The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Epithelial Ovarian Cancer
2.1. Alpha Smooth Muscle Actin (α-SMA) and Platelet Derived Growth Factor Beta Receptor (PDGFβR) Related Stroma
2.1.1. Tumor Growth and Metastasis Induction
2.1.2. Pericytes Draw the Architecture of Further Functional Vessels
2.1.3. Poor Prognosis and Translational Relevance
2.2. Leucocytes
2.2.1. Imbalance of the Lymphocytes’ Subtypes Correlates with immunosuppressive Environment and Angiogenesis
2.2.2. Imbalance of the Lymphocytes’ Subtypes Correlates with Poor Prognosis but Harbors a Highly Critical Translational Significance
2.2.3. The Role of TAMs in EOC
3. Endometrial Cancer
Mesenchymal Cells and their Autocrine and Paracrine Role in the Development of EC
4. Cervical Cancer
4.1. Inflammation, Pro-angiogenesis and Cancer-instructed Stromal Fibroblasts in CCx: Pathways and Translational Relevance
4.2. Synergic Effects of Epithelial High-risk HPV and Stromal ERα
5. The Role of TAMs in Endometrial and Cervical Cancer and Their Translational Relevance in Gynecological Malignancies
5.1. TAMs in EC and CCx
5.2. Translational Significance of TAMs in Gynecological Malignancies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EOC | Epithelial ovarian cancer |
| BM-MSCs | Bone-Marrow-Derived Mesenchymal Stem Cells |
| AOCS | Australian Ovarian Cancer Study |
| TME | Tumor Micro-Environment |
| EMT | Epithelial-Mesenchymal Transition |
| ECM | Extra-Cellular Matrix |
| CCL | Chemokine, C-C motif, Ligand |
| Th17 | T-helper 17 |
| HPV | Human Papilloma Virus |
| α-SMA | Alpha Smooth Muscle Actin |
| PDGFβR | Platelet Derived Growth Factor Beta Receptor |
| CAFs | Cancer-Associated Fibroblasts |
| FAP | Fibroblast Activation Protein |
| MCAM/CD146 | Melanoma Cell Adhesion Molecule/ Cluster of Differentiation 146 |
| CD | Cluster of Differentiation |
| FGF/FGFR | Fibroblast Growth Factor/ Fibroblast Growth Factor Receptor |
| VCAN | VersiCAN gene |
| VEGF-A | Vascular Endothelial Growth Factor-A |
| CXCL | Chemokine C-X-C motif ligand |
| PFS | Progression Free Survival |
| OS | Overall Survival |
| LCA | Leukocyte Common Antigen |
| TILs | Tumor-Infiltrating Lymphocytes |
| Tregs | T-regulatory cells |
| NK | Natural Killer |
| HSGC | High Serous Grade Carcinoma |
| TRM | Tissue Resident Memory |
| HLA | Human Leucocyte Antigens |
| IL | InterLeukin |
| INF γ | INterFeron γ |
| PD-L1 | PD- Ligand 1 |
| CTLA | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| MHC | Major Histocompatibility Complex |
| TP53 | Tumor Protein 53 |
| APCs | Antigen-Presenting-Cells |
| FOXP3 | Fork Head Box 3 |
| TGF-β | Tumor Grow Factor β |
| TAMs | Tumor-Associated Macrophages |
| VEGF | Vascular Endothelial Growth Factor |
| TNF | Tumor Necrosis Factor |
| PLCγ | PhosphoLipase Cγ |
| JAK/ STAT | JAnus Kinase/Signal Transducer and Activator of Transcription |
| PI3K | PhosphoInositide 3 Kinase |
| Tim-3 | T-cell immunoglobulin and mucin-domain-containing-3 |
| MIC A/B | Major histocompatibility complex class I-related chains (MIC) A and B |
| ULBPs | UL-16 binding proteins |
| NKG2D | Natural killer group 2, member D |
| EC | Endometrial Cancer |
| PTEN | Phosphatase and Tensin homolog |
| CTNNB1 | Catenin Beta 1 |
| PIK3CA | Phosphatidylinositol 3-kinase |
| MLH1 | MutL Homolog 1 |
| POLE | DNA POLymerase Epsilon |
| HNPCC | Hereditary Non-Polyposis Colon Cancer |
| ER α | Estrogen Receptor α |
| ESR1 | (gene) EStrogen Receptor 1 |
| E2 | 17β-estradiol |
| IGF1 | Insulin-like Growth Factor 1 |
| TGF | Tumor Growth Factor |
| MAD2L1 | Mitotic Arrest Deficient 2 Like 1 |
| CDKN1A | Cyclin-Dependent Kinase Inhibitor 1A |
| EH | Endometrial Hyperplasia |
| PR | Progesterone Receptor |
| CCx | Cervical cancer |
| pRB | Retinoblastoma protein |
| MCP1 | Monocyte Chemoattractant Protein 1 |
| MIP3A | Macrophage Inflammatory Protein-3α |
