Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease


1
Instituto de Investigación Sanitaria San Carlos (IdISSC), 28040 Madrid, Spain
2
Departamento de Bioquímica y Biología Molecular, CIBERNED, IRICYS. Facultad de Medicina, Universidad Complutense de Madrid, 28040 Madrid, Spain
3
Neuroscience and Ophthalmology Research Group, Institute of Inflammation and Ageing, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(10), 2450; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102450
Submission received: 12 March 2019
/
Revised: 11 April 2019
/
Accepted: 22 April 2019
/
Published: 17 May 2019
(This article belongs to the Special Issue Glutamate Receptors in Health and Disease)
Abstract
:Glia form a central component of the nervous system whose varied activities sustain an environment that is optimised for healthy development and neuronal function. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA)-type glutamate receptors (AMPAR) are a central mediator of glutamatergic excitatory synaptic transmission, yet they are also expressed in a wide range of glial cells where they influence a variety of important cellular functions. AMPAR enable glial cells to sense the activity of neighbouring axons and synapses, and as such many aspects of glial cell development and function are influenced by the activity of neural circuits. However, these AMPAR also render glia sensitive to elevations of the extracellular concentration of glutamate, which are associated with a broad range of pathological conditions. Excessive activation of AMPAR under these conditions may induce excitotoxic injury in glial cells, and trigger pathophysiological responses threatening other neural cells and amplifying ongoing disease processes. The aim of this review is to gather information on AMPAR function from across the broad diversity of glial cells, identify their contribution to pathophysiological processes, and highlight new areas of research whose progress may increase our understanding of nervous system dysfunction and disease.
Keywords:
glia; AMPA receptor; astrocyte; microglia; glutamate; excitotoxicity; inflammation; hypoxia; ischemia; multiple sclerosis; neurodegeneration1. Introduction
Glutamatergic signaling through alpha-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptors (AMPAR) forms a major component of excitatory synaptic transmission in the central nervous system (CNS). However, glutamate release in the CNS is not exclusive to synaptic terminals, but also arises from unmyelinated axons [1] and non-neuronal glial cells [2] under both physiological and pathophysiological conditions. Glutamate is therefore present at varying concentrations in a number of extra-synaptic locations where it influences non-neuronal AMPAR, particularly those expressed by CNS glia cells, leading to influences on a range of critical functions. Importantly, glutamate release is enhanced under a number of pathological conditions [3,4,5] leading to concentrations of extracellular glutamate that trigger excitotoxic processes that threatens glial cell viability, and set in motion cellular and molecular processes that initiate and intensify pathophysiological conditions. In this review we provide an overview of AMPAR expression in glial cells of the CNS and peripheral nervous system (PNS), describing functions for these receptors in physiological conditions, highlighting their involvement in glial responses to pathophysiological conditions, and hypothesising on additional roles that glial AMPAR may perform in the context of nervous system injury and disease. The review will also indicate areas for future research, including cannabinoid AMPAR interactions, and AMPAR-stimulated transcriptional regulation, whose investigation promises to stimulate new knowledge on mechanisms regulating injury and disease processes, and identify new targets for CNS protection and repair.
