peri-biliary plexus |
| PBC | primary biliary cholangitis |
| PCNA | protein cellular nuclear antigen |
| PI3K | phosphatidylinositol 3-kinases |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PSC | primary sclerosing cholangitis |
| ROS | reactive oxygen species |
| SCT | secretin |
| S1PR2 | Sphingosine 1-phosphate receptor 2 |
| SR | secretin receptor |
| TCA | Taurocholic acid |
| TGR5 | Transmembrane G protein coupled receptor |
| THCA | 3 a,7a,12a-trihydroxy-5b-cholestanoic acid |
| TLCA | Taurolithocholic acid |
| TUDCA | Tauroursodeoxycholic acid |
| UDCA | Ursodeoxycholic acid |
| VEGF | vascular endothelial growth factor |
| VLCS | very long-chain acyl CoA synthetase |
References
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Axelson, M.; Ellis, E.; Mork, B.; Garmark, K.; Abrahamsson, A.; Bjorkhem, I.; Ericzon, B.G.; Einarsson, C. Bile acid synthesis in cultured human hepatocytes: Support for an alternative biosynthetic pathway to cholic acid. Hepatology 2000, 31, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Axelson, M.; Sjovall, J. Potential bile acid precursors in plasma—Possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J. Steroid. Biochem. 1990, 36, 631–640. [Google Scholar] [CrossRef]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Mazer, N.A.; Benedek, G.B.; Carey, M.C. Quasielastic light-scattering studies of aqueous biliary lipid systems. Mixed micelle formation in bile salt-lecithin solutions. Biochemistry 1980, 19, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [PubMed]
- Kubitz, R.; Droge, C.; Stindt, J.; Weissenberger, K.; Haussinger, D. The bile salt export pump (bsep) in health and disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. The enterohepatic circulation of bile acids in mammals: Form and functions. Front. Biosci. (Landmark Ed.) 2009, 14, 2584–2598. [Google Scholar] [CrossRef]
- Dawson, P.A.; Haywood, J.; Craddock, A.L.; Wilson, M.; Tietjen, M.; Kluckman, K.; Maeda, N.; Parks, J.S. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem. 2003, 278, 33920–33927. [Google Scholar] [CrossRef]
- Dawson, P.A.; Hubbert, M.; Haywood, J.; Craddock, A.L.; Zerangue, N.; Christian, W.V.; Ballatori, N. The heteromeric organic solute transporter alpha-beta, ostalpha-ostbeta, is an ileal basolateral bile acid transporter. J. Biol. Chem. 2005, 280, 6960–6968. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Molecular cloning, chromosomal localization, and functional characterization of a human liver na+/bile acid cotransporter. J. Clin. Invest. 1994, 93, 1326–1331. [Google Scholar] [CrossRef]
