Non-Cryogenic Structure and Dynamics of HIV-1 Integrase Catalytic Core Domain by X-ray Free-Electron Lasers

 , , and
, , and
Abstract
:1. Introduction
2. Results
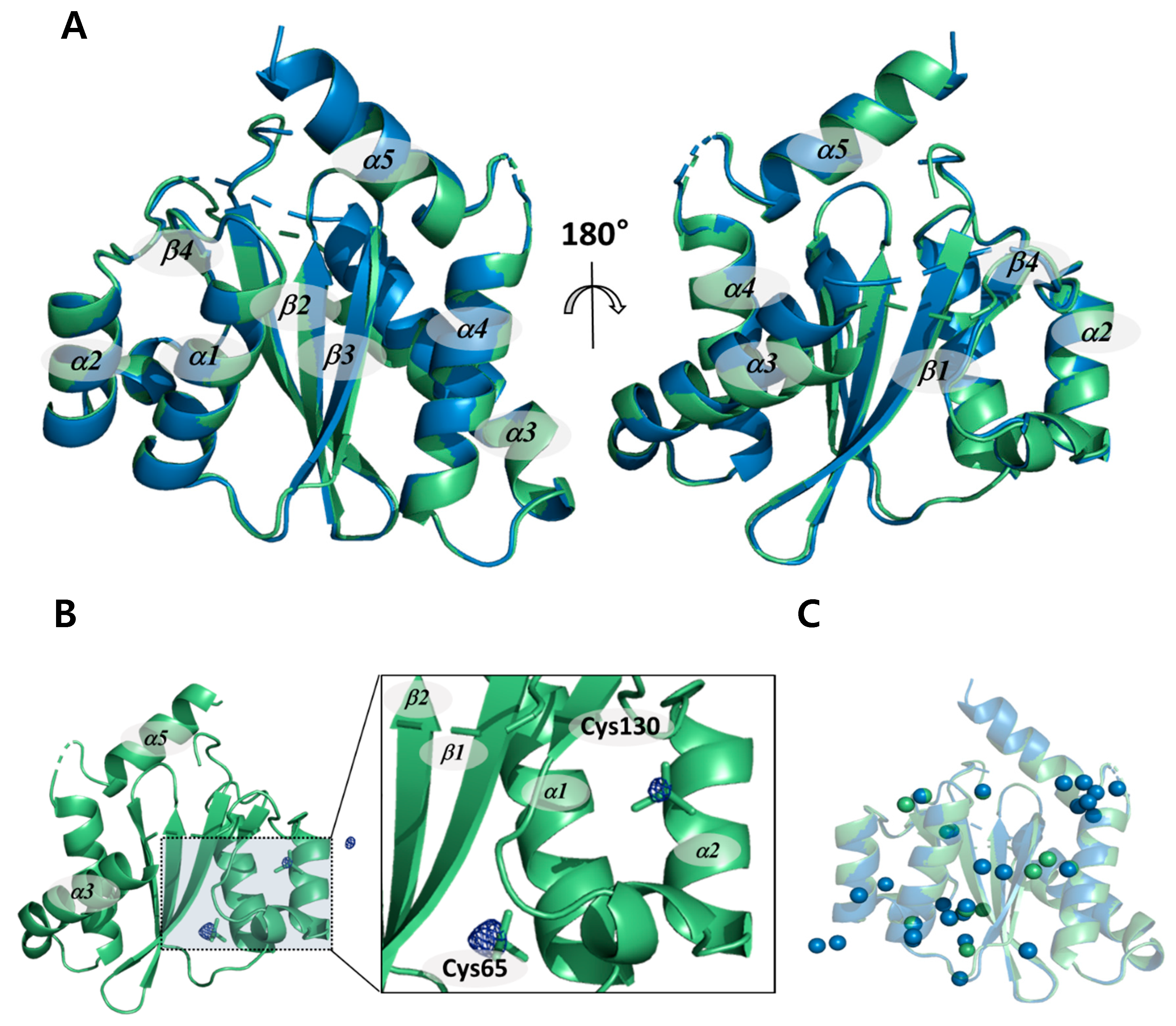
2.1. Non-Cryogenic and Cryogenic Structures
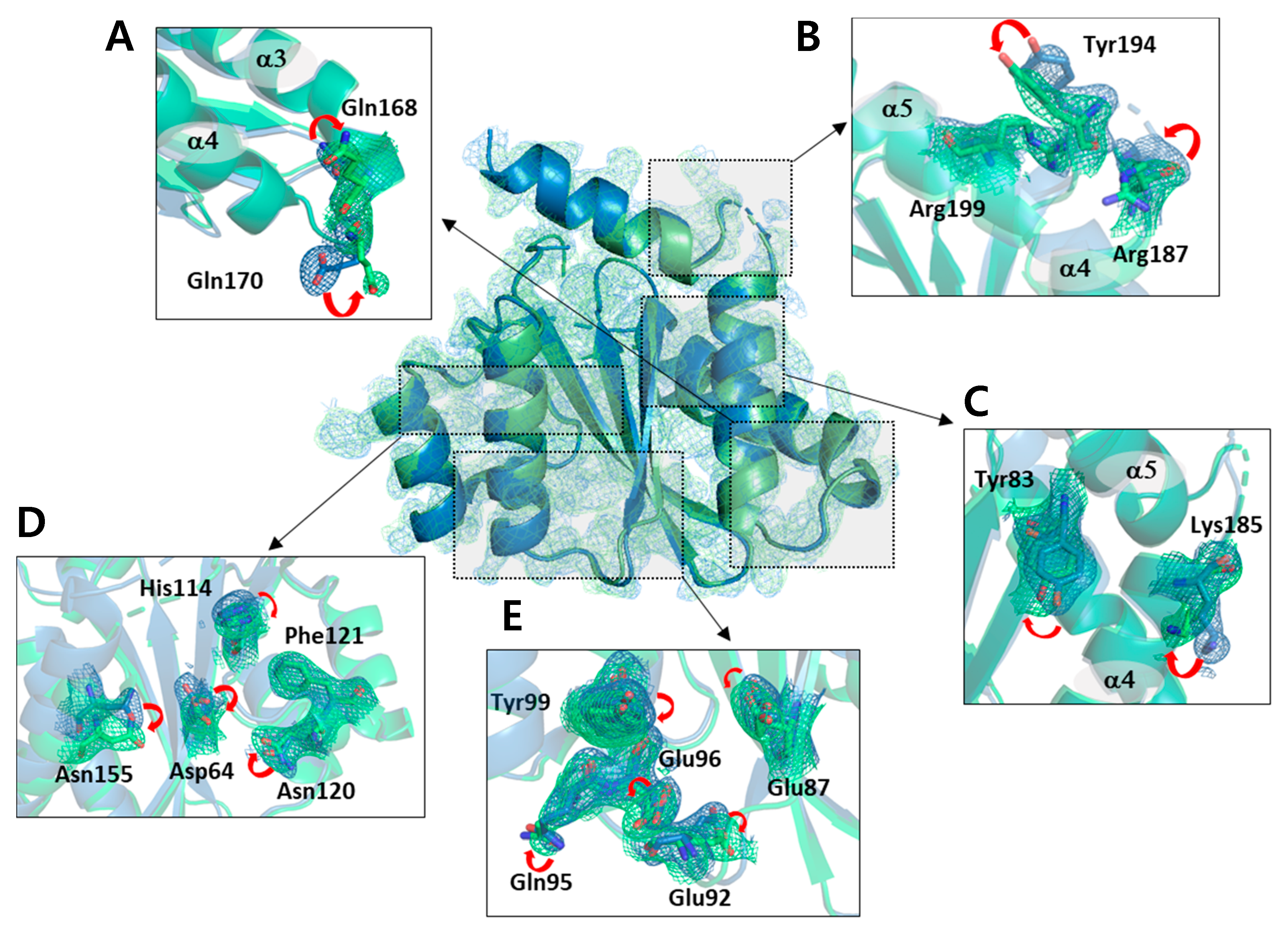
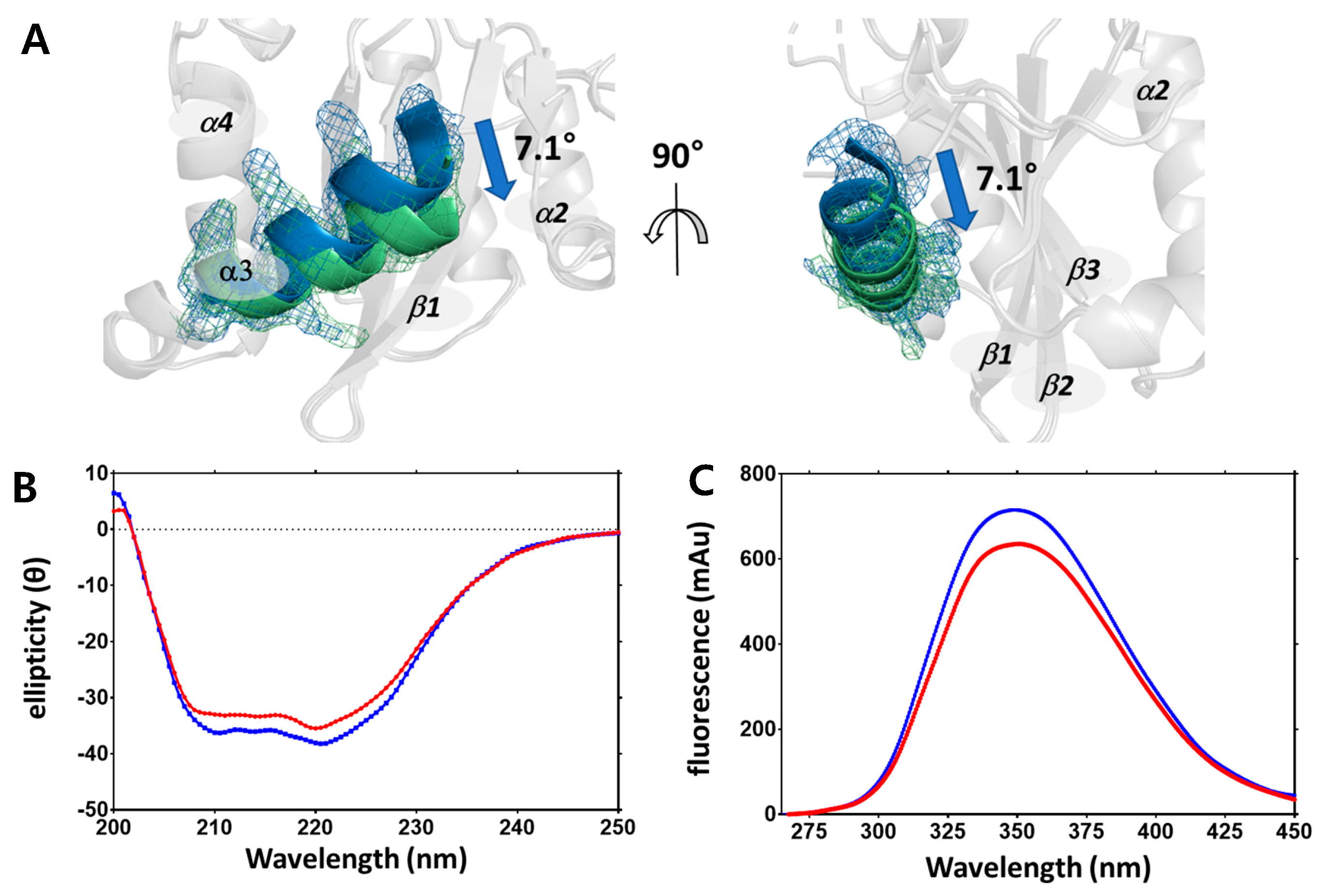
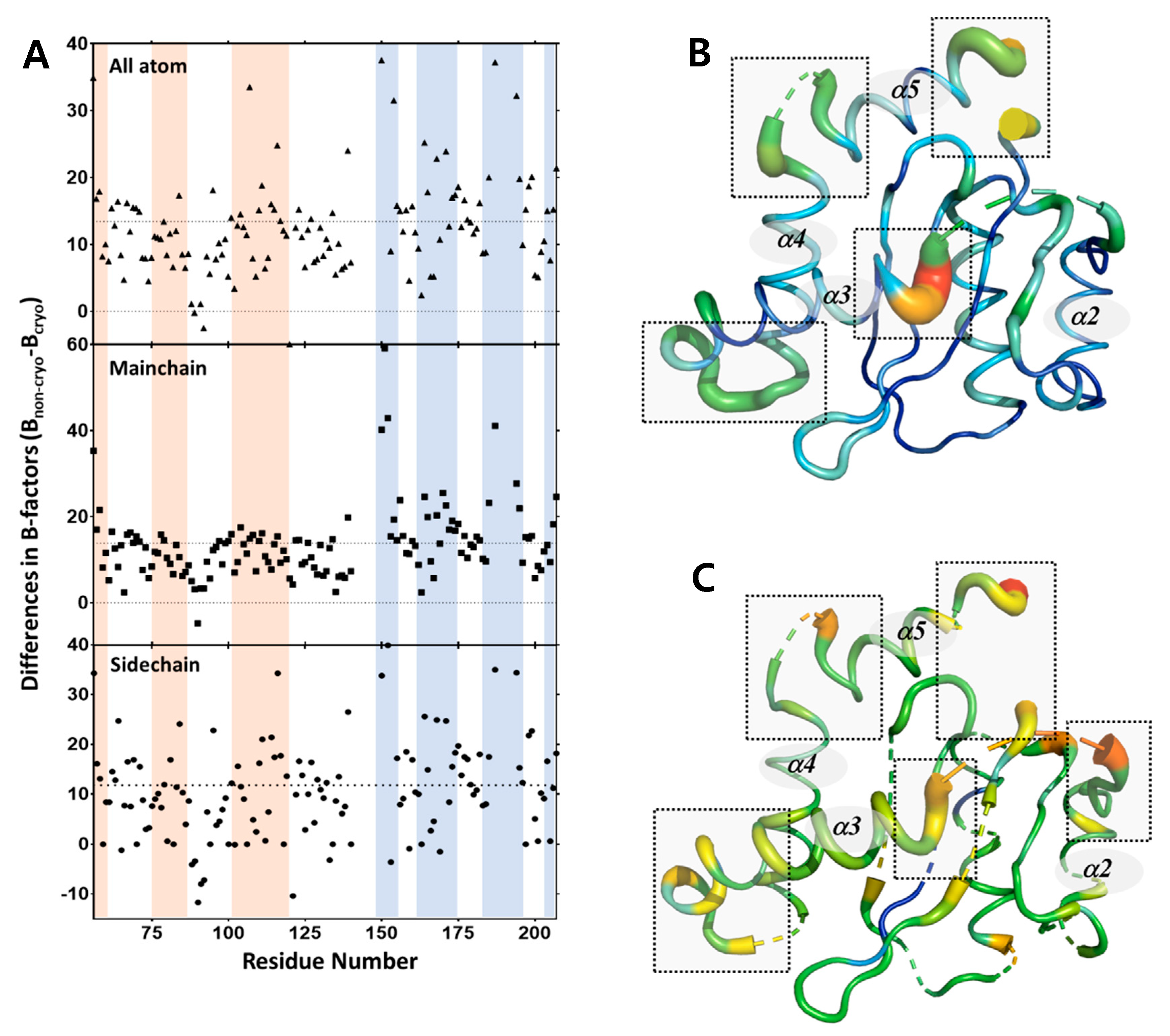
2.2. Structural Differences upon Temperature Difference
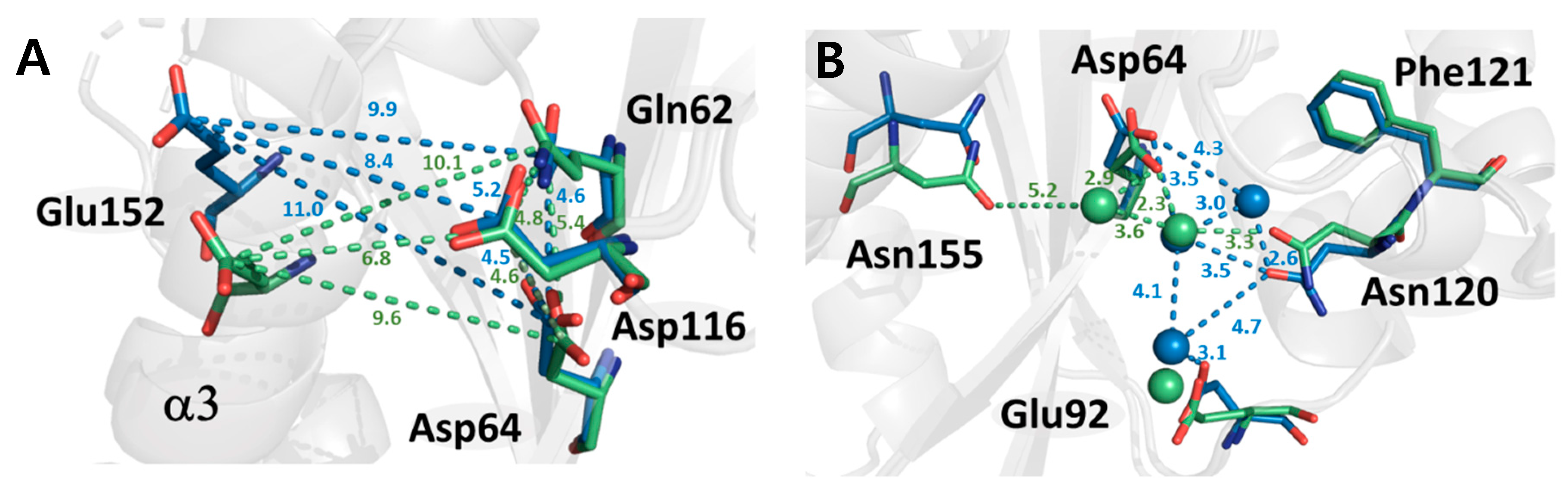
2.3. Structural Properties of HIV-1 Integrase Catalytic Core Region
2.4. Protein Dynamics of IN-CCD by SFX-XFELs
3. Discussion
4. Materials and Methods
4.1. Protein Purification and Crystallization
4.2. Data collection and Processing at PAL
4.3. Structure Determination and Refinement
4.4. Circular Dichroism and Fluorescence Spectroscopy
4.5. 15N-{1H} NOE Experiment
4.6. DPI Analysis for Non-Cryogenic and Cryogenic Structures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Kupitz, C.; OlmosJr, J.L.; Holl, M.; Tremblay, L.; Pande, K.; Pandey, S.; Oberthür, D.; Hunter, M.; Liang, M.; Aquila, A.; et al. Structural enzymology using X-ray free electron lasers. Struct. Dyn. 2017, 4, 044003. [Google Scholar] [CrossRef]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.M.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Adv. Condens. Matter Phys. 2013, 2013, 10. [Google Scholar] [CrossRef]