| HSIL | High-grade Squamous Intraepithelial Lesion |
| CCR | C-C motif Chemokine Receptor |
| LSIL | Low-grade Squamous Intraepithelial Lesion |
| C/EBPβ | CCAAT Enhancer Binding Protein Beta |
| MMP-9 | Matrix-Metalloproteinase |
| FIGO | International Federation of Gynaecology and Obstetrics |
| THBS | Thrombospondins |
| EGFR | Epidermal growth factor receptor |
| HBEGF | Heparin Binding EGF Like Growth Factor |
| FGF9 | Fibroblast Growth Factor 9 |
| PUFAs | Polyunsaturated Fatty Acids |
| Evs | Extracellular Vesicles |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| EGF | Endothelial Growth Factor |
| EGFR | Endothelial Growth Factor Receptor |
| PCX | Paclitaxel |
| TLR | Toll-Like Receptor |
| SIRPα | Signal-Regulatory Protein Alpha |
| CSF-1 | Colony-Stimulating Factor 1 |
| CSF1-R | Colony-Stimulating Factor 1 Receptor |
| LVSI | LymphoVascular Space Invasion |
| MET | Mesenchymal-epithelial transition |
References
- Klymenko, Y.; Nephew, K.P. Epigenetic crosstalk between the tumor microenvironment and ovarian cancer cells: A therapeutic road less traveled. Cancers 2018, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Corvigno, S.; Wisman, G.B.; Mezheyeuski, A.; van der Zee, A.G.; Nijman, H.W.; Avall-Lundqvist, E.; Ostman, A.; Dahlstrand, H. Markers of fibroblast-rich tumor stroma and perivascular cells in serous ovarian cancer: Inter- and intra-patient heterogeneity and impact on survival. Oncotarget 2016, 7, 18573–18584. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Chong, L.; George, J.; Schluter, H.; Monchgesang, S.; Mills, S.; Li, J.; Parish, C.; Bowtell, D.; Kaur, P.; et al. Pericytes promote malignant ovarian cancer progression in mice and predict poor prognosis in serous ovarian cancer patients. Clin. Cancer Res. 2016, 22, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.A.; Simitsidellis, I.; Collins, F.; Saunders, P.T.K. Endometrial intracrinology: Oestrogens, androgens and endometrial disorders. Int. J. Mol. Sci. 2018, 19, 3276. [Google Scholar] [CrossRef]
- Senol, S.; Sayar, I.; Ceyran, A.B.; Ibiloglu, I.; Akalin, I.; Firat, U.; Kosemetin, D.; Engin Zerk, P.; Aydin, A. Stromal clues in endometrial carcinoma: Loss of expression of beta-catenin, epithelial-mesenchymal transition regulators and estrogen-progesterone receptor. Int. J. Gynecol. Pathol. 2016, 35, 238–248. [Google Scholar] [CrossRef]
- Winuthayanon, W.; Lierz, S.L.; Delarosa, K.C.; Sampels, S.R.; Donoghue, L.J.; Hewitt, S.C.; Korach, K.S. Juxtacrine activity of estrogen receptor alpha in uterine stromal cells is necessary for estrogen-induced epithelial cell proliferation. Sci. Rep. 2017, 7, 8377. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Park, Y.; Chung, S.H. Epithelial oestrogen receptor alpha is dispensable for the development of oestrogen-induced cervical neoplastic diseases. J. Pathol. 2018, 245, 147–152. [Google Scholar] [CrossRef]
- Walch-Ruckheim, B.; Mavrova, R.; Henning, M.; Vicinus, B.; Kim, Y.J.; Bohle, R.M.; Juhasz-Boss, I.; Solomayer, E.F.; Smola, S. Stromal fibroblasts induce ccl20 through il6/c/ebpbeta to support the recruitment of th17 cells during cervical cancer progression. Cancer Res. 2015, 75, 5248–5259. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Q.; Lin, Y.; Wu, L.; Wu, X.; Wang, K.; He, Q.; Xu, C.; Wan, X.; Wang, X. Crosstalk between tems and endothelial cells modulates angiogenesis and metastasis via igf1-igf1r signalling in epithelial ovarian cancer. Br. J. Cancer 2017, 117, 1371–1382. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Loizzi, V.; Del Vecchio, V.; Gargano, G.; De Liso, M.; Kardashi, A.; Naglieri, E.; Resta, L.; Cicinelli, E.; Cormio, G. Biological pathways involved in tumor angiogenesis and bevacizumab based anti-angiogenic therapy with special references to ovarian cancer. Int. J. Mol. Sci. 2017, 18, 1967. [Google Scholar] [CrossRef] [PubMed]