- Martinez-Augustin, O.; Sanchez de Medina, F. Intestinal bile acid physiology and pathophysiology. World J. Gastroenterol. 2008, 14, 5630–5640. [Google Scholar] [CrossRef] [Green Version]
- Carulli, N.; Bertolotti, M.; Carubbi, F.; Concari, M.; Martella, P.; Carulli, L.; Loria, P. Review article: Effect of bile salt pool composition on hepatic and biliary functions. Aliment. Pharmacol Ther 2000, 14, 14–18. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Carey, M.C. The hydrophobic-hydrophilic balance of bile salts. Inverse correlation between reverse-phase high performance liquid chromatographic mobilities and micellar cholesterol-solubilizing capacities. J. Lipid Res. 1982, 23, 70–80. [Google Scholar]
- Scholmerich, J.; Becher, M.S.; Schmidt, K.; Schubert, R.; Kremer, B.; Feldhaus, S.; Gerok, W. Influence of hydroxylation and conjugation of bile salts on their membrane-damaging properties—Studies on isolated hepatocytes and lipid membrane vesicles. Hepatology 1984, 4, 661–666. [Google Scholar] [CrossRef]
- El-Agamy, D.S.; Almaramhy, H.H.; Ahmed, N.; Bojan, B.; Alrohily, W.D.; Elkablawy, M.A. Anti-inflammatory effects of vardenafil against cholestatic liver damage in mice: A mechanistic study. Cell. Physiol. Biochem. 2018, 47, 523–534. [Google Scholar] [CrossRef]
- Tan, K.P.; Wood, G.A.; Yang, M.; Ito, S. Participation of nuclear factor (erythroid 2-related), factor 2 in ameliorating lithocholic acid-induced cholestatic liver injury in mice. Br. J. Pharmacol. 2010, 161, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.J.; Bae, S.; Kang, J.S.; Yoon, J.H.; Cho, E.J.; Lee, J.H.; Kim, Y.J.; Lee, W.J.; Kim, C.Y.; Lee, H.S. Hepatoprotective effect of vitamin c on lithocholic acid-induced cholestatic liver injury in gulo(-/-) mice. Eur. J. Pharmacol. 2015, 762, 247–255. [Google Scholar] [CrossRef]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 1989, 30, 719–730. [Google Scholar]
- Alpini, G.; McGill, J.M.; Larusso, N.F. The pathobiology of biliary epithelia. Hepatology 2002, 35, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Tabibian, J.H.; Masyuk, A.I.; Masyuk, T.V.; O’Hara, S.P.; LaRusso, N.F. Physiology of cholangiocytes. Compr. Physiol. 2013, 3, 541–565. [Google Scholar]
- Barton, R.T.; Ucmakli, A. Treatment of squamous cell carcinoma of the floor of the mouth. Surg. Gynecol. Obstet. 1977, 145, 21–27. [Google Scholar]
- Schaffner, F.; Popper, H. Electron microscopic studies of normal and proliferated bile ductules. Am. J. Pathol. 1961, 38, 393–410. [Google Scholar]
