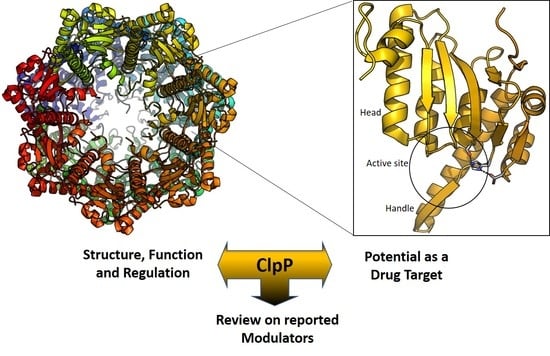
ClpP Protease, a Promising Antimicrobial Target

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
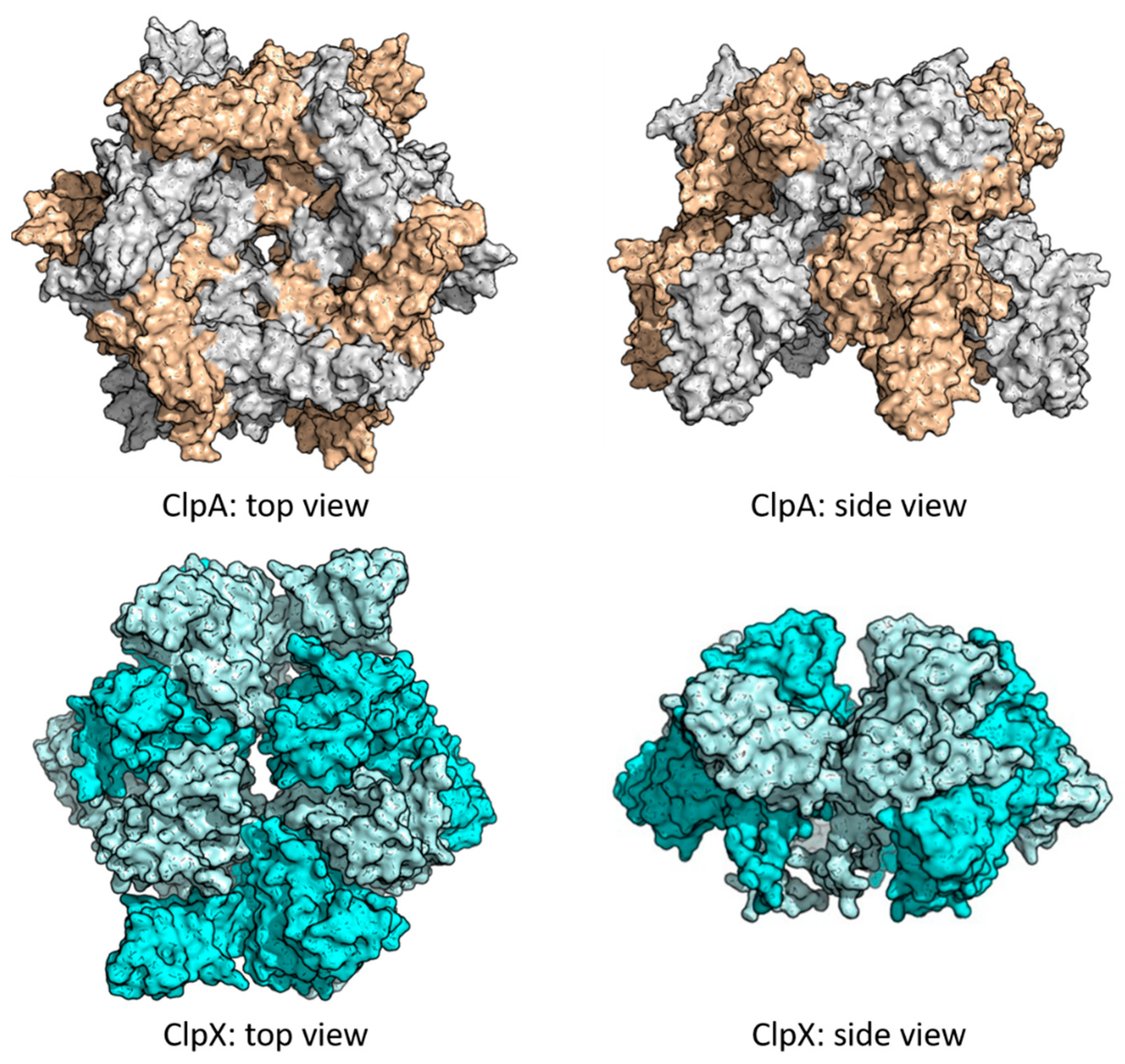{kind=link}
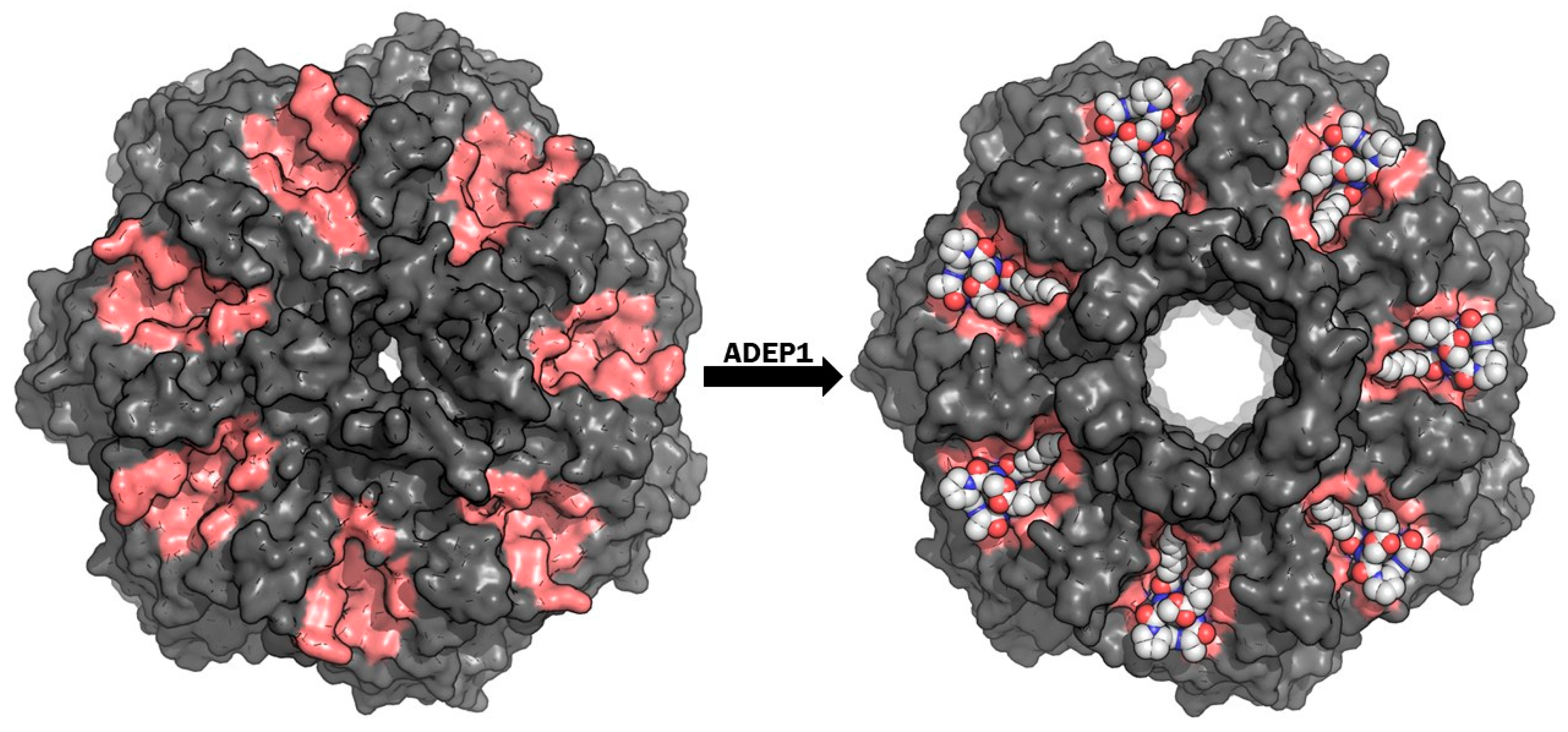{kind=link}
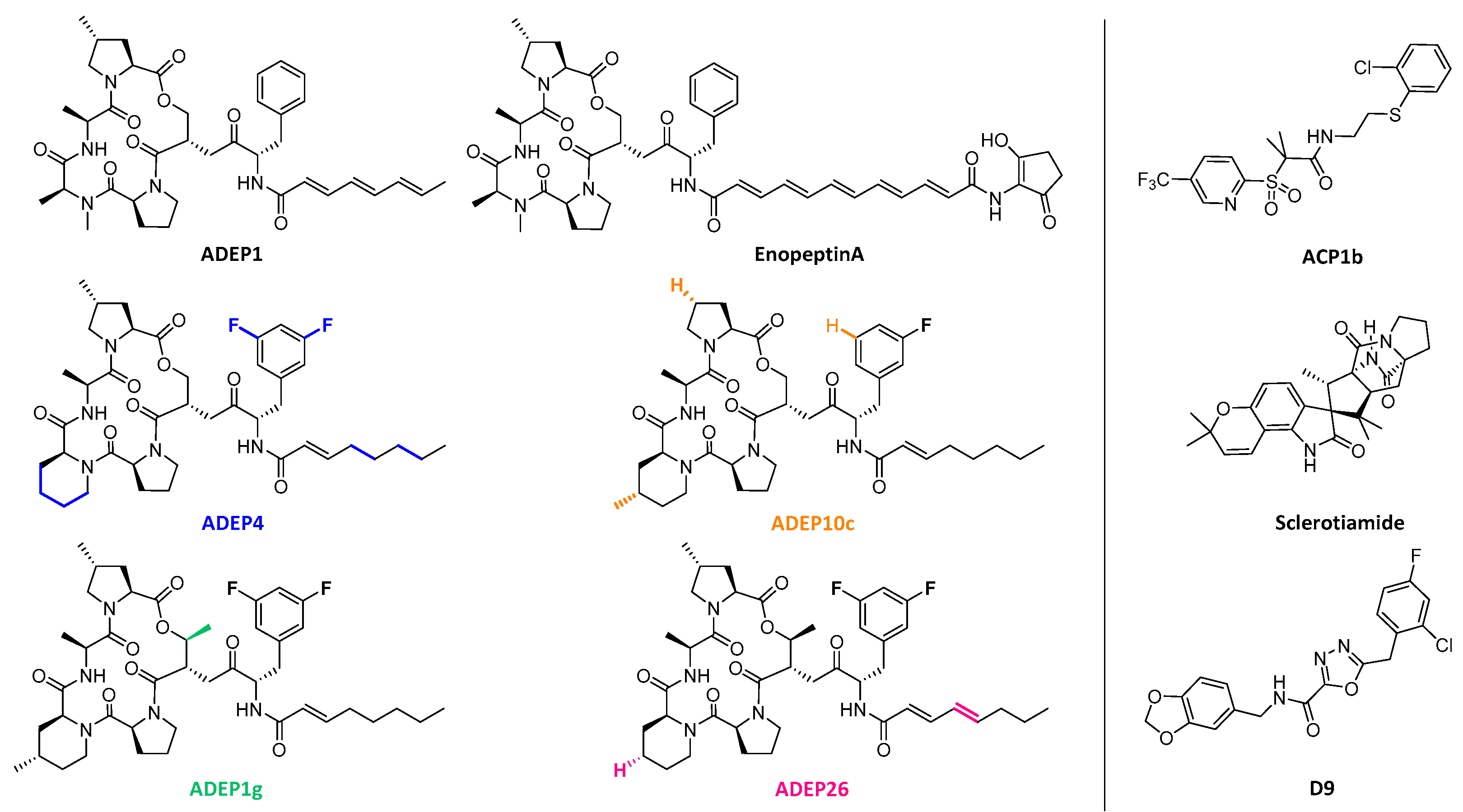{kind=link}
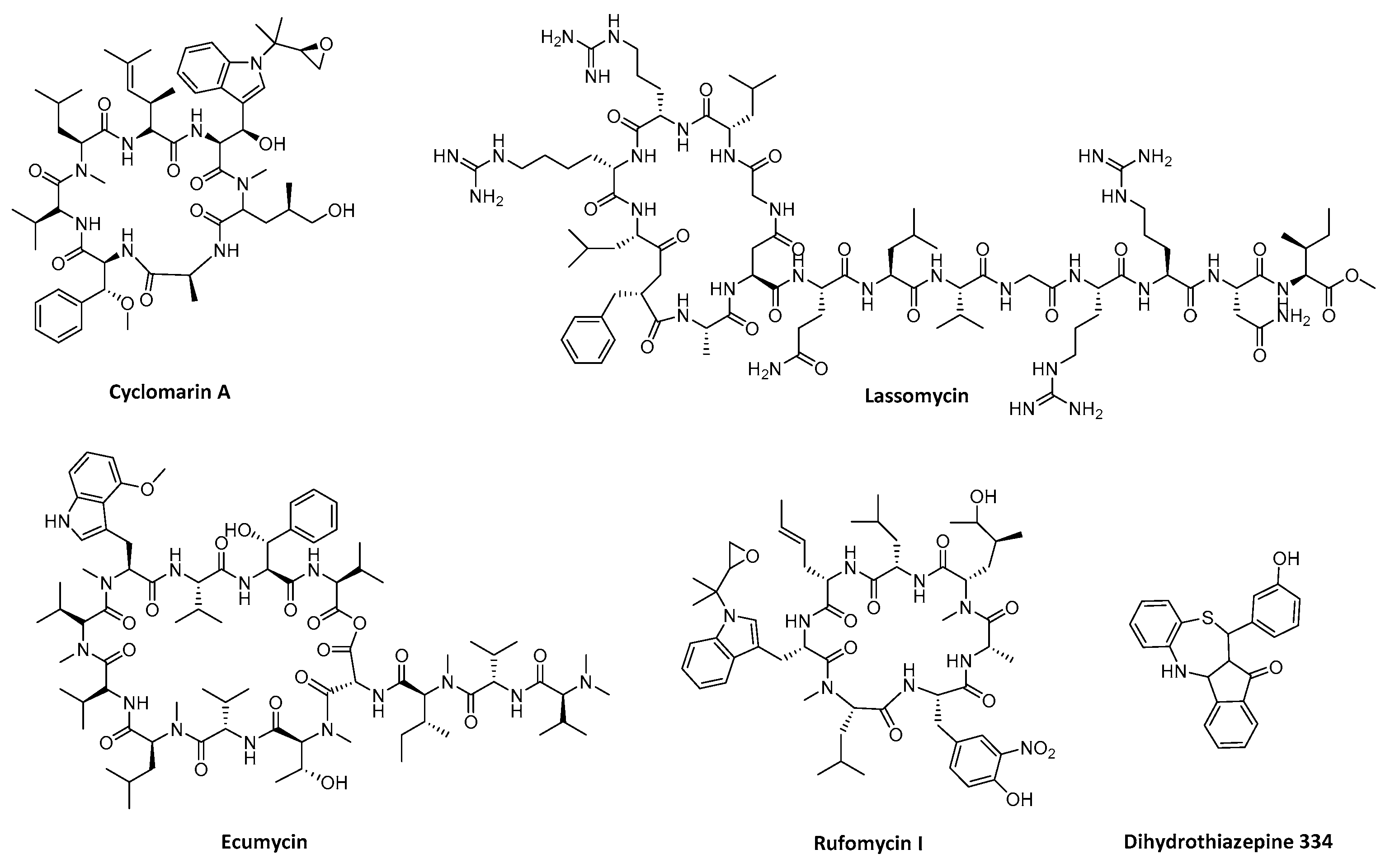{kind=link}
{kind=link}
{kind=link}
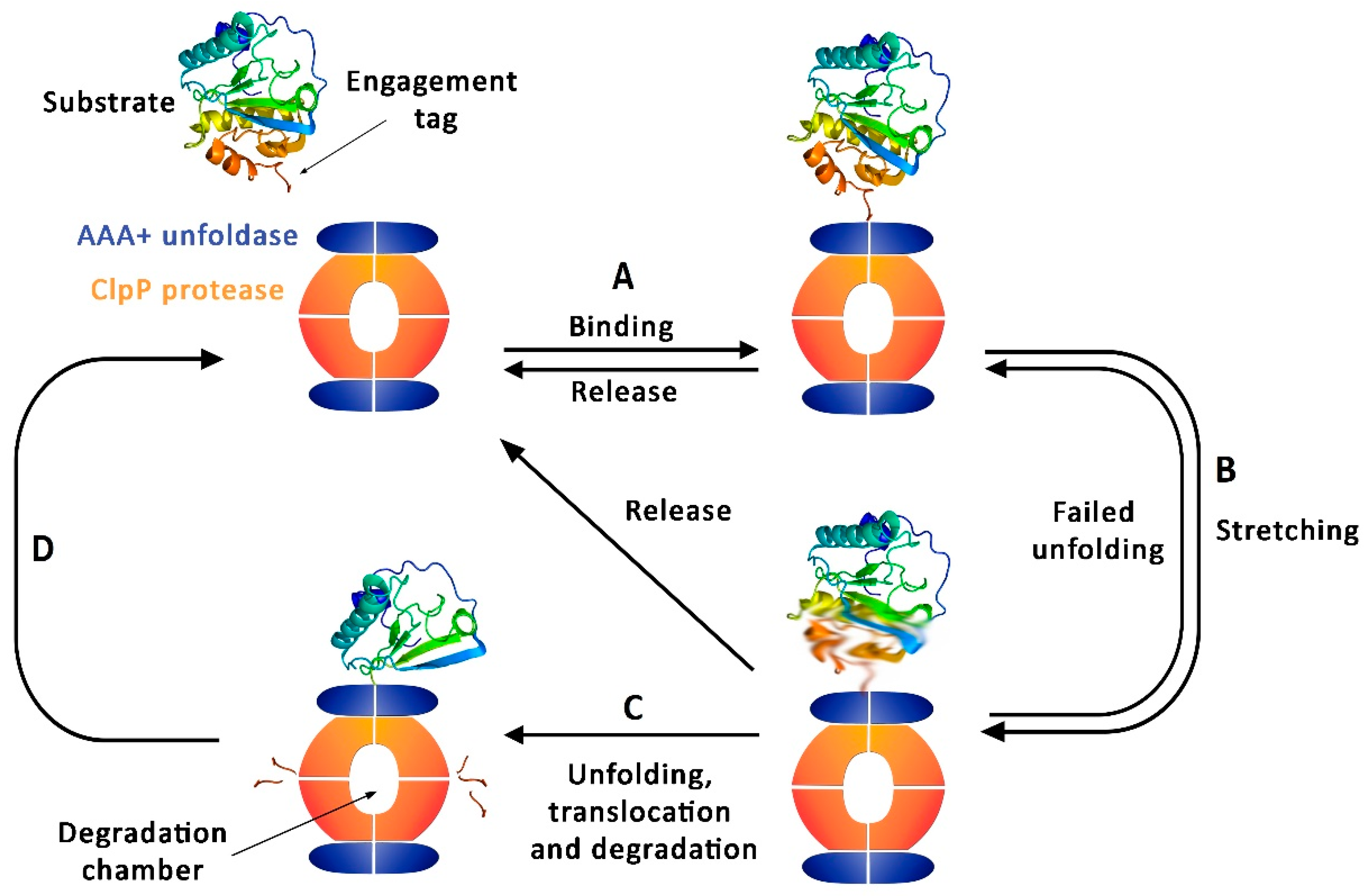
1. ClpP Function and Regulation
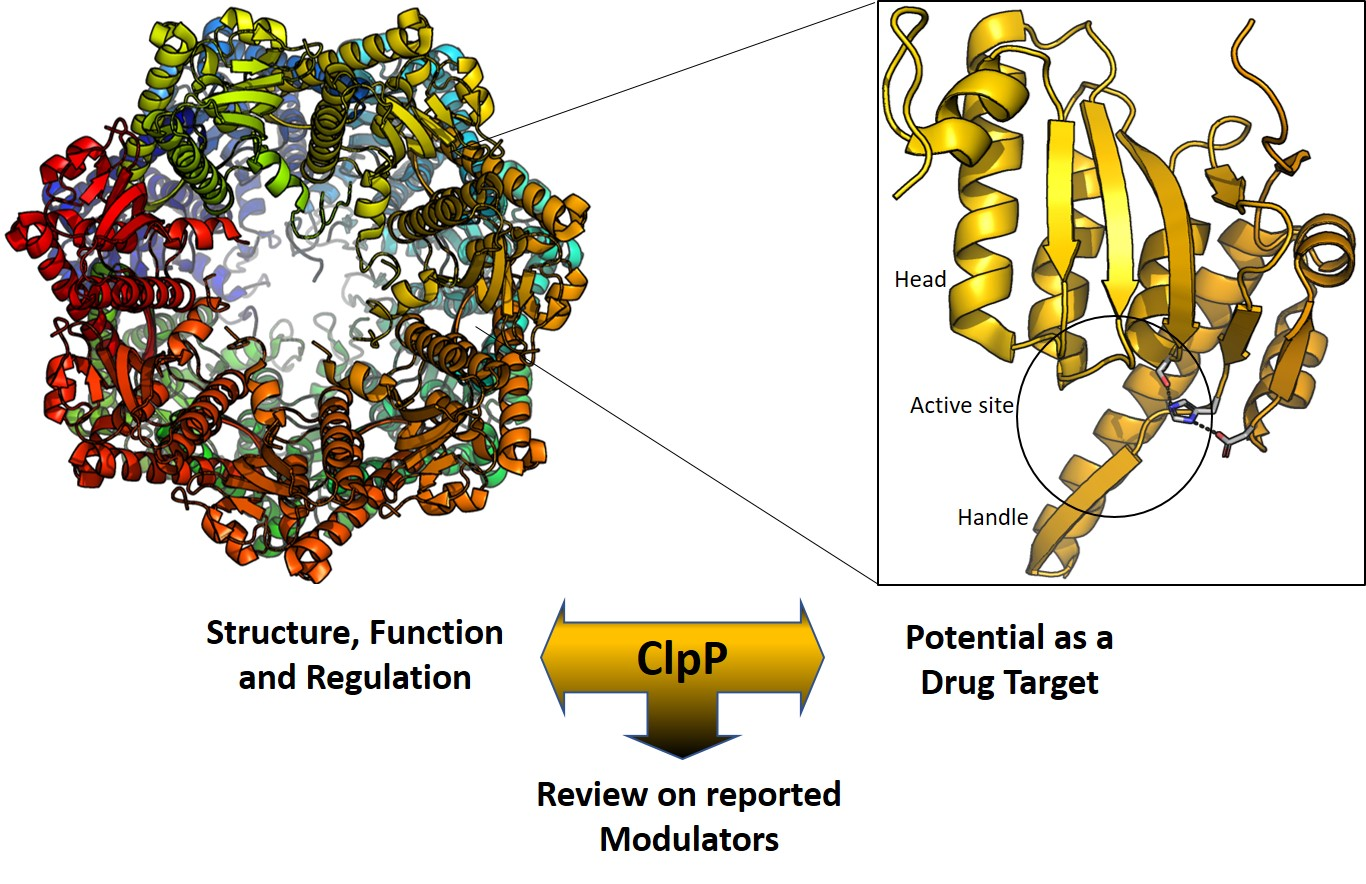
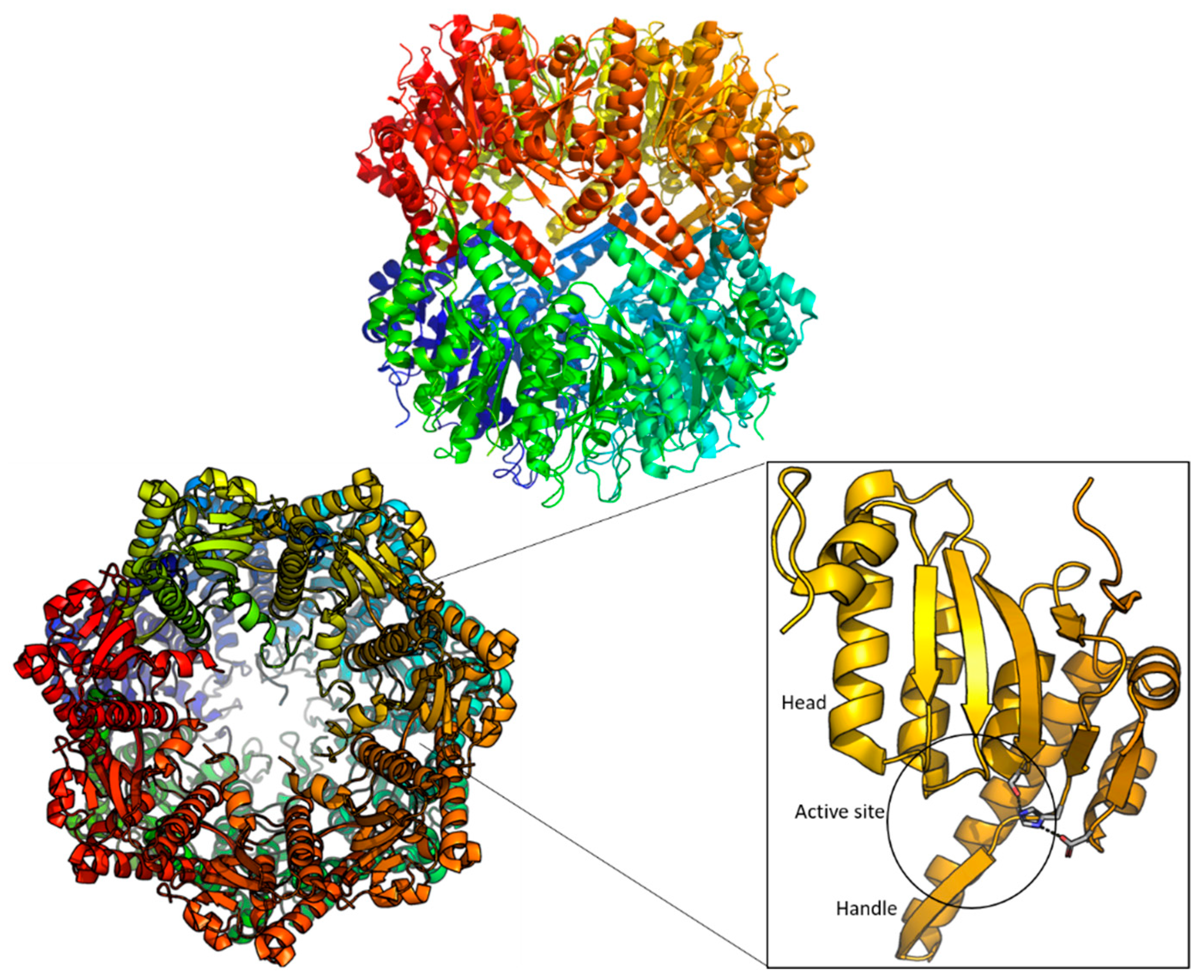
2. Structure of ClpP
3. ClpP as a Drug Target
3.1. ClpP in Bacteria and Parasites
3.2. ClpP in Human Mitochondria
4. ClpP Modulation
4.1. Targeting ClpP-ATPases Interaction
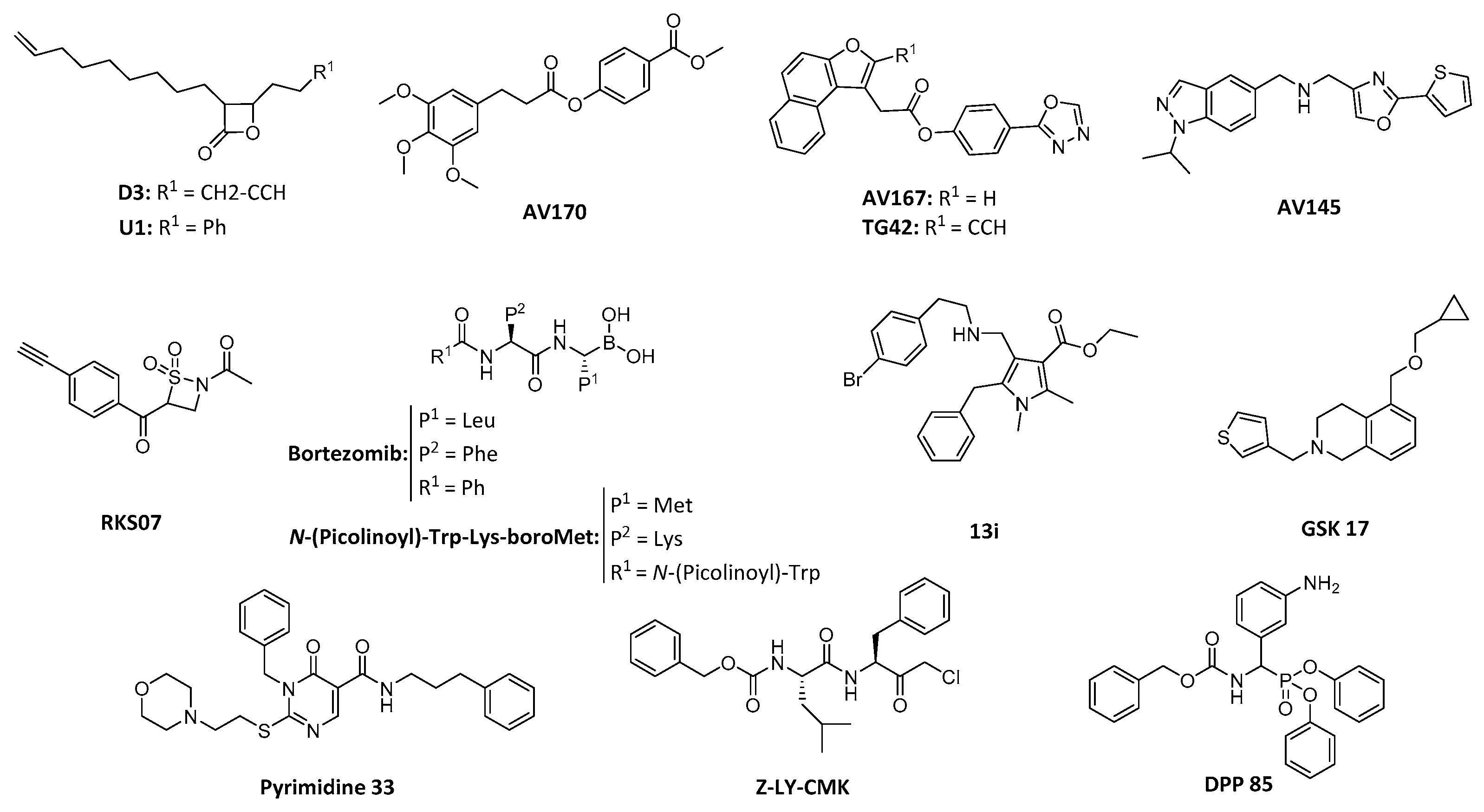
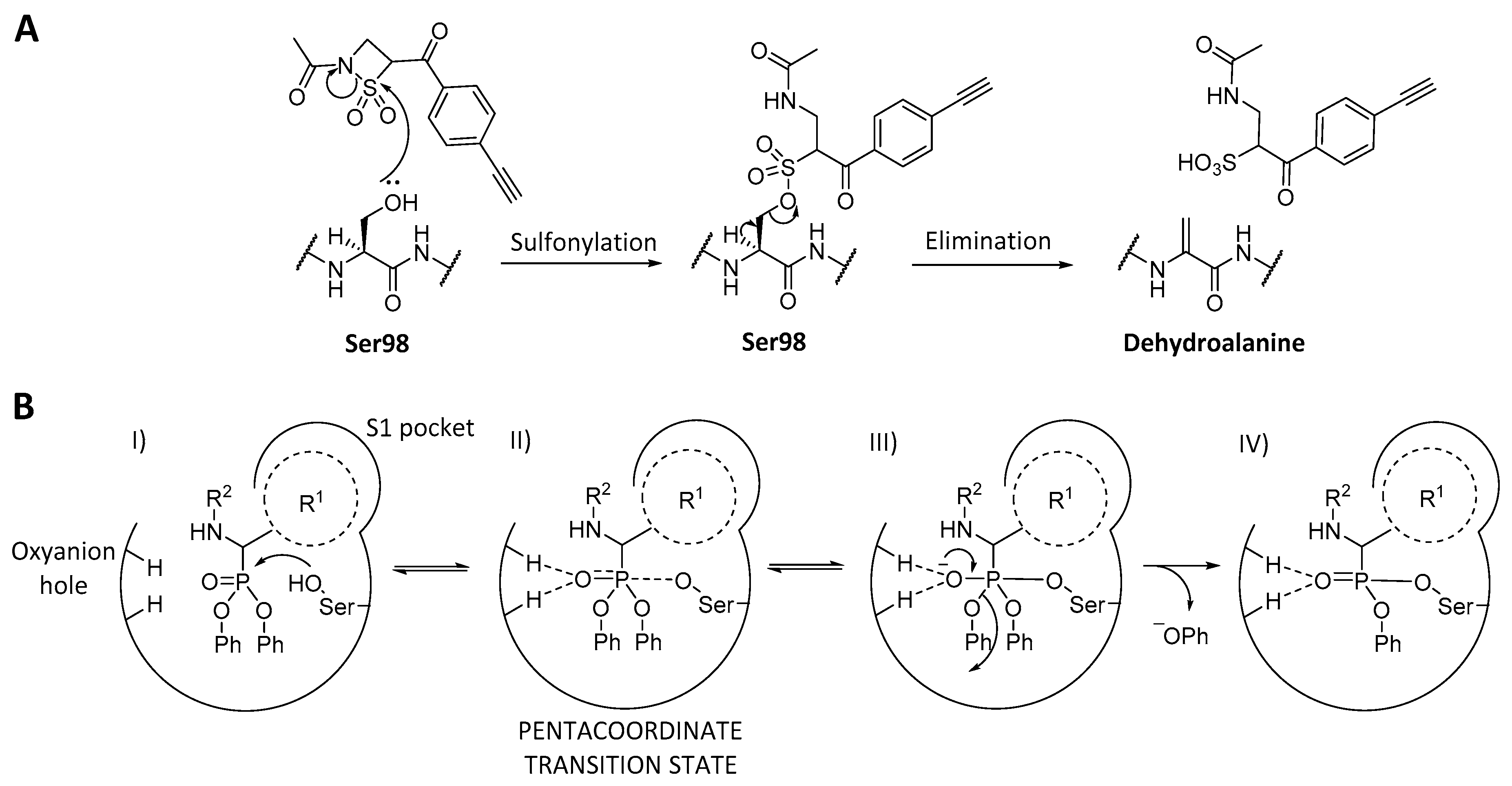
4.2. ClpP Inhibition
5. Remarks and Future Challenges
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAA+ | ATPases associated with diverse cellular activities |
| ADEP | Acyldepsipeptide |
| agr | Accessory gene regulator |
| AML | Acute myeloid leukemia |
| B. subtilis | Bacillus subtilis |
| ClpA | Caseinolytic protease subunit A |
| ClpC | Caseinolytic protease subunit C |
| ClpP | Caseinolytic protease proteolytic subunit |
| ClpS | Caseinolytic protease subunit S |
| ClpX | Caseinolytic protease subunit X |
| CSS | Cell surface signaling |
| E. coli | Escherichia coli |
| E. faecalis | Enterococcus faecalis |
| hClpP | Human ClpP |
| hClpX | Human ClpX |
| HTS | High-throughput screening |
| Hsp90 | Heat shock protein-90 |
| Isd | Iron-regulated determinants |
| L. monocytogenes | Listeria monocytogenes |
| L. pneumophila | Legionella pneumophila |
| M. tuberculosis | Mycobacterium tuberculosis |
| Mtb | Mycobacterium tuberculosis |
| Mpa | Mycobacterium proteasome-associated ATPase |
| N. gonorrhoeae | Neisseria gonorrhoeae |
| N. meningitidis | Neisseria meningitidis |
| P. aeruginosa | Pseudomonas aeruginosa |
| P. falciparum | Plasmodium falciparum |
| Pan | Proteasome-activating nucleotidase |
| ROS | Reactive oxygen species |
| S. aureus | Staphylococcus aureus |
| S. pneumoniae | Streptococcus pneumoniae |
| S. Typhimurium | Salmonella Typhimurium |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| WT | Wild-type |
| Z-LY-CMK | Benzyl ((S)-1-(((S)-4-chloro-1-(4-hydroxyphenyl)-3-oxobutan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate |
| ΔclpP | clpP-defective mutant |
References
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Martin, A. Design principles of a universal protein degradation machine. J. Mol. Biol. 2013, 425, 199–213. [Google Scholar] [CrossRef]
- Yedidi, R.S.; Wendler, P.; Enenkel, C. AAA-ATPases in protein degradation. Front. Mol. Biosci. 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Barthelme, D.; Sauer, R.T. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science 2012, 337, 843–846. [Google Scholar] [CrossRef]