- Levison, P.A.H.a.D.A. Review: Assessment of cell proliferation in histological material. J. Clin. Pathol. 1990, 43, 184–192. [Google Scholar]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; Van Kooten, C.; Müller, G. Tgf-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (fgf-2). Kidney Int. 2001, 59, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Leinster, D.A.; Kulbe, H.; Everitt, G.; Thompson, R.; Perretti, M.; Gavins, F.N.; Cooper, D.; Gould, D.; Ennis, D.P.; Lockley, M.; et al. The peritoneal tumour microenvironment of high-grade serous ovarian cancer. J. Pathol. 2012, 227, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Marech, I.; Leporini, C.; Ammendola, M.; Porcelli, M.; Gadaleta, C.D.; Russo, E.; De Sarro, G.; Ranieri, G. Classical and non-classical proangiogenic factors as a target of antiangiogenic therapy in tumor microenvironment. Cancer Lett. 2016, 380, 216–226. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence and survival in epithelial ovarian cancer. New Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Powell, D.J., Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Lagana, A.S.; Sofo, V.; Vitale, S.G.; Triolo, O. Epithelial ovarian cancer inherent resistance: May the pleiotropic interaction between reduced immunosurveillance and drug-resistant cells play a key role? Gynecol. Oncol. Rep. 2016, 18, 57–58. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker cd103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. Ifn-gamma from lymphocytes induces pd-l1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef]
- Nielsen, J.S.; Nelson, B.H. Tumor-infiltrating b cells and t cells: Working together to promote patient survival. Oncoimmunology 2012, 1, 1623–1625. [Google Scholar] [CrossRef]
- Ranieri, G. Biological basis of tumor angiogenesis and therapeutic intervention: Past, present and future. Int. J. Mol. Sci. 2018, 19, 1655. [Google Scholar] [CrossRef]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of pd-1 and ctla-4 combined with tumor vaccine effectively restores t-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, D.; Xia, Z.; Luan, M.; Wu, L.; Wang, G.; Zhang, S. Combined tim-3 blockade and cd137 activation affords the long-term protection in a murine model of ovarian cancer. J. Transl. Med. 2013, 11, 215. [Google Scholar] [CrossRef]
- Li, K.; Mandai, M.; Hamanishi, J.; Matsumura, N.; Suzuki, A.; Yagi, H.; Yamaguchi, K.; Baba, T.; Fujii, S.; Konishi, I. Clinical significance of the nkg2d ligands, mica/b and ulbp2 in ovarian cancer: High expression of ulbp2 is an indicator of poor prognosis. Cancer Immunol. Immunother. 2009, 58, 641–652. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized m2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Marech, I.; Ammendola, M.; Sacco, R.; Sammarco, G.; Zuccala, V.; Zizzo, N.; Leporini, C.; Luposella, M.; Patruno, R.; Filippelli, G.; et al. Tumour-associated macrophages correlate with microvascular bed extension in colorectal cancer patients. J. Cell Mol. Med. 2016, 20, 1373–1380. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. Tameless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Pluddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef]
- Kawamura, K.; Komohara, Y.; Takaishi, K.; Katabuchi, H.; Takeya, M. Detection of m2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol. Int. 2009, 59, 300–305. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.-M.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of cd163 expression, cytokine levels and early relapse. Int. J. Cancer 2013, 134, 32–42. [Google Scholar] [CrossRef]
- Colvin, E.K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high m1/m2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef]