- Afroze, S.; Meng, F.; Jensen, K.; McDaniel, K.; Rahal, K.; Onori, P.; Gaudio, E.; Alpini, G.; Glaser, S.S. The physiological roles of secretin and its receptor. Ann. Transl. Med. 2013, 1, 29. [Google Scholar]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and structural features of cholangiocytes in health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef]
- Jones, H.; Alpini, G.; Francis, H. Bile acid signaling and biliary functions. Acta Pharm Sin. B 2015, 5, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Masyuk, A.I.; Huang, B.Q.; Radtke, B.N.; Gajdos, G.B.; Splinter, P.L.; Masyuk, T.V.; Gradilone, S.A.; LaRusso, N.F. Ciliary subcellular localization of tgr5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1013–G1024. [Google Scholar] [CrossRef]
- McCain, J. The mapk (erk) pathway: Investigational combinations for the treatment of braf-mutated metastatic melanoma. P T 2013, 38, 96–108. [Google Scholar]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. Tgr5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef]
- Keitel, V.; Cupisti, K.; Ullmer, C.; Knoefel, W.T.; Kubitz, R.; Haussinger, D. The membrane-bound bile acid receptor tgr5 is localized in the epithelium of human gallbladders. Hepatology 2009, 50, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary hco(3)(-) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Barrena, M.G.; Barcena-Varela, M.; Banales, J.M. New evidence supporting the biliary bicarbonate umbrella theory. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 126–128. [Google Scholar] [CrossRef]
- Erice, O.; Labiano, I.; Arbelaiz, A.; Santos-Laso, A.; Munoz-Garrido, P.; Jimenez-Aguero, R.; Olaizola, P.; Caro-Maldonado, A.; Martin-Martin, N.; Carracedo, A.; et al. Differential effects of fxr or tgr5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; York, J.P.; Wang, L.; Yang, C.; Zhang, A.; Francis, H.L.; Webb, P.; McKeehan, W.L.; Alpini, G.; Lesage, G.D.; et al. Fxr-induced secretion of fgf15/19 inhibits cyp27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch. 2014, 466, 1011–1019. [Google Scholar] [CrossRef]
- Alpini, G.; Glaser, S.S.; Rodgers, R.; Phinizy, J.L.; Robertson, W.E.; Lasater, J.; Caligiuri, A.; Tretjak, Z.; LeSage, G.D. Functional expression of the apical na+-dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology 1997, 113, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Pham, L.; Tietz, P.; Marinelli, R.A.; deGroen, P.C.; Levine, S.; Dawson, P.A.; LaRusso, N.F. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J. Clin. Invest. 1997, 100, 2714–2721. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. Current concepts of biliary secretion. Dig. Dis. Sci. 1989, 34, 16s–20s. [Google Scholar] [CrossRef]