- Maurizi, M.R.; Thompson, M.W.; Singh, S.K.; Kim, S.-H. Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol. 1994, 244, 314–331. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Moerschell, R.P.; Chung, C.H.; Maurizi, M.R. ATP-dependent protease La (Lon) from Escherichia Coli. Methods Enzymol. 1994, 244, 350–375. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Neher, S.B.; Kim, Y.-I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- Kock, H.; Gerth, U.; Hecker, M. The ClpP peptidase is the major determinant of bulk protein turnover in Bacillus Subtilis. J. Bacteriol. 2004, 186, 5856–5864. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Qazi, S.N.; Hill, P.J.; Ingmer, H. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol. Microbiol. 2003, 48, 1565–1578. [Google Scholar] [CrossRef]
- Frees, D.; Gerth, U.; Ingmer, H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Qiu, D.; Eisinger, V.M.; Head, N.E.; Pier, G.B.; Yu, H.D. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 2008, 154, 2119–2130. [Google Scholar] [CrossRef]
- Raju, R.M.; Unnikrishnan, M.; Rubin, D.H.; Krishnamoorthy, V.; Kandror, O.; Akopian, T.N.; Goldberg, A.L.; Rubin, E.J. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 2012, 8, e1002511. [Google Scholar] [CrossRef]
- Cassenego, A.P.; de Oliveira, N.E.; Laport, M.S.; Abranches, J.; Lemos, J.A.; Giambiagi-deMarval, M. The CtsR regulator controls the expression of ClpC, ClpE and ClpP and is required for the virulence of Enterococcus faecalis in an invertebrate model. Antonie Van Leeuwenhoek 2016, 109, 1253–1259. [Google Scholar] [CrossRef]
- Zhao, B.B.; Li, X.H.; Zeng, Y.L.; Lu, Y.J. ClpP-deletion impairs the virulence of Legionella pneumophila and the optimal translocation of effector proteins. BMC Microbiol. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70, 366–377. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. ClpP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef]
- Fischer, F.; Langer, J.D.; Osiewacz, H.D. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Hansen, J.; Corydon, T.J.; Palmfeldt, J.; Durr, A.; Fontaine, B.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Bross, P. Decreased expression of the mitochondrial matrix proteases Lon and ClpP in cells from a patient with hereditary spastic paraplegia (SPG13). Neuroscience 2008, 153, 474–482. [Google Scholar] [CrossRef]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [Green Version]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 2016, 14, 33–44. [Google Scholar] [CrossRef]
- Gerth, U.; Kock, H.; Kusters, I.; Michalik, S.; Switzer, R.L.; Hecker, M. Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis. J. Bacteriol. 2008, 190, 321–331. [Google Scholar] [CrossRef]
- Gur, E.; Ottofueling, R.; Dougan, D.A. Regulated Proteolysis in Microrganisms, 1st ed.; Springer: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Flynn, J.M.; Levchenko, I.; Sauer, R.T.; Baker, T.A. Modulating substrate choice: The SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004, 18, 2292–2301. [Google Scholar] [CrossRef]
- Neher, S.B.; Sauer, R.T.; Baker, T.A. Distinct peptide signals in the UmuD and UmuD’ subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease. Proc. Natl. Acad. Sci. USA 2003, 100, 13219–13224. [Google Scholar] [CrossRef]