- Calvey, G.D.; Katz, A.M.; Schaffer, C.B.; Pollack, L. Mixing injector enables time-resolved crystallography with high hit rate at X-ray free electron lasers. Struct. Dyn. 2016, 3, 054301. [Google Scholar] [CrossRef]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2016, 541, 242. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.; Fujiwara, T.; Craigie, R. Retroviral DNA integration directed by HIV integration protein in vitro. Science 1990, 249, 1555–1558. [Google Scholar] [CrossRef] [Green Version]
- LaFemina, R.L.; Schneider, C.L.; Robbins, H.L.; Callahan, P.L.; LeGrow, K.; Roth, E.; Schleif, W.A.; Emini, E.A. Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cells. J. Virol. 1992, 66, 7414–7419. [Google Scholar] [PubMed]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 208. [Google Scholar] [CrossRef]
- Brown, P.O.; Bowerman, B.; Varmus, H.E.; Bishop, J.M. Retroviral integration: Structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc. Natl. Acad. Sci. USA 1989, 86, 2525–2529. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Fujiwara, T.; Bushman, F. The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 1990, 62, 829–837. [Google Scholar] [CrossRef]
- Sherman, P.A.; Fyfe, J.A. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. USA 1990, 87, 5119–5123. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Merkel, G.; Kulkosky, J.; Leis, J.; Skalka, A.M. The avian retroviral IN protein is both necessary and sufficient for integrative recombination in vitro. Cell 1990, 63, 87–95. [Google Scholar] [CrossRef]