- Xia, X.; Francis, H.; Glaser, S.; Alpini, G.; LeSage, G. Bile acid interactions with cholangiocytes. World J. Gastroenterol. 2006, 12, 3553–3563. [Google Scholar] [CrossRef]
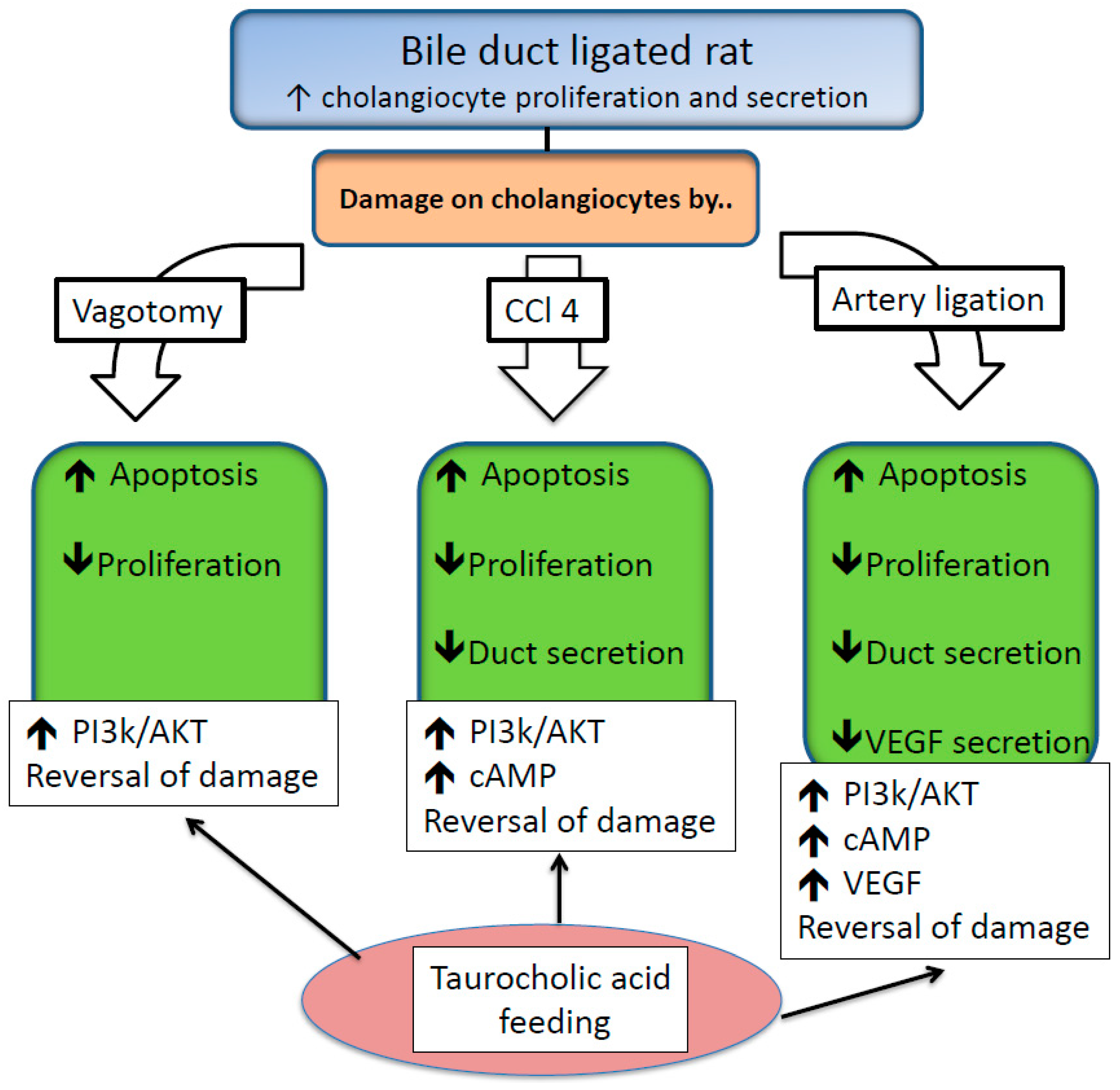
- Alpini, G.; Glaser, S.; Alvaro, D.; Ueno, Y.; Marzioni, M.; Francis, H.; Baiocchi, L.; Stati, T.; Barbaro, B.; Phinizy, J.L.; et al. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology 2002, 123, 1226–1237. [Google Scholar] [CrossRef]
- Marzioni, M.; LeSage, G.D.; Glaser, S.; Patel, T.; Marienfeld, C.; Ueno, Y.; Francis, H.; Alvaro, D.; Tadlock, L.; Benedetti, A.; et al. Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct-ligated rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G837–G852. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, S.G.; Wagner, A.J.; Conzen, S.D.; Jordan, J.; Bellacosa, A.; Tsichlis, P.N.; Hay, N. The pi 3-kinase/akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997, 11, 701–713. [Google Scholar] [CrossRef]
- LeSage, G.D.; Glaser, S.S.; Marucci, L.; Benedetti, A.; Phinizy, J.L.; Rodgers, R.; Caligiuri, A.; Papa, E.; Tretjak, Z.; Jezequel, A.M.; et al. Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from bdl rat liver. Am. J. Physiol. 1999, 276, G1289–G1301. [Google Scholar] [CrossRef]
- Boll, M.; Weber, L.W.; Becker, E.; Stampfl, A. Mechanism of carbon tetrachloride-induced hepatotoxicity. Hepatocellular damage by reactive carbon tetrachloride metabolites. Z. Naturforsch. C 2001, 56, 649–659. [Google Scholar] [CrossRef]
- Clawson, G.A. Mechanisms of carbon tetrachloride hepatotoxicity. Pathol. Immunopathol. Res. 1989, 8, 104–112. [Google Scholar] [CrossRef]
- Marucci, L.; Alpini, G.; Glaser, S.S.; Alvaro, D.; Benedetti, A.; Francis, H.; Phinizy, J.L.; Marzioni, M.; Mauldin, J.; Venter, J.; et al. Taurocholate feeding prevents ccl4-induced damage of large cholangiocytes through pi3-kinase-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G290–G301. [Google Scholar] [CrossRef]
- Batts, K.P. Ischemic cholangitis. Mayo. Clin. Proc. 1998, 73, 380–385. [Google Scholar] [CrossRef]
- Ben-Ari, Z.; Pappo, O.; Mor, E. Intrahepatic cholestasis after liver transplantation. Liver Transpl. 2003, 9, 1005–1018. [Google Scholar] [CrossRef] [Green Version]
