HER2 Upregulates ATF4 to Promote Cell Migration via Activation of ZEB1 and Downregulation of E-Cadherin


Abstract
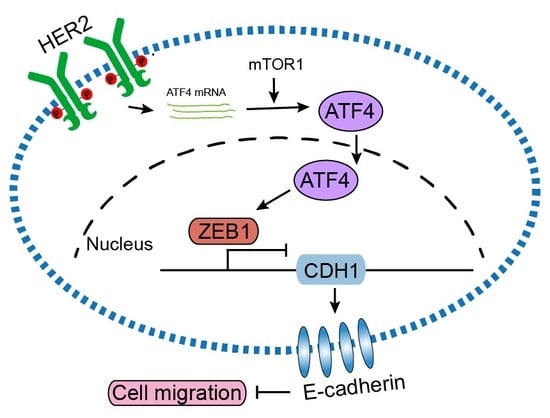
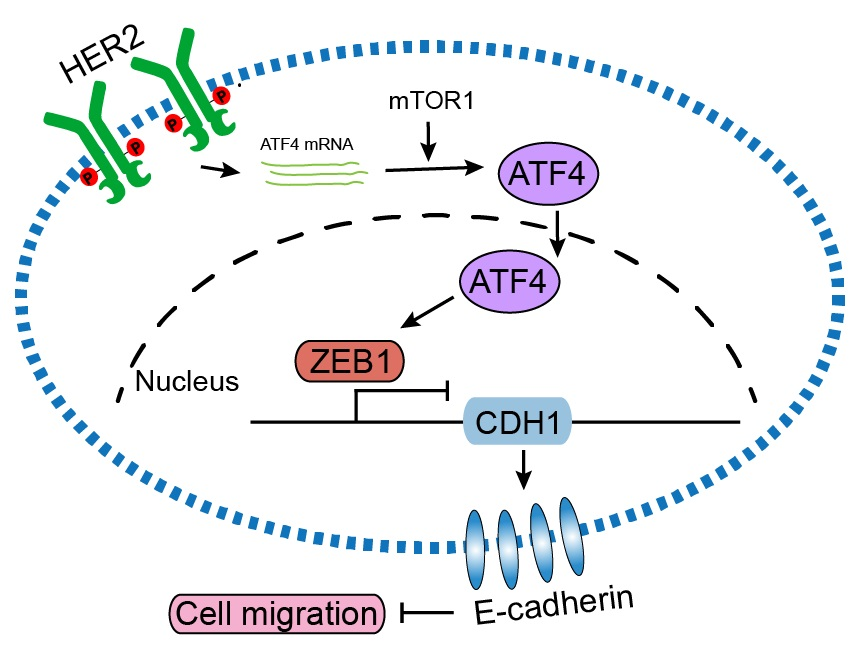
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. HER2 Induces Cell Migration through Upregulation of ATF4 and Downregulation of E-Cadherin
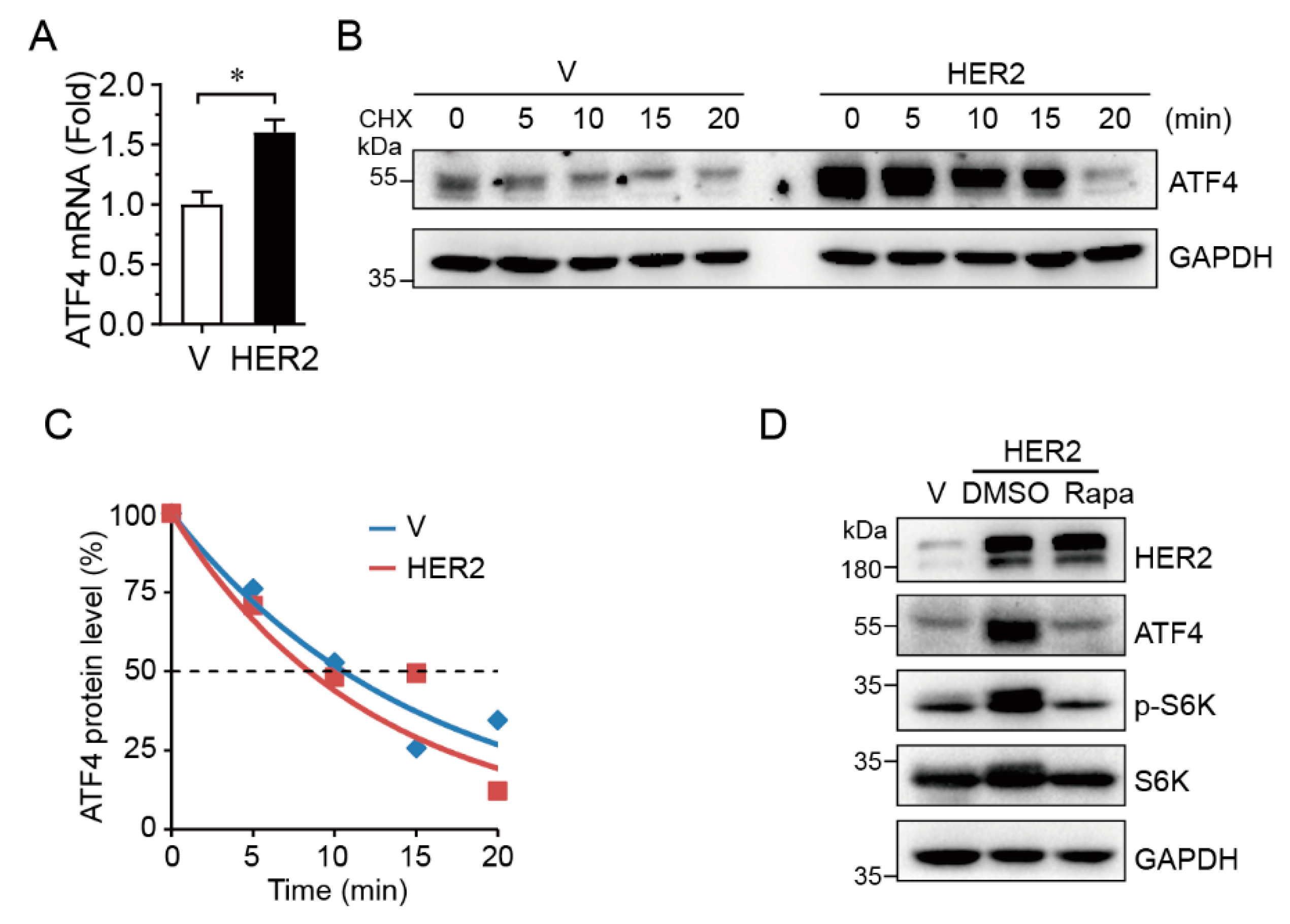
2.2. HER2 Upregulates ATF4 Transcription and Promotes its Protein Expression in an mTOR-Dependent Manner
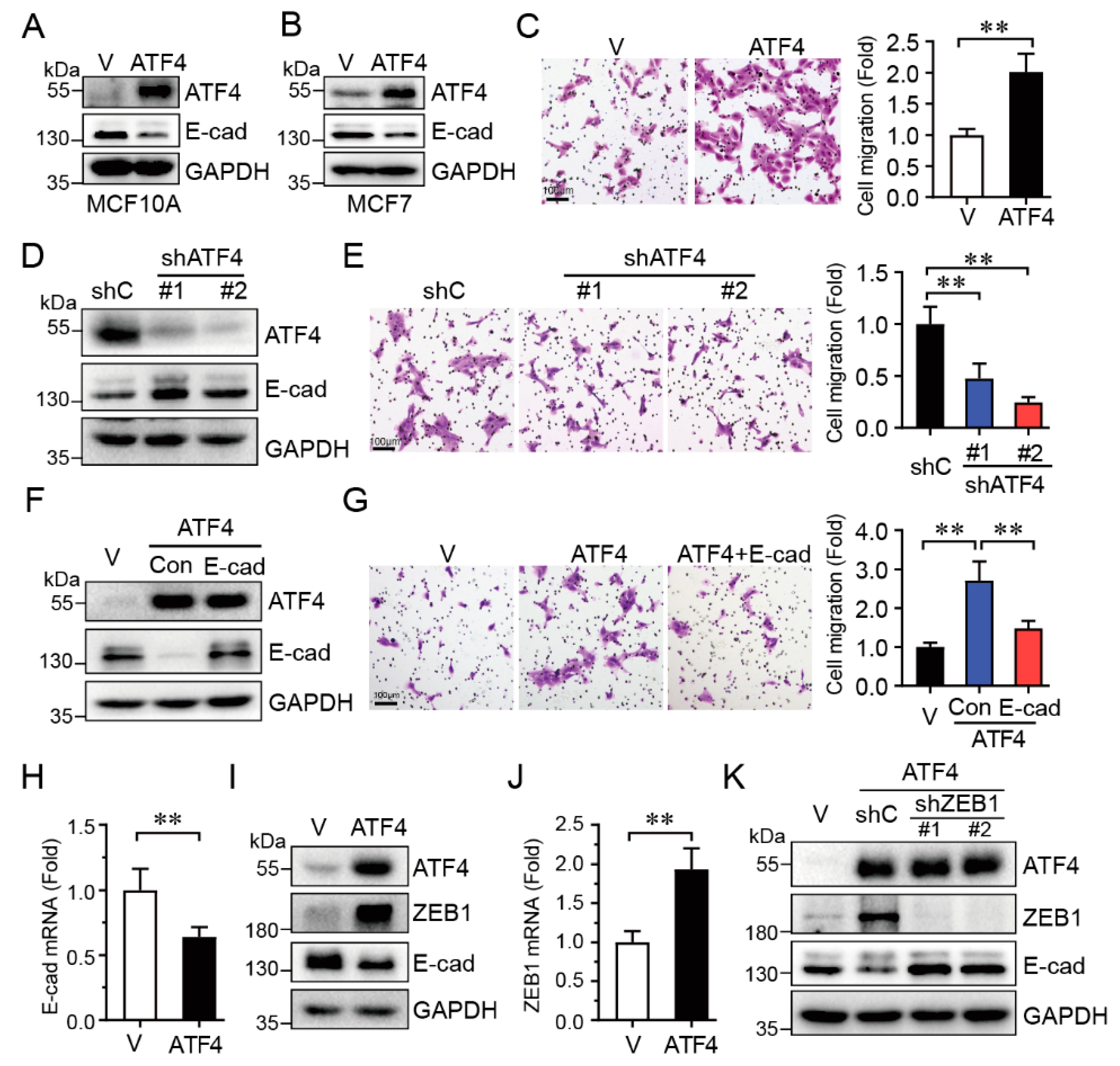
2.3. ATF4 Promotes Cell Migration via Suppressing E-Cadherin Expression
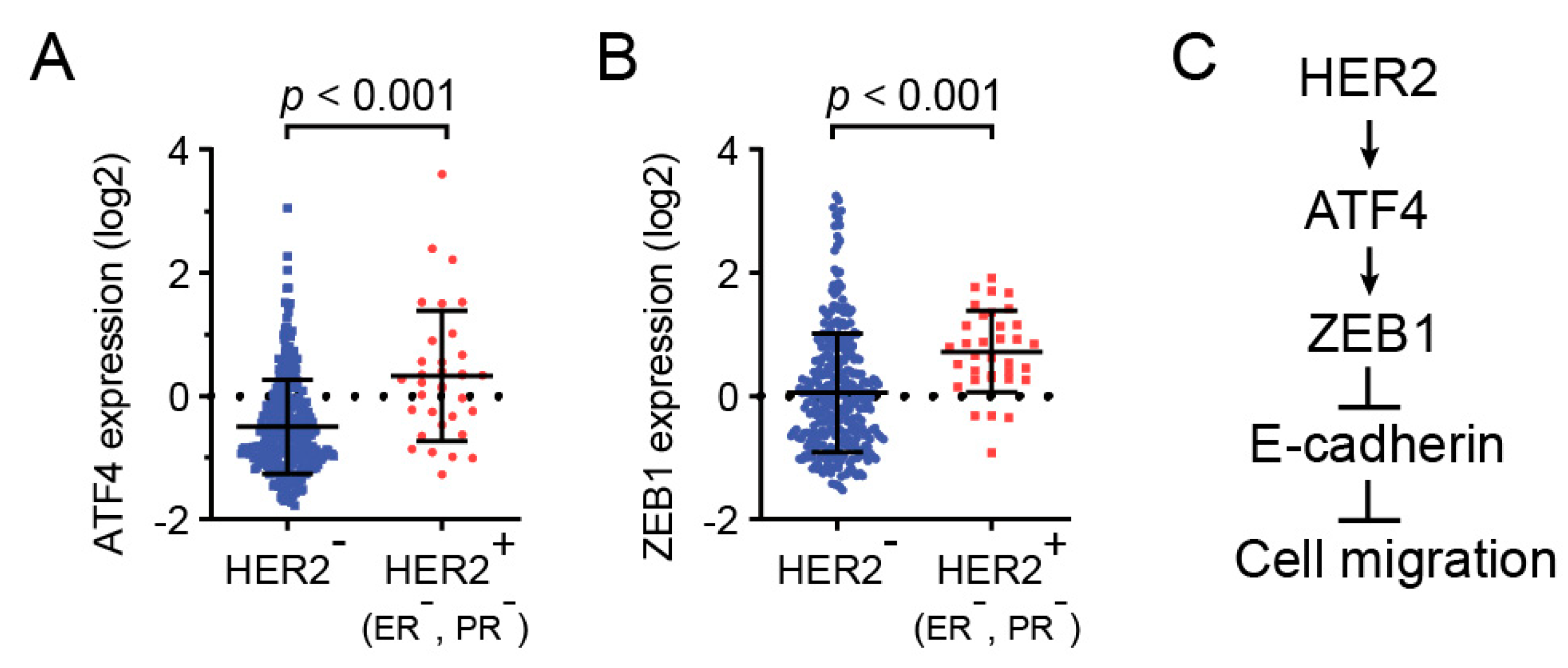
2.4. HER2, ZEB1, and ATF4 Expression is Correlated in Human Breast Cancers
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Cell Morphology, and Cell Proliferation Assays
4.2. Plasmid Construction and Lentivirus Infection
4.3. Western Blot Analysis
4.4. Transwell Assays
4.5. Q-PCR
4.6. Protein Half-life
4.7. Bioinformatics Analysis
4.8. Statistical analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF4 | activating transcription factor |
| ER stress | endoplasmic reticulum stress |
| UPR | unfolded protein response |
| EMT | epithelial-to-mesenchymal transition |
References
- Koeppen, H.K.; Wright, B.D.; Burt, A.D.; Quirke, P.; McNicol, A.M.; Dybdal, N.O.; Sliwkowski, M.X.; Hillan, K.J. Overexpression of her2/neu in solid tumours: An immunohistochemical survey. Histopathology 2001, 38, 96–104. [Google Scholar] [CrossRef]