- Bushman, F.D.; Craigie, R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: Specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. USA 1991, 88, 1339–1343. [Google Scholar] [CrossRef]
- Markowitz, M.; Morales-Ramirez, J.O.; Nguyen, B.-Y.; Kovacs, C.M.; Steigbigel, R.T.; Cooper, D.A.; Liporace, R.; Schwartz, R.; Isaacs, R.; Gilde, L.R.; et al. Antiretroviral Activity, Pharmacokinetics, and Tolerability of MK-0518, a Novel Inhibitor of HIV-1 Integrase, Dosed As Monotherapy for 10 Days in Treatment-Naive HIV-1-Infected Individuals. JAIDS J. Acquir. Immune Defic. Syndr. 2006, 43, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarino, A. A historical sketch of the discovery and development of HIV-1 integrase inhibitors. Expert Opin. Investig. Drugs 2006, 15, 1507–1522. [Google Scholar] [CrossRef]
- Sax, P.E.; DeJesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J.E.; Liu, H.C.; Zhong, L.; Yale, K.; et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 2012, 379, 2439–2448. [Google Scholar] [CrossRef]
- Esposito, D.; Craigie, R. HIV Integrase Structure and Function. In Advances in Virus Research; Rlaramorosch, K., Murphy, F.A., Shawn, A.J., Eds.; Academic Press: Cambridge, MA, USA, 1999; Volume 52, pp. 319–333. [Google Scholar]