1.1. Glia: A Brief Overview of Diversity in Form and Function
Glia are a diverse group of neural cells whose principle uniting features are a non-neuronal identity, and the performance of functions essential to the normal operation of the nervous system. Glia can be divided into two categories based on their developmental origins: macroglia derived from the ectoderm; and microglia derived from hemopoietic stem cells originating from the yolk sac during early embryonic development [6]. The principle CNS macroglia are the astrocytes and oligodendrocytes (OL). Astrocytes are a heterogenous group of cells that, in addition to a well-defined role in optimising extracellular conditions for neuronal function via the uptake of neurotransmitters and ions, may also contribute to the regulation of a number of other functions including synaptic function, cerebral blood flow and maintenance of the blood brain barrier [7]. OL are exclusively involved in myelin generation in the CNS [8]. However, it is now appreciated that myelination provides benefits to their neuronal targets that extend beyond the enhancement of axonal conduction velocities to encompass the provision of trophic and metabolic support [9], and potentially an involvement in information processing and and learning [10,11]. CNS macroglia also include NG2-glia, radial glia, and ependymal cells. NG2- glia are characterised as multi-process bearing NG2/PDGF receptor α expressing cells capable of generating OL and astrocytes (and possibly neurons, although this is the subject of intense debate [12]) in both the embryonic and postnatal CNS [13]. However, their extensive distribution throughout the adult CNS, and synaptic integration with neural circuity, suggest functions that extend beyond the role of a glial progenitor [13,14,15]. Radial glia are astrocyte-like neural progenitors that sequentially give rise to neurons, astrocytes and OL during embryonic development [16]. They are also considered to be the progenitors for the CNS ependymal cells (see below) [17]. Structurally, radial glia are polarised cells with a cell body located close to the ventricle, a short “endfoot” oriented towards the ventricle wall, and a longer process that extends to make contact with the pial surface [18]. This morphological arrangement provides a scaffold that guides newly born neurons as they migrate into the developing cerebral cortex [19]. Ependymal cells cover the brain’s ventricles and the central canal of the spinal cord. Examples of ependymal cells include the cerebral spinal fluid secreting choroid plexus epithelial cells [20], and thalamic tanycytes. Tanycytes are radial glia-like cells located in the walls of the 3rd and 4th ventricle, that exhibit a neuro- and gliogenic capacity, and through their function as glucosensors, are proposed to play a role in regulating feeding and energy balance [21]. Microglia, which are restricted to the CNS, present a dualistic identity, providing protective and modulatory influences under healthy conditions through the release of pro-survival trophic factors, and by tidying CNS tissues through the removal of dead cells and the pruning of excess synapses. However, when activated under pathological conditions they play a central role in the progression of CNS injury and disease states through the release of inflammatory and cytotoxic mediators [22]. Thus, microglia are essential to both the maintenance of a healthy CNS environment, and the pathophysiological processes that threaten it.
PNS macroglia include Schwann cells, satellite cells and enteric glia. Schwann cells exhibit two forms, the myelinating variety which form compact myelin sheaths on PNS axons, and non-myelinating forms whose interactions with multiple axons form the Remak fibres [23]. Under pathological conditions Schwann cells are capable of de-differentiating into an immature phenotype reported to exhibit pro-demyelination characteristics, while also expressing neurotrophic factors involved in neuronal survival and axonal regeneration [24]. Satellite glia interact with the soma of neurons in the sensory, sympathetic and parasympathetic ganglia where they are considered to play a similar role to astrocytes in the CNS. Finally, enteric glia are located in the ganglia of the gastrointestinal tract and are considered the most similar of PNS glia to astrocytes due to their multi-process morphology, including “end feet” connected to blood vessels, and their coupling via gap junctions to form a glial syncytium [25].