- He, Y.-F.; Zhang, M.-Y.; Wu, X.; Sun, X.-J.; Xu, T.; He, Q.-Z.; Di, W. High muc2 expression in ovarian cancer is inversely associated with the m1/m2 ratio of tumor-associated macrophages and patient survival time. PLoS ONE 2013, 8, e79769. [Google Scholar] [CrossRef]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar tumor-associated macrophages in ovarian cancer as targets for therapy. Cancers 2018, 10, 366. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Wang, D.; Ali, L.; Lele, S.; Huba, M.A.; Liu, S.; Odunsi, K. Intraepithelial t cells and tumor-associated macrophages in ovarian cancer patients. Cancer Immun. 2013, 13, 1. [Google Scholar]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef]
- No, J.H.; Moon, J.M.; Kim, K.; Kim, Y.-B. Prognostic significance of serum soluble cd163 level in patients with epithelial ovarian cancer. Gynecol. Obstet. Investig. 2013, 75, 263–267. [Google Scholar] [CrossRef]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of m2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Rancourt, C.; Piché, A. Prognostic significance of il-6 and il-8 ascites levels in ovarian cancer patients. BMC Cancer 2011, 11. [Google Scholar] [CrossRef]
- Scambia, G.; Testa, U.; Benedetti Panici, P.; Foti, E.; Martucci, R.; Gadducci, A.; Perillo, A.; Facchini, V.; Peschle, C.; Mancuso, S. Prognostic significance of interleukin 6 serum levels in patients with ovarian cancer. Br. J. Cancer 1995, 71, 354–356. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-W.; Chen, M.-W.; Hsiao, M.; Wang, S.; Chen, C.-A.; Hsiao, S.-M.; Chang, J.-S.; Lai, T.-C.; Rose-John, S.; Kuo, M.-L.; et al. Il-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2010, 71, 424–434. [Google Scholar] [CrossRef]
- Yanaihara, N.; Anglesio, M.S.; Ochiai, K.; Hirata, Y.; Saito, M.; Nagata, C.; Iida, Y.; Takakura, S.; Yamada, K.; Tanaka, T.; et al. Cytokine gene expression signature in ovarian clear cell carcinoma. Int. J. Oncol. 2012, 41, 1094–1100. [Google Scholar] [CrossRef]
- Finkernagel, F.; Reinartz, S.; Lieber, S.; Adhikary, T.; Wortmann, A.; Hoffmann, N.; Bieringer, T.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Müller, R. The unique molecular and cellular microenvironment of ovarian cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Le Page, C.; Marineau, A.; Bonza, P.K.; Rahimi, K.; Cyr, L.; Labouba, I.; Madore, J.; Delvoye, N.; Mes-Masson, A.-M.; Provencher, D.M.; et al. Btn3a2 expression in epithelial ovarian cancer is associated with higher tumor infiltrating t cells and a better prognosis. PLoS ONE 2012, 7, e38541. [Google Scholar] [CrossRef]
- Pogge von Strandmann, E.; Reinartz, S.; Wager, U.; Müller, R. Tumor–host cell interactions in ovarian cancer: Pathways to therapy failure. Trends Cancer 2017, 3, 137–148. [Google Scholar] [CrossRef]
- Klimp, A.H.; Hollema, H.; Kempinga, C.; van der Zee, A.G.; de Vries, E.G.; Daemen, T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res. 2001, 61, 7305–7309. [Google Scholar]
- Wang, X.; Deavers, M.; Patenia, R.; Bassett, R.L.; Mueller, P.; Ma, Q.; Wang, E.; Freedman, R.S. Monocytes/macrophage and t-cell infiltrates in peritoneum of patients with ovarian cancer or benign pelvic disease. J. Transl. Med. 2006, 4, 30. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Laplante, C.; Garde-Granger, P.; Carignan, A.; Bessette, P.; Rancourt, C.; Piché, A. Ccl18 from ascites promotes ovarian cancer cell migration through proline-rich tyrosine kinase 2 signaling. Mol. Cancer 2016, 15, 58. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory t cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Valls, A.F.; Shen, Y.; Schmid, T. A core of macrophages facilitates ovarian cancer metastases. Transl. Cancer Res. 2017, 6, S189–S196. [Google Scholar] [CrossRef]