- Chiu, T.K.; Davies, D.R. Structure and Function of HIV-1 Integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef]
- Kulkosky, J.; Jones, K.S.; Katz, R.A.; Mack, J.P.; Skalka, A.M. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 1992, 12, 2331–2338. [Google Scholar] [CrossRef]
- Dyda, F.; Hickman, A.; Jenkins, T.; Engelman, A.; Craigie, R.; Davies, D. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef]
- Goldgur, Y.; Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Craigie, R.; Davies, D.R. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc. Natl. Acad. Sci. USA 1998, 95, 9150–9154. [Google Scholar] [CrossRef] [Green Version]
- Goldgur, Y.; Craigie, R.; Cohen, G.H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D.R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef]
- Hare, S.; Vos, A.M.; Clayton, R.F.; Thuring, J.W.; Cummings, M.D.; Cherepanov, P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232. [Google Scholar] [CrossRef]
- Hare, S.; Shun, M.-C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A Novel Co-Crystal Structure Affords the Design of Gain-of-Function Lentiviral Integrase Mutants in the Presence of Modified PSIP1/LEDGF/p75. PLOS Pathog. 2009, 5, e1000259. [Google Scholar] [CrossRef]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Bordo, D. How many water molecules can be detected by protein crystallography? Acta Crystallogr. Sect. D 1999, 55, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Delelis, O.; Carayon, K.; Saïb, A.; Deprez, E.; Mouscadet, J.-F. Integrase and integration: Biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef]
- Marchand, C.; Kiselev, E.; Marler, L.; Métifiot, M.; Pommier, Y.; Johnson, B.C.; Hughes, S.H.; Burke, T.R., Jr.; Zhao, X.Z. Selectivity for strand-transfer over 3′-processing and susceptibility to clinical resistance of HIV-1 integrase inhibitors are driven by key enzyme–DNA interactions in the active site. Nucleic Acids Res. 2016, 44, 6896–6906. [Google Scholar]
- Hare, S.; Maertens, G.N.; Cherepanov, P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 31, 3020–3028. [Google Scholar] [CrossRef] [Green Version]
- Cherepanov, P.; Ambrosio, A.L.B.; Rahman, S.; Ellenberger, T.; Engelman, A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef] [Green Version]
- Fitzkee, N.C.; Masse, J.E.; Shen, Y.; Davies, D.R.; Bax, A. Solution Conformation and Dynamics of the HIV-1 Integrase Core Domain. J. Biol. Chem. 2010, 285, 18072–18084. [Google Scholar] [CrossRef]
- Jenkins, T.M.; Engelman, A.; Ghirlando, R.; Craigie, R. A Soluble Active Mutant of HIV-1 Integrase: INVOLVEMENT OF BOTH THE CORE AND CARBOXYL-TERMINAL DOMAINS IN MULTIMERIZATION. J. Biol. Chem. 1996, 271, 7712–7718. [Google Scholar] [CrossRef]