- Peterson, C.N.; Ruiz, N.; Silhavy, T.J. RpoS proteolysis is regulated by a mechanism that does not require the SprE (RssB) response regulator phosphorylation site. J. Bacteriol. 2004, 186, 7403–7410. [Google Scholar] [CrossRef]
- Flynn, J.M.; Levchenko, I.; Seidel, M.; Wickner, S.H.; Sauer, R.T.; Baker, T.A. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc. Natl. Acad. Sci. USA 2001, 98, 10584–10589. [Google Scholar] [CrossRef] [Green Version]
- Kirstein, J.; Schlothauer, T.; Dougan, D.A.; Lilie, H.; Tischendorf, G.; Mogk, A.; Bukau, B.; Turgay, K. Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J. 2006, 25, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Battesti, A.; Hoskins, J.R.; Tong, S.; Milanesio, P.; Mann, J.M.; Kravats, A.; Tsegaye, Y.M.; Bougdour, A.; Wickner, S.; Gottesman, S. Anti-adaptors provide multiple modes for regulation of the RssB adaptor protein. Genes Dev. 2013, 27, 2722–2735. [Google Scholar] [CrossRef] [Green Version]
- Battesti, A.; Gottesman, S. Roles of adaptor proteins in regulation of bacterial proteolysis. Curr. Opin. Microbiol. 2013, 16, 140–147. [Google Scholar] [CrossRef]
- Wang, J.; Hartling, J.A.; Flanagan, J.M. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 1997, 91, 447–456. [Google Scholar] [CrossRef]
- Alexopoulos, J.A.; Guarne, A.; Ortega, J. ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 2012, 179, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Akopian, T.; Kandror, O.; Raju, R.M.; Unnikrishnan, M.; Rubin, E.J.; Goldberg, A.L. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012, 31, 1529–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; van Wijk, K.J. Organization, function and substrates of the essential Clp protease system in plastids. Biochim. Biophys. Acta 2015, 1847, 915–930. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.M.; Breidenstein, E.B.M.; de la Fuente-Nunez, C.; Reffuveille, F.; Mawla, G.D.; Hancock RE, W.; Baker, T.A. Two isoforms of Clp peptidase in Pseudomonas aeruginosa control distinct aspects of cellular physiology. J. Bacteriol. 2017, 199, e00568-16. [Google Scholar] [CrossRef]
- Schelin, J.; Lindmark, F.; Clarke, A.K. The ClpP multigene family for the ATP-dependent Clp protease in the cyanobacterium Synechococcus. Microbiology 2002, 148, 2255–2265. [Google Scholar] [CrossRef]
- Ma, W.; Tang, C.; Lai, L. Specificity of trypsin and chymotrypsin: Loop-motion-controlled dynamic correlation as a determinant. Biophys. J. 2005, 89, 1183–1193. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer TA, P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinf. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, K.K. Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J. Biol. Chem. 2003, 278, 50664–50670. [Google Scholar] [CrossRef]
- Yu, A.Y.; Houry, W.A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuron, F.; Maurizi, M.R.; Belnap, D.M.; Kocsis, E.; Booy, F.P.; Kessel, M.; Steven, A.C. At sixes and sevens: Characterization of the symmetry mismatch of the ClpAP chaperone- assisted protease. J. Struct. Biol. 1998, 123, 248–259. [Google Scholar] [CrossRef]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanical Protein Unfolding and Degradation. Annu. Rev. Physiol. 2018, 80, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ologbenla, A.; Houry, W.A. Dynamics of the ClpP serine protease: A model for self-compartmentalized proteases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 400–412. [Google Scholar] [CrossRef]