- Sica, G.L.; Choi, I.-H.; Zhu, G.; Tamada, K.; Wang, S.-D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-h4, a molecule of the b7 family, negatively regulates t cell immunity. Immunity 2003, 18, 849–861. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Zhu, G.; Myers, L.; Mottram, P.; Cheng, P.; Chen, L.; Coukos, G.; Zou, W. Relationship between b7-h4, regulatory t cells and patient outcome in human ovarian carcinoma. Cancer Res. 2007, 67, 8900–8905. [Google Scholar] [CrossRef]
- Gottlieb, C.E.; Mills, A.M.; Cross, J.V.; Ring, K.L. Tumor-associated macrophage expression of pd-l1 in implants of high grade serous ovarian carcinoma: A comparison of matched primary and metastatic tumors. Gynecol. Oncol. 2017, 144, 607–612. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes released from tumor-associated macrophages transfer mirnas that induce a treg/th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an m1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and tam functions-it takes two to tango. FEBS J. 2017, 285, 700–716. [Google Scholar] [CrossRef]
- Castegna, A.; Menga, A. Glutamine synthetase: Localization dictates outcome. Genes 2018, 9, 108. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; Gonzalez-Martin, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. Esmo-esgo-estro consensus conference on endometrial cancer: Diagnosis, treatment and follow-up. Int. J. Gynecol. Cancer 2016, 26, 2–30. [Google Scholar] [CrossRef]
- Mazzocca, A.; Schonauer, L.M.; De Nola, R.; Lippolis, A.; Marrano, T.; Loverro, M.; Sabba, C.; Di Naro, E. Autotaxin is a novel molecular identifier of type i endometrial cancer. Med. Oncol. 2018, 35, 157. [Google Scholar] [CrossRef]
- Ning, C.; Xie, B.; Zhang, L.; Li, C.; Shan, W.; Yang, B.; Luo, X.; Gu, C.; He, Q.; Jin, H.; et al. Infiltrating macrophages induce erα expression through an il17a-mediated epigenetic mechanism to sensitize endometrial cancer cells to estrogen. Cancer Res. 2016, 76, 1354–1366. [Google Scholar] [CrossRef]
- Jiang, X.-F.; Tang, Q.-I.; Li, H.-G.; Shen, X.-M.; Luo, X.; Wang, X.-Y.; Lin, Z.-Q. Tumor-associated macrophages correlate with progesterone receptor loss in endometrial endometrioid adenocarcinoma. J. Obstet. Gynaecol. Res. 2012, 39, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.W.; Schwab, K.E.; Gargett, C.E. Clonogenicity of human endometrial epithelial and stromal cells. Biol. Reprod. 2004, 70, 1738–1750. [Google Scholar] [CrossRef]
- Busca, A.; Djordjevic, B.; Giassi, A.; Parra-Herran, C. Ifitm1 is superior to cd10 as a marker of endometrial stroma in the evaluation of myometrial invasion by endometrioid adenocarcinoma. Am. J. Clin. Pathol. 2016, 145, 486–496. [Google Scholar] [CrossRef]
- Cho, H.; Chung, J.Y.; Kim, S.; Braunschweig, T.; Kang, T.H.; Kim, J.; Chung, E.J.; Hewitt, S.M.; Kim, J.H. Mica/b and ulbp1 nkg2d ligands are independent predictors of good prognosis in cervical cancer. BMC Cancer 2014, 14, 957. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Townsend, J.S.; Steele, C.B.; Hayes, N.; Bhatt, A.; Moore, A.R. Human papillomavirus vaccine as an anticancer vaccine: Collaborative efforts to promote human papillomavirus vaccine in the national comprehensive cancer control program. J. Womens Health (Larchmt) 2017, 26, 200–206. [Google Scholar] [CrossRef]
- Krishnan, V.; Schaar, B.; Tallapragada, S.; Dorigo, O. Tumor associated macrophages in gynecologic cancers. Gynecol. Oncol. 2018, 149, 205–213. [Google Scholar] [CrossRef]
- Weber, S.K.; Sauerwald, A.; Pölcher, M.; Braun, M.; Debald, M.; Serce, N.B.; Kuhn, W.; Brunagel-Walgenbach, G.; Rudlowski, C. Detection of lymphovascular invasion by d2-40 (podoplanin) immunoexpression in endometrial cancer. Int. J. Gynecol. Cancer 2012, 22, 1442–1448. [Google Scholar] [CrossRef]