- Gaudio, E.; Barbaro, B.; Alvaro, D.; Glaser, S.; Francis, H.; Ueno, Y.; Meininger, C.J.; Franchitto, A.; Onori, P.; Marzioni, M.; et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology 2006, 130, 1270–1282. [Google Scholar] [CrossRef]
- Gaudio, E.; Barbaro, B.; Alvaro, D.; Glaser, S.; Francis, H.; Franchitto, A.; Onori, P.; Ueno, Y.; Marzioni, M.; Fava, G.; et al. Administration of r-vegf-a prevents hepatic artery ligation-induced bile duct damage in bile duct ligated rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G307–G317. [Google Scholar] [CrossRef]
- Glaser, S.; Onori, P.; Gaudio, E.; Ueno, Y.; Pannarale, L.; Franchitto, A.; Francis, H.; Mancinelli, R.; Carpino, G.; Venter, J.; et al. Taurocholic acid prevents biliary damage induced by hepatic artery ligation in cholestatic rats. Dig. Liver Dis. 2010, 42, 709–717. [Google Scholar] [CrossRef]
- Easl clinical practice guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [CrossRef]
- Corpechot, C.; Carrat, F.; Bahr, A.; Chretien, Y.; Poupon, R.E.; Poupon, R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology 2005, 128, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Carrat, F.; Bonnand, A.M.; Poupon, R.E.; Poupon, R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology 2000, 32, 1196–1199. [Google Scholar] [CrossRef]
- Wunsch, E.; Trottier, J.; Milkiewicz, M.; Raszeja-Wyszomirska, J.; Hirschfield, G.M.; Barbier, O.; Milkiewicz, P. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014, 60, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Beuers, U. Management of cholestatic disease in 2017. Liver Int. 2017, 37 (Suppl. 1), 123–129. [Google Scholar] [CrossRef]
- Alpini, G.; Baiocchi, L.; Glaser, S.; Ueno, Y.; Marzioni, M.; Francis, H.; Phinizy, J.L.; Angelico, M.; Lesage, G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of bdl rats through activation of pkc alpha. Hepatology 2002, 35, 1041–1052. [Google Scholar] [CrossRef]
- Frey, M.R.; Clark, J.A.; Leontieva, O.; Uronis, J.M.; Black, A.R.; Black, J.D. Protein kinase c signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J. Cell Biol. 2000, 151, 763–778. [Google Scholar] [CrossRef]
- Beuers, U.; Probst, I.; Soroka, C.; Boyer, J.L.; Kullak-Ublick, G.A.; Paumgartner, G. Modulation of protein kinase c by taurolithocholic acid in isolated rat hepatocytes. Hepatology 1999, 29, 477–482. [Google Scholar] [CrossRef]
- Jones, B.A.; Rao, Y.P.; Stravitz, R.T.; Gores, G.J. Bile salt-induced apoptosis of hepatocytes involves activation of protein kinase c. Am. J. Physiol. 1997, 272, G1109–G1115. [Google Scholar] [CrossRef]