- Buza, N.; Roque, D.M.; Santin, A.D. Her2/neu in endometrial cancer: A promising therapeutic target with diagnostic challenges. Arch. Pathol. Lab. Med. 2014, 138, 343–350. [Google Scholar] [CrossRef]
- Verri, E.; Guglielmini, P.; Puntoni, M.; Perdelli, L.; Papadia, A.; Lorenzi, P.; Rubagotti, A.; Ragni, N.; Boccardo, F. Her2/neu oncoprotein overexpression in epithelial ovarian cancer: Evaluation of its prevalence and prognostic significance. Clinical study. Oncology 2005, 68, 154–161. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Escriva-de-Romani, S.; Arumi, M.; Bellet, M.; Saura, C. Her2-positive breast cancer: Current and new therapeutic strategies. Breast 2018, 39, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Jean, D.; Tellez, C.; Huang, S.; Davis, D.W.; Bruns, C.J.; McConkey, D.J.; Hinrichs, S.H.; Bar-Eli, M. Inhibition of tumor growth and metastasis of human melanoma by intracellular anti-atf-1 single chain fv fragment. Oncogene 2000, 19, 2721–2730. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.; Feng, Y.; Claps, G.; Fukuda, M.N.; Perlina, A.; Donn, D.; Jilaveanu, L.; Kluger, H.; Freeze, H.H.; Ronai, Z.A. The transcription factor atf2 promotes melanoma metastasis by suppressing protein fucosylation. Sci. Signal. 2015, 8, ra124. [Google Scholar] [CrossRef]
- Yin, X.; Dewille, J.W.; Hai, T. A potential dichotomous role of atf3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wolford, C.C.; Chang, Y.S.; McConoughey, S.J.; Ramsey, S.A.; Aderem, A.; Hai, T. Atf3, an adaptive-response gene, enhances tgfβ signaling and cancer-initiating cell features in breast cancer cells. J. Cell Sci. 2010, 123, 3558–3565. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Li, L.; Zhu, L.J.; Smith, T.W.; Demers, A.; Ross, A.H.; Moser, R.P.; Green, M.R. A genome-wide rna interference screen reveals an essential creb3l2-atf5-mcl1 survival pathway in malignant glioma with therapeutic implications. Nat. Med. 2010, 16, 671. [Google Scholar] [CrossRef]
- Coleman, O.I.; Lobner, E.M.; Bierwirth, S.; Sorbie, A.; Waldschmitt, N.; Rath, E.; Berger, E.; Lagkouvardos, I.; Clavel, T.; McCoy, K.D.; et al. Activated atf6 induces intestinal dysbiosis and innate immune response to promote colorectal tumorigenesis. Gastroenterology 2018, 155, 1539–1552.e1512. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. Atf6α-rheb-mtor signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Hu, X.; Miao, J.; Zhang, M.; Wang, X.; Wang, Z.; Han, J.; Tong, D.; Huang, C. Mirna-103a-3p promotes human gastric cancer cell proliferation by targeting and suppressing atf7 in vitro. Mol. Cells 2018, 41, 390–400. [Google Scholar]
- Chen, H.; Yuan, R.; Zhang, Y.; Zhang, X.; Chen, L.; Zhou, X.; Yuan, Z.; Nie, Y.; Li, M.; Mo, D.; et al. Atf4 regulates srebp1c expression to control fatty acids synthesis in 3t3-l1 adipocytes differentiation. BBA-Gene Regul. Mech. 2016, 1859, 1459–1469. [Google Scholar] [CrossRef]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Molecular Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Singleton, D.C.; Harris, A.L. Targeting the atf4 pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 1189–1202. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the perk/atf4/lamp3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. Atf4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in emt and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Grooteclaes, M.L.; Frisch, S.M. Evidence for a function of ctbp in epithelial gene regulation and anoikis. Oncogene 2000, 19, 3823–3828. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. Zeb1 represses e-cadherin and induces an emt by recruiting the swi/snf chromatin-remodeling protein brg1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional activation of zeb1 by slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Jnvestigative Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between snail1 and twist in the regulation of zeb1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The emt-activator zeb1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by deltaef1 proteins in epithelial mesenchymal transition induced by tgf-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. Nf-kb represses e-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of zeb-1 and zeb-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef]
- Hu, L.; Liang, S.; Chen, H.; Lv, T.; Wu, J.; Chen, D.; Wu, M.; Sun, S.; Zhang, H.; You, H.; et al. △np63α is a common inhibitory target in oncogenic pi3k/ras/her2-induced cell motility and tumor metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E3964–E3973. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic pik3ca mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic kras regulates amino acid homeostasis and asparagine biosynthesis via atf4 and alters sensitivity to l-asparaginase. Cancer Cell 2018, 33, 91–107. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, A.; Munoz-Muela, E.; Marchal, J.A.; Cara, F.E.; Molina, M.P.; Cruz-Lozano, M.; Jimenez, G.; Verma, A.; Ramirez, A.; Qian, W.; et al. Activating transcription factor 4 modulates tgfbeta-induced aggressiveness in triple-negative breast cancer via smad2/3/4 and mtorc2 signaling. Clin. Cancer Res. 2018, 24, 5697–5709. [Google Scholar] [CrossRef]
- Park, Y.; Reyna-Neyra, A.; Philippe, L.; Thoreen, C.C. Mtorc1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of atf4. Cell Reports 2017, 19, 1083–1090. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing e-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of atf4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, P.; Sun, S.; Li, R.; Xiao, Z.-X.; Chen, H. HER2 Upregulates ATF4 to Promote Cell Migration via Activation of ZEB1 and Downregulation of E-Cadherin. Int. J. Mol. Sci. 2019, 20, 2223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092223
Zeng P, Sun S, Li R, Xiao Z-X, Chen H. HER2 Upregulates ATF4 to Promote Cell Migration via Activation of ZEB1 and Downregulation of E-Cadherin. International Journal of Molecular Sciences. 2019; 20(9):2223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092223
Chicago/Turabian StyleZeng, Peng, Shengnan Sun, Rui Li, Zhi-Xiong Xiao, and Hu Chen. 2019. "HER2 Upregulates ATF4 to Promote Cell Migration via Activation of ZEB1 and Downregulation of E-Cadherin" International Journal of Molecular Sciences 20, no. 9: 2223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092223