- Salvesen, H.B.; Akslen, L.A. Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int. J. Cancer 1999, 84, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Soeda, S.; Nakamura, N.; Ozeki, T.; Nishiyama, H.; Hojo, H.; Yamada, H.; Abe, M.; Sato, A. Tumor-associated macrophages correlate with vascular space invasion and myometrial invasion in endometrial carcinoma. Gynecol. Oncol. 2008, 109, 122–128. [Google Scholar] [CrossRef]
- Ding, H.; Cai, J.; Mao, M.; Fang, Y.; Huang, Z.; Jia, J.; Li, T.; Xu, L.; Wang, J.; Zhou, J.; et al. Tumor-associated macrophages induce lymphangiogenesis in cervical cancer via interaction with tumor cells. APMIS 2014, 122, 1059–1069. [Google Scholar] [CrossRef]
- Petrillo, M.; Zannoni, G.F.; Martinelli, E.; Pedone Anchora, L.; Ferrandina, G.; Tropeano, G.; Fagotti, A.; Scambia, G. Polarisation of tumor-associated macrophages toward m2 phenotype correlates with poor response to chemoradiation and reduced survival in patients with locally advanced cervical cancer. PLoS ONE 2015, 10, e0136654. [Google Scholar] [CrossRef]
- Heeren, A.M.; Punt, S.; Bleeker, M.C.G.; Gaarenstroom, K.N.; van der Velden, J.; Kenter, G.G.; de Gruijl, T.D.; Jordanova, E.S. Prognostic effect of different pd-l1 expression patterns in squamous cell carcinoma and adenocarcinoma of the cervix. Mod. Pathol. 2016, 29, 753–763. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colon, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an m1- profile in a tlr4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Miao, C.; Wang, Y.; Xu, Y.; Dong, R.; Zhang, Z.; Griffin, B.B.; Yuan, C.; Yan, S.; et al. Anti-angiogenesis effect of neferine via regulating autophagy and polarization of tumor-associated macrophages in high-grade serous ovarian carcinoma. Cancer Lett. 2018, 432, 144–155. [Google Scholar] [CrossRef]
- Jeong, M.; Kim, H.M.; Ahn, J.-H.; Lee, K.-T.; Jang, D.S.; Choi, J.-H. 9-hydroxycanthin-6-one isolated from stem bark of ailanthus altissima induces ovarian cancer cell apoptosis and inhibits the activation of tumor-associated macrophages. Chem. Biol. Interact. 2018, 280, 99–108. [Google Scholar] [CrossRef]
- Lee, K.; Ahn, J.-H.; Lee, K.-T.; Jang, D.; Choi, J.-H. Deoxyschizandrin, isolated from schisandra berries, induces cell cycle arrest in ovarian cancer cells and inhibits the protumoural activation of tumour-associated macrophages. Nutrients 2018, 10, 91. [Google Scholar] [CrossRef]
- Negus, R.P.; Stamp, G.W.; Relf, M.G.; Burke, F.; Malik, S.T.; Bernasconi, S.; Allavena, P.; Sozzani, S.; Mantovani, A.; Balkwill, F.R. The detection and localization of monocyte chemoattractant protein-1 (mcp-1) in human ovarian cancer. J. Clin. Investig. 1995, 95, 2391–2396. [Google Scholar] [CrossRef]
- Parente-Pereira, A.C.; Shmeeda, H.; Whilding, L.M.; Zambirinis, C.P.; Foster, J.; van der Stegen, S.J.C.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.K.; et al. Adoptive immunotherapy of epithelial ovarian cancer with vγ9vδ2 t cells, potentiated by liposomal alendronic acid. J. Immunol. 2014, 193, 5557–5566. [Google Scholar] [CrossRef]
- Moughon, D.L.; He, H.; Schokrpur, S.; Jiang, Z.K.; Yaqoob, M.; David, J.; Lin, C.; Iruela-Arispe, M.L.; Dorigo, O.; Wu, L. Macrophage blockade using csf1r inhibitors reverses the vascular leakage underlying malignant ascites in late-stage epithelial ovarian cancer. Cancer Res. 2015, 75, 4742–4752. [Google Scholar] [CrossRef]
- Murata, Y.; Kotani, T.; Ohnishi, H.; Matozaki, T. The cd47-sirp signalling system: Its physiological roles and therapeutic application. J. Biochem. 2014, 155, 335–344. [Google Scholar] [CrossRef]
- Disis, M.L.; Patel, M.R.; Pant, S.; Hamilton, E.P.; Lockhart, A.C.; Kelly, K.; Beck, J.T.; Gordon, M.S.; Weiss, G.J.; Taylor, M.H.; et al. Avelumab (msb0010718c; anti-pd-l1) in patients with recurrent/refractory ovarian cancer from the javelin solid tumor phase ib trial: Safety and clinical activity. J. Clin. Oncol. 2016, 34, 5533. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti–pd-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Pathogenetic Role | Translational Possibilities and Hypothesis |