- Mei, J.M.; Nourbakhsh, F.; Ford, C.W.; Holden, D.W. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature-tagged mutagenesis. Mol. Microbiol. 1997, 26, 399–407. [Google Scholar] [CrossRef]
- Gaillot, O.; Pellegrini, E.; Bregenholt, S.; Nair, S.; Berche, P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 2000, 35, 1286–1294. [Google Scholar] [CrossRef]
- Hensel, M.; Shea, J.E.; Gleeson, C.; Jones, M.D.; Dalton, E.; Holden, D.W. Simultaneous identification of bacterial virulence genes by negative selection. Science 1995, 269, 400–403. [Google Scholar] [CrossRef]
- Farrand, A.J.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Regulation of host hemoglobin binding by the Staphylococcus aureus Clp proteolytic system. J. Bacteriol. 2013, 195, 5041–5050. [Google Scholar] [CrossRef]
- Gaillot, O.; Bregenholt, S.; Jaubert, F.; Di Santo, J.P.; Berche, P. Stress-induced ClpP serine protease of Listeria monocytogenes is essential for induction of listeriolysin O-dependent protective immunity. Infect. Immun. 2001, 69, 4938–4943. [Google Scholar] [CrossRef] [PubMed]
- Beauregard, K.E.; Lee, K.-D.; Collier, R.J.; Swanson, J.A. pH-dependent perforation of macrophage phagosomes by Listeriolysin O from Listeria monocytogenes. J. Exp. Med. 1997, 186, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.Y.; Ogunniyi, A.D.; Choi, M.H.; Pyo, S.N.; Rhee, D.K.; Paton, J.C. The ClpP protease of Streptococcus pneumoniae modulates virulence gene expression and protects against fatal pneumococcal challenge. Infect. Immun. 2004, 72, 5646–5653. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, E.H.; Choi, S.Y.; Tran, T.D.; Kim, I.H.; Kim, S.N.; Pyo, S.; Rhee, D.K. Virulence attenuation of Streptococcus pneumoniae clpP mutant by sensitivity to oxidative stress in macrophages via an NO-mediated pathway. J. Microbiol. 2010, 48, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sashinami, H.; Takaya, A.; Tomoyasu, T.; Matsui, H.; Kikuchi, Y.; Hanawa, T.; Kamiya, S.; Nakane, A. Disruption of the genes for ClpXP protease in Salmonella enterica serovar Typhimurium results in persistent infection in mice, and development of persistence requires endogenous gamma interferon and tumor necrosis factor alpha. Infect. Immun. 2001, 69, 3164–3174. [Google Scholar] [CrossRef]
- Knudsen, G.M.; Olsen, J.E.; Aabo, S.; Barrow, P.; Rychlik, I.; Thomsen, L.E. ClpP deletion causes attenuation of Salmonella Typhimurium virulence through mis-regulation of RpoS and indirect control of CsrA and the SPI genes. Microbiology 2013, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Bishop, T.F.; Martin, L.W.; Lamont, I.L. Activation of a cell surface signaling pathway in Pseudomonas aeruginosa requires ClpP protease and new sigma factor synthesis. Front. Microbiol. 2017, 8, 2442. [Google Scholar] [CrossRef]