- Marzioni, M.; Francis, H.; Benedetti, A.; Ueno, Y.; Fava, G.; Venter, J.; Reichenbach, R.; Mancino, M.G.; Summers, R.; Alpini, G.; et al. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am. J. Pathol. 2006, 168, 398–409. [Google Scholar] [CrossRef]
- Qiao, L.; Yacoub, A.; Studer, E.; Gupta, S.; Pei, X.Y.; Grant, S.; Hylemon, P.B.; Dent, P. Inhibition of the mapk and pi3k pathways enhances udca-induced apoptosis in primary rodent hepatocytes. Hepatology 2002, 35, 779–789. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.; Moshage, H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Kennedy, L.; Hargrove, L.; Demieville, J.; Jones, H.; Madeka, T.; Karstens, A.; Chappell, K.; Alpini, G.; Sybenga, A.; et al. Ursodeoxycholate inhibits mast cell activation and reverses biliary injury and fibrosis in mdr2(-/-) mice and human primary sclerosing cholangitis. Lab. Invest. 2018, 98, 1465–1477. [Google Scholar] [CrossRef]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis mdr2(-/-) mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef]
- Attili, A.F.; Angelico, M.; Cantafora, A.; Alvaro, D.; Capocaccia, L. Bile acid-induced liver toxicity: Relation to the hydrophobic-hydrophilic balance of bile acids. Med. Hypotheses 1986, 19, 57–69. [Google Scholar] [CrossRef]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef]
- Benedetti, A.; Alvaro, D.; Bassotti, C.; Gigliozzi, A.; Ferretti, G.; La Rosa, T.; Di Sario, A.; Baiocchi, L.; Jezequel, A.M. Cytotoxicity of bile salts against biliary epithelium: A study in isolated bile ductule fragments and isolated perfused rat liver. Hepatology 1997, 26, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol. 2006, 168, 410–422. [Google Scholar] [CrossRef]
- Wagner, M.; Fickert, P.; Zollner, G.; Fuchsbichler, A.; Silbert, D.; Tsybrovskyy, O.; Zatloukal, K.; Guo, G.L.; Schuetz, J.D.; Gonzalez, F.J.; et al. Role of farnesoid x receptor in determining hepatic abc transporter expression and liver injury in bile duct-ligated mice. Gastroenterology 2003, 125, 825–838. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Li, F.; Xie, Y.; Farhood, A.; Fickert, P.; Trauner, M.; Jaeschke, H. Lithocholic acid feeding results in direct hepato-toxicity independent of neutrophil function in mice. Toxicol. Lett. 2014, 228, 56–66. [Google Scholar] [CrossRef]
- Gujral, J.S.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 2003, 38, 355–363. [Google Scholar] [CrossRef] [Green Version]
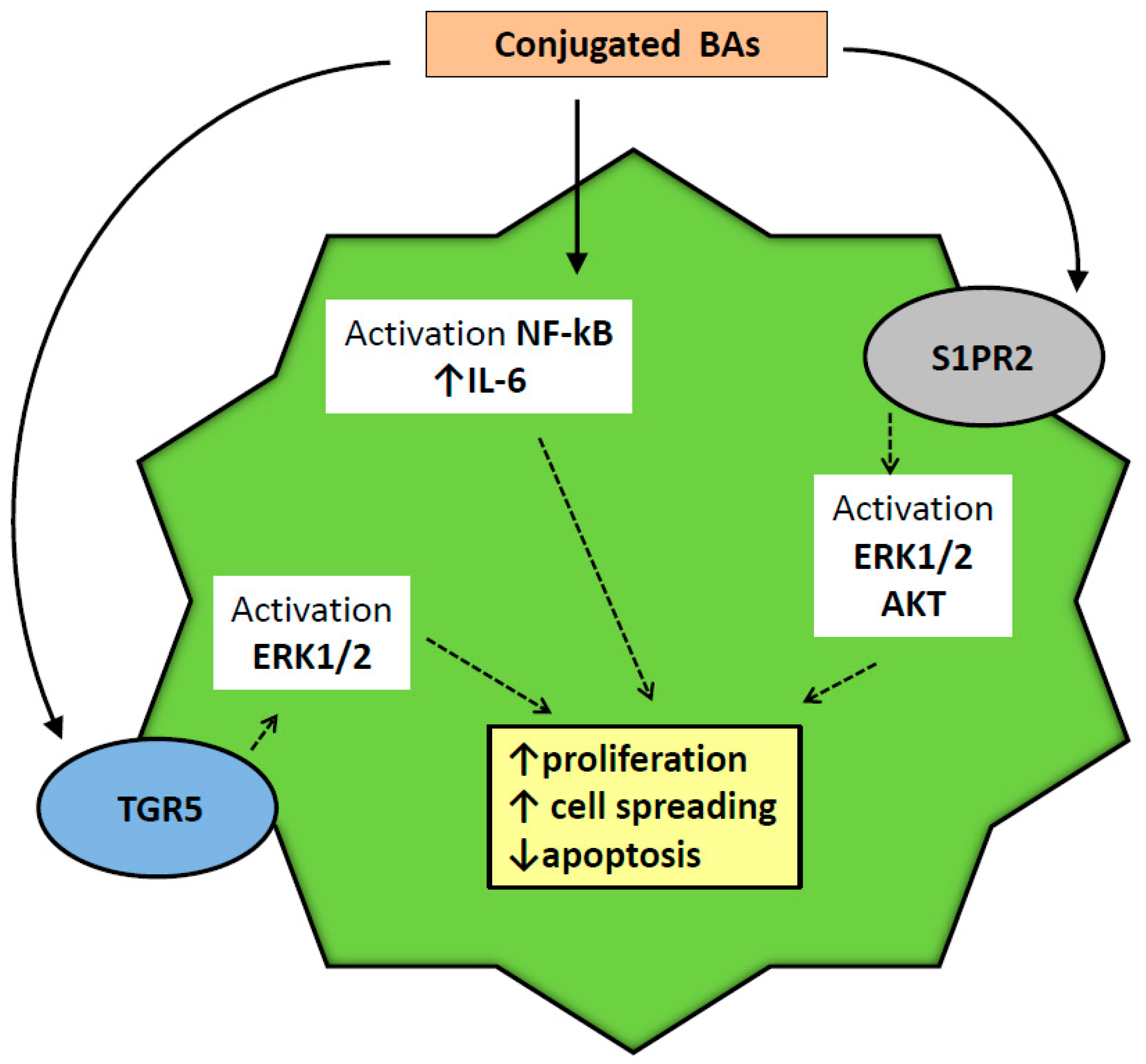