|---|---|---|
| CAFs | Recruited by PDGFβ and activated by TGFβ/VCAN, CXCL. | Inhibiting PDGF- β signaling. |
| They promote EOC cells’ motility, overgrowth, neo-angiogenesis, and invasion. | Inhibiting pro-angiogenic factors VEGF, FGF-2. | |
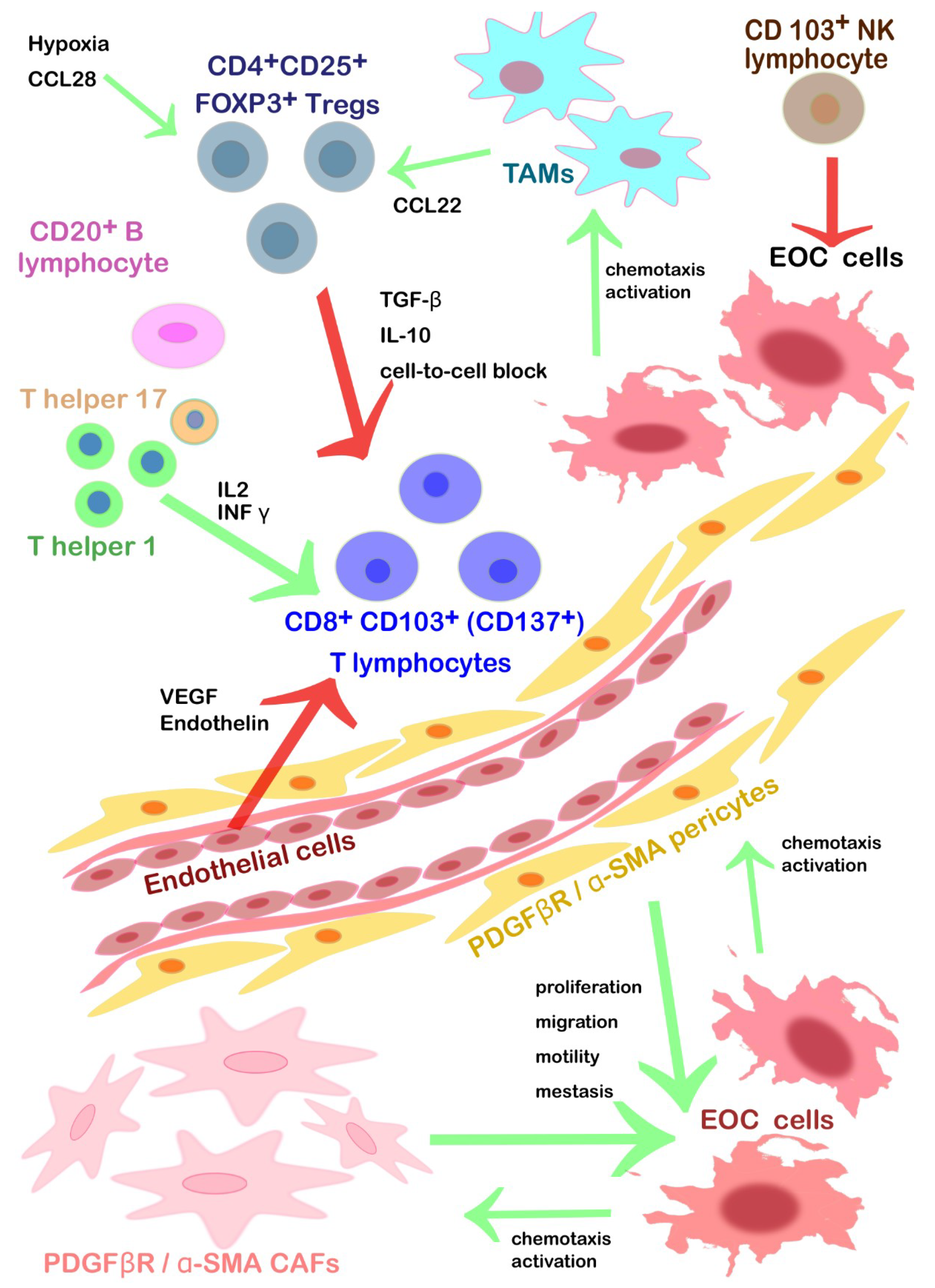
| α-SMA+ pericytes | Recruited by the PDGFβ, their rate and genetic signature correlate with proliferation, migration and cell motility of EOC. They reduce cell-to-cell adhesion without affecting angiogenesis significantly. They build a structural guide for the cancer new vessels. | Inhibiting of PDGF-β signaling. |
| CD103+ NK lymphocytes | Tumor growth restriction, innate immunity. Activated by NKG2DLs. | Neutral competitor ligand for NKG2D to prevent the NK cells’ anergy. |
| High levels of circulating ULBP2 (NKG2DL) fragments could down-regulate the EOC’ expression of NKG2D | Inhibitors of ULBP2 to prevent the impairment of the NK cells’ cytotoxic activity | |
| T helper 1 lymphocytes | Tumor growth restriction mediated by CD8+ activation. Diapedesis and differentiation promoted by INFγ, IL-2, and lymphocyte-attracting chemokines. They secrete in turn interferon γ (INF γ) and IL-2 to chemoattract cytotoxic TILs. | |
| Diapedesis inhibited by endothelin and VEGF. | Possible chemoattraction and diapedesis within TEM via inhibition of endothelin and VEGF | |
| T helper 17 lymphocytes | Pro-inflammatory TILs that stimulate CD8+ activation. | |
| Diapedesis inhibited by endothelin and VEGF. | Possible chemoattraction and diapedesis within TEM via inhibition of endothelin and VEGF. | |
| CD8+ CD103+ (CD137+) cytotoxic T lymphocytes | Tumor growth restriction, acquired immunity, cytotoxic activity after interaction with T helper. Presence in serous > endometrioid > clear cell > mucinous histological subtype. Diapedesis promoted by INFγ, IL-2, and lymphocyte-attracting chemokines. | Contemporary inhibition of CTLA-4 and PD-1, associated with vaccination. Agonistic antibodies specific for CD137 alone or in association with inhibition TIM3. Autologous CD8+ TILs cultured with IL2, expanded and then infused. Chimeric T cell receptor restricted for HLA-A2 that can bind a specific epitope of HER2. |
| Diapedesis inhibited by endothelin and VEGF. | Possible chemoattraction and diapedesis within TEM via inhibition of endothelin and VEGF. | |
| CD20+ B lymphocytes | Tumor growth restriction. They increase the survival rate of CD8+ TILs. CD20+ TILs might act as APCs (positivity for MHC I/II, CD80, CD86, and CD40) → antigen reservoir preventing CD8+ anergy form persistent stimulation due to tumor lysis activity. | |
| Diapedesis inhibited by endothelin and VEGF. | Possible chemoattraction and diapedesis within TEM via inhibition of endothelin and VEGF. | |
| CD4+CD25+ FOXP3+Tregs | They inhibit the cytotoxic functions of TILs releasing inhibitory cytokines (TGF-β and IL-10) or via a direct cell-to-cell block. | Tregs depletion or shift into T helper 17 (IL2). |
| Activated by CCL28 under hypoxia condition and in the presence of B7H4+ TAMs. | Inhibition of CCL28; immunotherapy against TAMs. | |
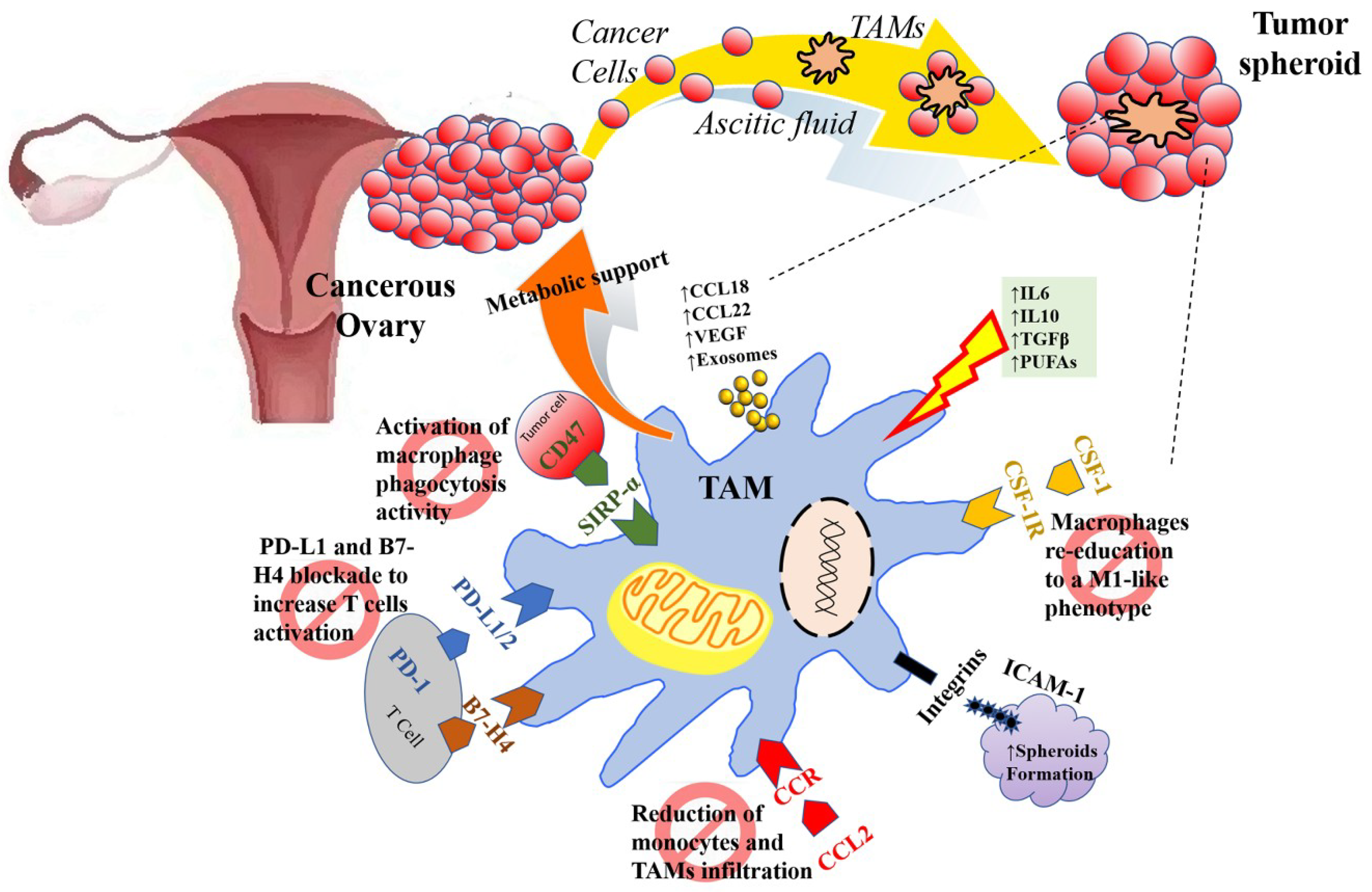
| TAMs | CCL2/CCR recruited. TAMs promote immunosuppressive activity: Tregs trafficking to TEM (CCL22) and inhibit T-cell cytotoxicity (B7-H4, PD-L1). | Reducing monocytes chemoattraction within TEM (bisphosphonates, inhibitors CCL2 antibodies). Inhibiting the PD-L1/2 checkpoint to reactivate cytotoxic T cells. Inhibition of PD-1/PD-L1 axis using target antibodies (pembrolizumab, nivolumab, avelumab) to promote survival, activation, and proliferation of cytotoxic T cells. |
| TAM offer metabolic support for EOC cells (glutamine). | Depleting extracellular glutamine. | |