- Robinson, J.L.; Brynildsen, M.P. An ensemble-guided approach identifies ClpP as a major regulator of transcript levels in nitric oxide-stressed Escherichia Coli. Metab. Eng. 2015, 31, 22–34. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.; McFadden, G.I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. B 2010, 365, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Rathore, S.; Sinha, D.; Asad, M.; Bottcher, T.; Afrin, F.; Chauhan, V.S.; Gupta, D.; Sieber, S.A.; Mohmmed, A. A cyanobacterial serine protease of Plasmodium falciparum is targeted to the apicoplast and plays an important role in its growth and development. Mol. Microbiol. 2010, 77, 873–890. [Google Scholar] [CrossRef]
- Kang, S.G.; Maurizi, M.R.; Thompson, M.; Mueser, T.; Ahvazi, B. Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J. Struct. Biol. 2004, 148, 338–352. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.G.; Dimitrova, M.N.; Ortega, J.; Ginsburg, A.; Maurizi, M.R. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 2005, 280, 35424–35432. [Google Scholar] [CrossRef]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic. Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef]
- Ahmed, S.; Jelani, M.; Alrayes, N.; Mohamoud, H.S.; Almramhi, M.M.; Anshasi, W.; Ahmed, N.A.; Wang, J.; Nasir, J.; Al-Aama, J.Y. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J. Neurol. Sci. 2015, 353, 149–154. [Google Scholar] [CrossRef]
- Dursun, F.; Mohamoud, H.S.; Karim, N.; Naeem, M.; Jelani, M.; Kirmizibekmez, H. A novel missense mutation in the CLPP gene causing Perrault Syndrome type 3 in a Turkish family. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 472–477. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Pryde, K.R.; Taanman, J.W.; Schapira, A.H. A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell. Rep. 2016, 17, 2522–2531. [Google Scholar] [CrossRef]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Maurizi, M.R. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim. Biophys. Acta 2016, 1862, 252–264. [Google Scholar] [CrossRef]
- Brotz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef]
- Michel, K.H.; Kastner, R.E. A54556 Antibiotics and Process for Production Thereof. U.S. Patent 4492650, 8 January 1985. [Google Scholar]
- Osada, H.; Yano, T.; Koshino, H.; Isono, K. Enopeptin A, a novel depsipeptide antibiotic with anti-bacteriophage activity. J. Antibiot. 1991, 44, 1463–1466. [Google Scholar] [CrossRef]
- Li, D.H.; Chung, Y.S.; Gloyd, M.; Joseph, E.; Ghirlando, R.; Wright, G.D.; Cheng, Y.Q.; Maurizi, M.R.; Guarne, A.; Ortega, J. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol. 2010, 17, 959–969. [Google Scholar] [CrossRef]
- Lee, B.G.; Park, E.Y.; Lee, K.E.; Jeon, H.; Sung, K.H.; Paulsen, H.; Rubsamen-Schaeff, H.; Brotz-Oesterhelt, H.; Song, H.K. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 2010, 17, 471–478. [Google Scholar] [CrossRef]
- Malik, I.T.; Brotz-Oesterhelt, H. Conformational control of the bacterial Clp protease by natural product antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [Green Version]