- Choi, S.; Attri, P.; Lee, I.; Oh, J.; Yun, J.-H.; Park, J.H.; Choi, E.H.; Lee, W. Structural and functional analysis of lysozyme after treatment with dielectric barrier discharge plasma and atmospheric pressure plasma jet. Sci. Rep. 2017, 7, 1027. [Google Scholar] [CrossRef] [PubMed]
- Asante-Appiah, E.; Skalka, A.M. A Metal-induced Conformational Change and Activation of HIV-1 Integrase. J. Biol. Chem. 1997, 272, 16196–16205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, D.A. Thermal Vibrations in Crystallography. Phys. Bull. 1975, 26, 498–499. [Google Scholar] [CrossRef]
- Barty, A.; Caleman, C.; Aquila, A.; Timneanu, N.; Lomb, L.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 2011, 6, 35. [Google Scholar] [CrossRef]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial Femtosecond Crystallography of G Protein–Coupled Receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Fromme, R.; Ishchenko, A.; Metz, M.; Chowdhury, S.R.; Basu, S.; Boutet, S.; Fromme, P.; White, T.A.; Barty, A.; Spence, J.C.H.; et al. Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ 2015, 2, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.-S.; Min, C.-K.; Heo, H.; Kim, C.; Yang, H.; Kim, G.; Nam, I.; Baek, S.Y.; Choi, H.-J.; Mun, G.; et al. Hard X-ray free-electron laser with femtosecond-scale timing jitter. Nat. Photonics 2017, 11, 708–713. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.; Nam, K.-H.; Kim, B.; Ko, I.S. Current status of the CXI beamline at the PAL-XFEL. J. Korean Phys. Soc. 2016, 69, 1089–1093. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.; Kim, S.; Nam, K.H. Multifarious injection chamber for molecular structure study (MICOSS) system: Development and application for serial femtosecond crystallography at Pohang Accelerator Laboratory X-ray Free-Electron Laser. J. Synchrotron Radiat. 2018, 25, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.-Y.; Park, J.; Kim, S.; Kim, S.; Rah, S.; Lim, J.; Nam, K.H. Focusing X-ray free-electron laser pulses using Kirkpatrick-Baez mirrors at the NCI hutch of the PAL-XFEL. J. Synchrotron Radiat. 2018, 25, 289–292. [Google Scholar] [CrossRef]
- Mariani, V.; Morgan, A.; Yoon, C.H.; Lane, T.J.; White, T.A.; O’Grady, C.; Kuhn, M.; Aplin, S.; Koglin, J.; Barty, A.; et al. OnDA: Online data analysis and feedback for serial X-ray imagingThis article will form part of a virtual special issue of the journal on free-electron laser software. J. Appl. Crystallogr. 2016, 49, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Liu, H. Click: A Visualization-Based Program for Preprocessing of Serial Crystallography Data, GitHub Repository. Available online: https://github.com/LiuLab-CSRC/ClickX (accessed on 19 April 2019).