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the european network for the study of cholangiocarcinoma (ens-cca). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.W.; Peng, J.; Tang, X.; Ko, K.S.; Xia, M.; Aller, M.A. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 2011, 141, 378–388, 388.e1–388.e4. [Google Scholar]
- Dai, J.; Wang, H.; Dong, Y.; Zhang, Y.; Wang, J. Bile acids affect the growth of human cholangiocarcinoma via nf-kb pathway. Cancer Invest. 2013, 31, 111–120. [Google Scholar] [CrossRef]
- Mathema, V.B.; Chaijaroenkul, W.; Karbwang, J.; Na-Bangchang, K. Growth inhibitory effect of beta-eudesmol on cholangiocarcinoma cells and its potential suppressive effect on heme oxygenase-1 production, stat1/3 activation, and nf-kappab downregulation. Clin. Exp. Pharmacol. Physiol. 2017, 44, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.L.; Liang, Y.J.; Zheng, T.S.; Song, R.P.; Wang, J.B.; Sun, B.S.; Pan, S.H.; Qu, L.D.; Liu, J.R.; Jiang, H.C.; et al. Ef24 inhibits tumor growth and metastasis via suppressing nf-kappab dependent pathways in human cholangiocarcinoma. Sci. Rep. 2016, 6, 32167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.H.; Ren, H.Y.; Shen, J.X.; Zhang, X.Y.; Ye, H.M.; Shen, D.Y. Magnolol suppresses the proliferation and invasion of cholangiocarcinoma cells via inhibiting the nf-kappab signaling pathway. Biomed. Pharm. 2017, 94, 474–480. [Google Scholar]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Pares, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751.e8–761.e8. [Google Scholar]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baiocchi, L.; Zhou, T.; Liangpunsakul, S.; Lenci, I.; Santopaolo, F.; Meng, F.; Kennedy, L.; Glaser, S.; Francis, H.; Alpini, G. Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe? Int. J. Mol. Sci. 2019, 20, 1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081869
Baiocchi L, Zhou T, Liangpunsakul S, Lenci I, Santopaolo F, Meng F, Kennedy L, Glaser S, Francis H, Alpini G. Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe? International Journal of Molecular Sciences. 2019; 20(8):1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081869
Chicago/Turabian StyleBaiocchi, Leonardo, Tianhao Zhou, Suthat Liangpunsakul, Ilaria Lenci, Francesco Santopaolo, Fanyin Meng, Lindsey Kennedy, Shannon Glaser, Heather Francis, and Gianfranco Alpini. 2019. "Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe?" International Journal of Molecular Sciences 20, no. 8: 1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081869