- Hinzen, B.; Raddatz, S.; Paulsen, H.; Lampe, T.; Schumacher, A.; Habich, D.; Hellwig, V.; Benet-Buchholz, J.; Endermann, R.; Labischinski, H.; et al. Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. Chemmedchem 2006, 1, 689–693. [Google Scholar] [CrossRef]
- Famulla, K.; Sass, P.; Malik, I.; Akopian, T.; Kandror, O.; Alber, M.; Hinzen, B.; Ruebsamen-Schaeff, H.; Kalscheuer, R.; Goldberg, A.L.; et al. Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol. Microbiol. 2016, 101, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Socha, A.M.; Tan, N.Y.; LaPlante, K.L.; Sello, J.K. Diversity-oriented synthesis of cyclic acyldepsipeptides leads to the discovery of a potent antibacterial agent. Bioorg. Med. Chem. 2010, 18, 7193–7202. [Google Scholar] [CrossRef]
- Carney, D.W.; Schmitz, K.R.; Truong, J.V.; Sauer, R.T.; Sello, J.K. Restriction of the conformational dynamics of the cyclic acyldepsipeptide antibiotics improves their antibacterial activity. J. Am. Chem. Soc. 2014, 136, 1922–1929. [Google Scholar] [CrossRef]
- Goodreid, J.D.; Janetzko, J.; Santa Maria, J.P., Jr.; Wong, K.S.; Leung, E.; Eger, B.T.; Bryson, S.; Pai, E.F.; Gray-Owen, S.D.; Walker, S.; et al. Development and characterization of potent cyclic acyldepsipeptide analogues with increased antimicrobial activity. J. Med. Chem. 2016, 59, 624–646. [Google Scholar] [CrossRef]
- Leung, E.; Datti, A.; Cossette, M.; Goodreid, J.; McCaw, S.E.; Mah, M.; Nakhamchik, A.; Ogata, K.; El Bakkouri, M.; Cheng, Y.Q.; et al. Activators of cylindrical proteases as antimicrobials: Identification and development of small molecule activators of ClpP protease. Chem. Biol. 2011, 18, 1167–1178. [Google Scholar] [CrossRef]
- Lavey, N.P.; Coker, J.A.; Ruben, E.A.; Duerfeldt, A.S. Sclerotiamide: The first non-peptide-based natural product activator of bacterial caseinolytic protease P. J. Nat. Prod. 2016, 79, 1193–1197. [Google Scholar] [CrossRef]
- Stahl, M.; Korotkov, V.S.; Balogh, D.; Kick, L.M.; Gersch, M.; Pahl, A.; Kielkowski, P.; Richter, K.; Schneider, S.; Sieber, S.A. Selective activation of human caseinolytic protease P (ClpP). Angew. Chem. Int. Ed. Engl. 2018, 57, 14602–14607. [Google Scholar] [CrossRef]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. Engl. 2011, 50, 5889–5891. [Google Scholar] [CrossRef]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef]
- Choules, M.P.; Wolf, N.M.; Lee, H.; Anderson, J.R.; Grzelak, E.M.; Wang, Y.; Ma, R.; Gao, W.; McAlpine, J.B.; Jin, Y.Y.; et al. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob. Agents Chemother. 2019, 63, e02204-18. [Google Scholar] [CrossRef]
- Gao, W.; Kim, J.Y.; Anderson, J.R.; Akopian, T.; Hong, S.; Jin, Y.Y.; Kandror, O.; Kim, J.W.; Lee, I.A.; Lee, S.Y.; et al. The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 2015, 59, 880–889. [Google Scholar] [CrossRef]
- Fetzer, C.; Korotkov, V.S.; Thanert, R.; Lee, K.M.; Neuenschwander, M.; von Kries, J.P.; Medina, E.; Sieber, S.A. A chemical disruptor of the ClpX chaperone complex attenuates the virulence of multidrug-tesistant Staphylococcus aureus. Angew. Chem. Int. Ed. Engl. 2017, 56, 15746–15750. [Google Scholar] [CrossRef]