- Barty, A.; Kirian, R.A.; Maia, F.R.N.C.; Hantke, M.; Yoon, C.H.; White, T.A.; Chapman, H. Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014, 47, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Kirian, R.A.; Martin, A.V.; Aquila, A.; Nass, K.; Barty, A.; Chapman, H.N. CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Crystallogr. 2012, 45, 335–341. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Attri, P.; Venkatesu, P.; Kumar, A. Activity and stability of α-chymotrypsin in biocompatible ionic liquids: Enzyme refolding by triethyl ammonium acetate. Phys. Chem. Chem. Phys. 2011, 13, 2788–2796. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, M.; Shiratani, M.; Cho, A.E.; Choi, E.H.; Attri, P. Variation in structure of proteins by adjusting reactive oxygen and nitrogen species generated from dielectric barrier discharge jet. Sci. Rep. 2016, 6, 35883. [Google Scholar] [CrossRef] [Green Version]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef] [PubMed]
- Grzesiek, S.; Bax, A. The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 1993, 115, 12593–12594. [Google Scholar] [CrossRef]
- Kumar, K.S.D.; Gurusaran, M.; Satheesh, S.N.; Radha, P.; Pavithra, S.; Thulaa Tharshan, K.P.S.; Helliwell, J.R.; Sekar, K. Online_DPI: A web server to calculate the diffraction precision index for a protein structure. J. Appl. Crystallogr. 2015, 48, 939–942. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Cryogenic Structure of IN-CCD | Cryogenic Structure of IN-CCD | |
|---|---|---|
| Data collection | ||
| Temperature | 293 K | 100 K |
| Space group | P 31 2 1 | P 31 2 1 |
| Cell dimension | ||
| a, b, c (Å) | 73.275 73.275 66.71 | 72.08 72.08 65.192 |
| α, β, γ (°) | 90.00 90.00 20.00 | 90.00 90.00 120.00 |
| Number of collected images | 330,698 | N.A |
| Number of hits | 40,024 | N.A |
| Number of indexed patterns | 27,311 | N.A |
| Number of merged patterns | 27,308 | N.A |
| Indexing rate (%) | 68.2 | N.A |
| Resolution (Å) | 24.67–2.5 (2.61–2.5) * | 36.05–2.15 (2.25–2.15) * |
| Rmerge (%) | N.A | 2.89 (22.2) |
| Rsplit (%) | 9.80 (77.37) | N.A |
| Rpim (%) | N.A | 2.89 (22.2) |
| I/σ (I) | 7.26 (1.45) | 33.7 (4.4) |
| Completeness (%) | 99.85 (100) | 99.98 (100) |
| Multiplicity | 406.99 (275.68) | 1.9 (1.9) |
| CC1/2 (%) | 99.35 (55.06) | 99.9 (86.1) |
| Refinement | ||
| Resolution (Å) | 24.67–2.50 | 36.05–2.15 |
| Number of reflections | 7450 | 10,935 |
| Rwork/Rfree | 0.1766/0.2117 | 0.1994/0.2499 |
| Cruickshank DPI (Å) | 0.205 | 0.174 |
| Number of atoms | ||
| Protein | 1098 | 1106 |
| Water | 12 | 29 |
| B-factor (Å2) | ||
| Wilson B/Overall B | 57.46/67.36 | 41.39/54.02 |
| Root mean square deviations | ||
| Bond lengths | 0.009 | 0.009 |
| Bond angles | 0.47 | 0.60 |
| PDB code | 6JCG | 6JCF |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-H.; Yun, J.-H.; Shi, Y.; Han, J.; Li, X.; Jin, Z.; Kim, T.; Park, J.; Park, S.; Liu, H.; et al. Non-Cryogenic Structure and Dynamics of HIV-1 Integrase Catalytic Core Domain by X-ray Free-Electron Lasers. Int. J. Mol. Sci. 2019, 20, 1943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081943
Park J-H, Yun J-H, Shi Y, Han J, Li X, Jin Z, Kim T, Park J, Park S, Liu H, et al. Non-Cryogenic Structure and Dynamics of HIV-1 Integrase Catalytic Core Domain by X-ray Free-Electron Lasers. International Journal of Molecular Sciences. 2019; 20(8):1943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081943
Chicago/Turabian StylePark, Jae-Hyun, Ji-Hye Yun, Yingchen Shi, Jeongmin Han, Xuanxuan Li, Zeyu Jin, Taehee Kim, Jaehyun Park, Sehan Park, Haiguang Liu, and et al. 2019. "Non-Cryogenic Structure and Dynamics of HIV-1 Integrase Catalytic Core Domain by X-ray Free-Electron Lasers" International Journal of Molecular Sciences 20, no. 8: 1943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081943