| Activated by IL10, IL6, TGFβ, PUFAs acquiring an M2-like polarization state. TAM promote angiogenesis (VEGF). | Shifting the M2 to M1-like (pro-inflammatory and anti-angiogenetic) via TLR4 signaling (PCX), via inhibition of mTOR/p70S6K (neferine), via the inhibition of inhibitors of the CSF/CSF-1R pathway (GW2580, a selective CSF1R kinase inhibitor) or using 9-hydroxycanthin-6-one, deoxyschizandrin). | |
| TAMs promote metastasis dissemination thanks to the secretion of CCL18 and matrix support and growth factors (EGF) within EOC spheroids floating in the peritoneal fluid bound together by integrins and ICAM-1 via CD11b/c binding. | ||
| Re-activation of phagocytosis inhibiting CD47 (EOC cells “don’t eat me signal” that binds TAMs’ SIRPα). |
| Cell Type | Pathogenetic Role | Translational Possibilities |
|---|---|---|
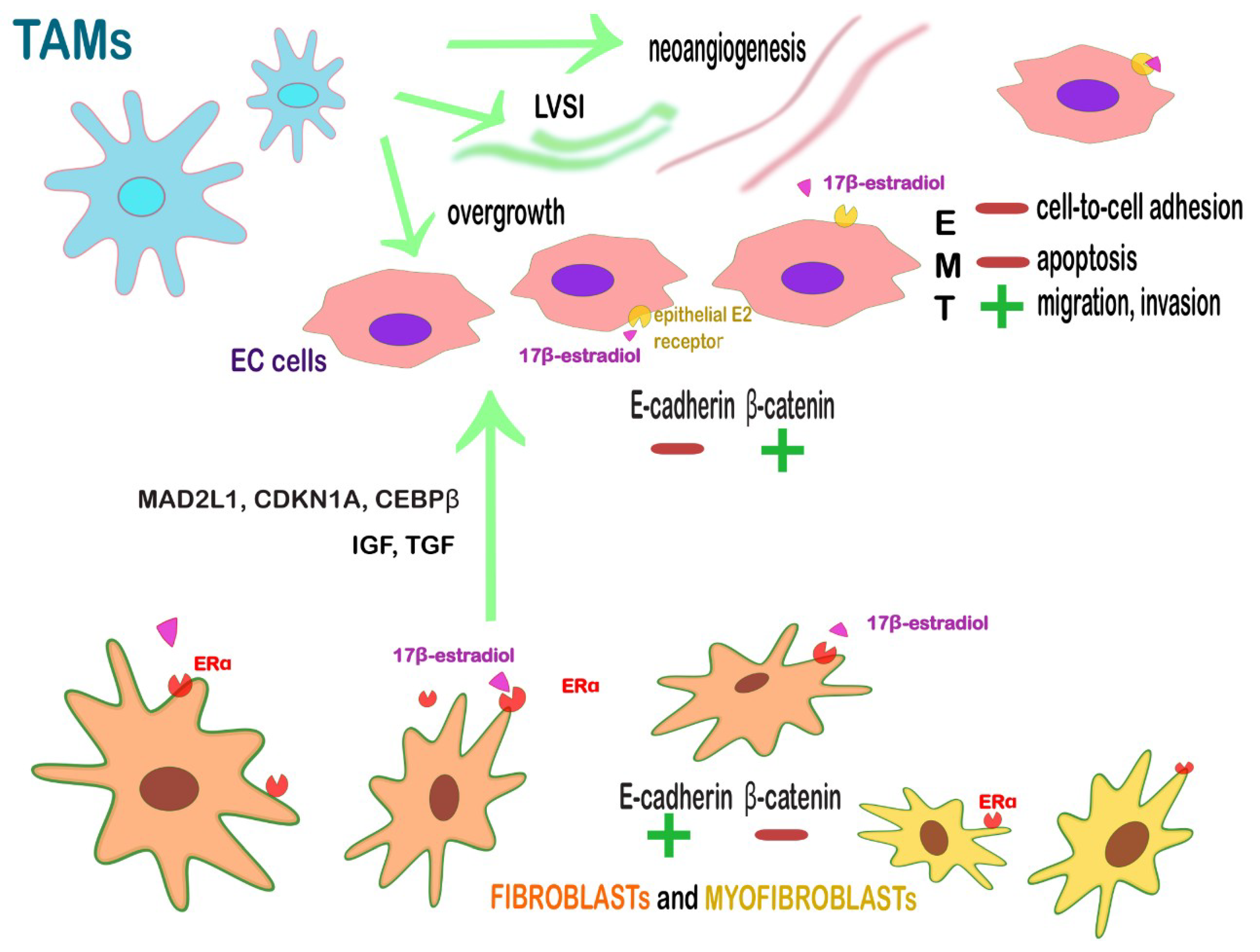
| ERα+ fibroblasts and myofibroblasts | Juxtacrine and paracrine action on endometrium with the secretion of anti-apoptotic and proliferative factors. Loss of β-catenin and EMT-associated proteins (TWIST, SNAIL-SLUG) in an opposite subset of EC cells → enable the near cancer cells to migrate, invade and to escape from apoptosis. | Targeting stromal ERα or the further cascade-molecules: IGF1, TGF and cell-cycle-related proteins, such as MAD2L1, CDKN1A, and CEBPβ.Targeting stromal ERα might revert also the multistep tumoral process since it is influenced by estrogens. |
| CD163+ M2 TAMs | Promote angiogenesis, LVSI, lymph node metastasis, tumor overgrowth. | Re-education toward an antitumor, immunostimulatory function (PCX); blocking monocytes migration to the TME; activate the phagocytic activity of TAMs; blockade of PD-L1 on TAMs (avelumab, nivolumab pembrolizumab). |
| Cell-Type | Pathogenetic Role | Translational Possibilities |
|---|---|---|
| Persistently high-risk HPV+ keratinocytes | Inhibition of inflammation in early stages; progressive chemoattraction for monocytes (MCP1 and MIP3A), Th17 (CCL20, IL6) but also NK cells in advanced stages | Targeting EGFR, CCL2 (also known as MCP1); CCL20, (also known as MIP3A); IL6 |
| ERα+ fibroblasts and myofibroblasts | Mesenchymal-epithelial transition Secretion of inflammatory chemokines, anti-apoptotic, pro-angiogenic factors and ECM enzymes | Targeting stromal ERα, IL1A and IL1B, FGF9, HBEGF, CXCR2 and its ligands CXCLs (mainly CXCL5 and CXCL1), MMP9. |
| CD163+ M2 TAMs | Promote angiogenesis, LVSI, lymph node metastasis, tumor overgrowth | Re-education toward an antitumor, immunostimulatory function (PCX); blocking monocytes migration to the TME; activate the phagocytic activity of TAMs; blockade of PD-L1 on TAMs (avelumab, nivolumab pembrolizumab) |
| Th17 lymphocytes | Chronic pro-inflammatory/pro-tumoral effect | Targeting CCL20 and IL6; re-education under IL2 stimuli |
| NK lymphocytes | Innate immune activity against tumoral cells expressing MICA and ULBP1 (NKG2DLs) | Clonal autologous expansion; vaccines against MICA and ULBP1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Nola, R.; Menga, A.; Castegna, A.; Loizzi, V.; Ranieri, G.; Cicinelli, E.; Cormio, G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. Int. J. Mol. Sci. 2019, 20, 2401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102401
De Nola R, Menga A, Castegna A, Loizzi V, Ranieri G, Cicinelli E, Cormio G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. International Journal of Molecular Sciences. 2019; 20(10):2401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102401
Chicago/Turabian StyleDe Nola, Rosalba, Alessio Menga, Alessandra Castegna, Vera Loizzi, Girolamo Ranieri, Ettore Cicinelli, and Gennaro Cormio. 2019. "The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication" International Journal of Molecular Sciences 20, no. 10: 2401. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102401