- Bottcher, T.; Sieber, S.A. beta-Lactones as privileged structures for the active-site labeling of versatile bacterial. Angew. Chem. Int. Ed. 2008, 47, 4600–4603. [Google Scholar] [CrossRef]
- Bottcher, T.; Sieber, S.A. beta-Lactones as specific inhibitors of CIpP attenuate the production of extracellular virulence factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008, 130, 14400–14401. [Google Scholar] [CrossRef]
- Bottcher, T.; Sieber, S.A. Structurally refined beta-lactones as potent inhibitors of devastating bacterial virulence factors. Chembiochem 2009, 10, 663–666. [Google Scholar] [CrossRef]
- Bottcher, T.; Sieber, S.A. beta-Lactones decrease the Intracellular virulence of Listeria monocytogenes in macrophages. Chemmedchem 2009, 4, 1260–1263. [Google Scholar] [CrossRef]
- Compton, C.L.; Schmitz, K.R.; Sauer, R.T.; Sello, J.K. Antibacterial Activity of and Resistance to Small Molecule Inhibitors of the ClpP Peptidase. ACS Chem. Biol. 2013, 8, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Hackl, M.W.; Lakemeyer, M.; Dahmen, M.; Glaser, M.; Pahl, A.; Lorenz-Baath, K.; Menzel, T.; Sievers, S.; Bottcher, T.; Antes, I.; et al. Phenyl esters are potent inhibitors of caseinolytic protease P and reveal a stereogenic switch for deoligomerization. J. Am. Chem. Soc. 2015, 137, 8475–8483. [Google Scholar] [CrossRef]
- Gronauer, T.F.; Mandl, M.M.; Lakemeyer, M.; Hackl, M.W.; Messner, M.; Korotkov, V.S.; Pachmayr, J.; Sieber, S.A. Design and synthesis of tailored human caseinolytic protease P inhibitors. Chem. Commun. 2018, 54, 9833–9836. [Google Scholar] [CrossRef]
- Pahl, A.; Lakemeyer, M.; Vielberg, M.T.; Hackl, M.W.; Vomacka, J.; Korotkov, V.S.; Stein, M.L.; Fetzer, C.; Lorenz-Baath, K.; Richter, K.; et al. Reversible Inhibitors Arrest ClpP in a defined conformational state that can be revoked by ClpX association. Angew. Chem. Int. Ed. 2015, 54, 15892–15899. [Google Scholar] [CrossRef]
- Gersch, M.; Kolb, R.; Alte, F.; Groll, M.; Sieber, S.A. Disruption of oligomerization and dehydroalanine formation as mechanisms for ClpP protease inhibition. J. Am. Chem. Soc. 2014, 136, 1360–1366. [Google Scholar] [CrossRef]
- Moreira, W.; Ngan, G.J.; Low, J.L.; Poulsen, A.; Chia, B.C.; Ang, M.J.; Yap, A.; Fulwood, J.; Lakshmanan, U.; Lim, J.; et al. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 2015, 6, e00253-15. [Google Scholar] [CrossRef]
- Liu, P.; Yang, Y.; Ju, Y.; Tang, Y.; Sang, Z.; Chen, L.; Yang, T.; An, Q.; Zhang, T.; Luo, Y. Design, synthesis and biological evaluation of novel pyrrole derivatives as potential ClpP1P2 inhibitor against Mycobacterium tuberculosis. Bioorg. Chem. 2018, 80, 422–432. [Google Scholar] [CrossRef]
- Fraga, H.; Rodriguez, B.; Bardera, A.; Cid, C.; Akopian, T.; Kandror, O.; Park, A.; Colmenarejo, G.; Lelievre, J.; Goldberg, A. Development of high throughput screening methods for inhibitors of ClpC1P1P2 from Mycobacteria tuberculosis. Anal. Biochem. 2019, 567, 30–37. [Google Scholar] [CrossRef]
- Mundra, S.; Thakur, V.; Bello, A.M.; Rathore, S.; Asad, M.; Wei, L.; Yang, J.; Chakka, S.K.; Mahesh, R.; Malhotra, P.; et al. A novel class of Plasmodial ClpP protease inhibitors as potential antimalarial agents. Bioorg. Med. Chem. 2017, 25, 5662–5677. [Google Scholar] [CrossRef]
- Szyk, A.; Maurizi, M.R. Crystal structure at 1.9A of E. coli ClpP with a peptide covalently bound at the active site. J. Struct. Biol. 2006, 156, 165–174. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Sassetti, E.; Salado, I.G.; Witt, G.; Benramdane, S.; Reinhardt, L.; DCruz, C.; Joossens, J.; Van der Veken, P.; Brötz-Oesterhelt, H.; et al. alpha-Amino diphenyl phosphonates as novel inhibitors of Escherichia coli ClpP protease. J. Med. Chem. 2019, 62, 774–797. [Google Scholar] [CrossRef]
- Lamden, L.A.; Bartlett, F.A. Aminoalkylphosphonofluoridate derivatives: Rapid and potentially selective inactivators of serine peptidases. Biochem. Biophys. Res. Commun. 1983, 112, 1085–1090. [Google Scholar] [CrossRef]
- Joossens, J.; Van der Veken, P.; Surpateanu, G.; Lambeir, A.M.; El-Sayed, I.; Ali, O.M.; Augustyns, K.; Haemers, A. Diphenyl phosphonate inhibitors for the urokinase-type plasminogen activator: Optimization of the P4 position. J. Med. Chem. 2006, 49, 5785–5793. [Google Scholar] [CrossRef]
- Van der Veken, P.; Soroka, A.; Brandt, I.; Chen, Y.S.; Maes, M.B.; Lambeir, A.M.; Chen, X.; Haemers, A.; Scharpe, S.; Augustyns, K.; et al. Irreversible inhibition of dipeptidyl peptidase 8 by dipeptide-derived diaryl phosphonates. J. Med. Chem. 2007, 50, 5568–5570. [Google Scholar] [CrossRef]
- Winiarski, L.; Oleksyszyn, J.; Sienczyk, M. Human neutrophil elastase phosphonic inhibitors with improved potency of action. J. Med. Chem. 2012, 55, 6541–6553. [Google Scholar] [CrossRef]
- Pietrusewicz, E.; Sienczyk, M.; Oleksyszyn, J. Novel diphenyl esters of peptidyl alpha-aminoalkylphosphonates as inhibitors of chymotrypsin and subtilisin. J. Enzyme Inhib. Med. Chem. 2009, 24, 1229–1236. [Google Scholar] [CrossRef]
- Burchacka, E.; Zdzalik, M.; Niemczyk, J.S.; Pustelny, K.; Popowicz, G.; Wladyka, B.; Dubin, A.; Potempa, J.; Sienczyk, M.; Dubin, G.; et al. Development and binding characteristics of phosphonate inhibitors of SplA protease from Staphylococcus aureus. Protein Sci. 2014, 23, 179–189. [Google Scholar] [CrossRef]
- Dickey, S.W.; Cheung GY, C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Defoirdt, T. Quorum-sensing systems as targets for antivirulence therapy. Trends Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van Der Veken, P.; De Winter, H.; Augustyns, K. ClpP Protease, a Promising Antimicrobial Target. Int. J. Mol. Sci. 2019, 20, 2232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092232
Moreno-Cinos C, Goossens K, Salado IG, Van Der Veken P, De Winter H, Augustyns K. ClpP Protease, a Promising Antimicrobial Target. International Journal of Molecular Sciences. 2019; 20(9):2232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092232
Chicago/Turabian StyleMoreno-Cinos, Carlos, Kenneth Goossens, Irene G. Salado, Pieter Van Der Veken, Hans De Winter, and Koen Augustyns. 2019. "ClpP Protease, a Promising Antimicrobial Target" International Journal of Molecular Sciences 20, no. 9: 2232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092232