1.2. AMPAR
AMPAR are one of three types of ionotropic glutamate receptors, the others being the N-methyl-d-aspartate receptors (NMDAR), known for their involvement in synaptic plasticity [26], and the kainate receptors (KR). KR are frequently grouped with AMPAR due to their similar pharmacological properties (e.g., agonised by kainate, and antagonised by several of the same drugs). However, KR are formed from distinct protein subunits that confer unique receptor properties, such as slower inactivation kinetics, that distinguish them from AMPAR [27,28]. Although glutamate can influence glia via activation of NMDAR and KR, these actions have been reviewed elsewhere [29,30] and will not be covered in the present work. Regarding AMPAR function in neuronal circuits, AMPAR activation mediates the majority of fast excitatory synaptic communication in the CNS. AMPAR are heterotetrameric complexes containing various combinations of the pore forming GluA subunits 1–4 (GluA1–4) whose assembly produces cation permeable receptors with physiological properties that differ depending on their specific subunit composition [28]. AMPAR channel activation permits the influx of Na+, and varying levels of Ca2+, depending on the subunits present in the complex. Cation influx induces excitatory post synaptic currents (EPSCs) with rapid kinetics that are characterised by pronounced receptor desensitisation and influenced by subunit composition [31,32]. The inclusion of GluA2 limits the permeability of AMPAR to Ca2+ due to codon editing at the so-called Q/R site located within the pore forming region. The vast majority of GluA2 subunits undergo Q/R editing leading to the presence of a positively charged arginine residue within this region that inhibits the permeation of divalent ions [33,34]. In this way, AMPAR lacking GluA2 show greater Ca2+ permeability than complexes containing this subunit. Variation in AMPAR desensitisation of all GluA subunits arises through alternative splicing to produce Flip and Flop variants that differ in their kinetics of desensitisation [28]. Importantly, most Flip variants exhibit slower, less pronounced, desensitisation, thus a dominance of these subunits over the Flop variant in AMPAR would be expected to increase vulnerability to excitotoxic injury. This scenario may occur in amyotrophic lateral sclerosis (ALS) where motoneurons in the cervical ventral horn show a decrease in Flop variants, while Flip expression is sustained [35]. Diversity in AMPAR receptor function is also provided by a range of auxiliary proteins, most notably the transmembrane AMPAR regulatory proteins (TARPs), whose inclusion in the complex influences AMPAR pharmacology and kinetics, and regulates GluA subunit trafficking [36,37].
AMPAR function in the nervous system is not restricted to neuronal cells. Indeed many glial cells exhibit functional AMPAR whose activation regulates a range of important cellular activities including cell migration [38,39], morphological development and re-structuring [40,41] proliferation and differentiation [42,43], transcriptional regulation [44,45], survival [46], and the regulation of various ion channels [42,47,48]. Importantly, overactivation of AMPAR plays a key role in mediating cellular injury to neuronal and glial cells alike. These excitotoxic actions, which largely depend on the excessive influx of Ca2+, are described in the following section.
1.3. AMPAR Involvement in Cellular Injury
Glutamatergic excitotoxic injury is defined by the overactivation of glutamate receptors due to high or prolonged glutamate exposure. Cell death induced by excitotoxicity is an important feature in either acute or neurodegenerative pathologies [4,49]. After an ischemic stroke the increase of glutamate level directly correlates to the severity of the stroke, infarct volume and poorer functional outcome of patients [50,51]. That correlation has also been observed in different rodent stroke models [52]. Similar results were observed in the cerebrospinal fluid (CSF) or cerebral areas of patients after a traumatic brain injury [53,54]. Higher levels of glutamate have also been described in the CSF of infants correlating with the severity of hypoxic-ischemic (H-I) encephalopathy [55], and indeed levels of glutamate remain elevated for days in animal models of pre-term H-I injury [56,57,58]. Glutamate excitotoxicity is also an important event in chronic neurodegenerative diseases [49]. Both Parkinson’s and Alzheimer’s diseases are characterized by a dysregulation of glutamate homeostasis that may damage neurons [59,60,61], and both serum and CSF from patients with Multiple Sclerosis (MS) and Amyotrophic Lateral Sclerosis (ALS) show higher levels of glutamate [5,62,63].
Excessive levels of glutamate induce cell death by several complex mechanisms that have been previously reviewed [49,60,64]. Importantly, sustained activation of AMPAR, NMDAR and KR under these conditions produce a large increase in intracellular Ca2+ [65,66,67] that triggers various cellular injury processes including endoplasmic reticulum (ER) stress, mitochondrial dysfunction and the production of reactive oxygen and nitrogen species [59,66,68,69,70,71,72,73,74,75]. Ca2+ influx also induces the activation of calcium-dependent enzymes that promote cell damage. This situation is exemplified by calpain, whose activation by elevated Ca2+ levels promotes the internalization of plasma membrane Ca2+ ATPase leading to further dysregulation of intracellular Ca2+ and oxidative stress [68,76,77]. These outcomes are related to ER stress since this organelle is highly sensitive to disturbances in calcium homeostasis and the reactive species that ensue under these conditions. Therefore, the ER is highly sensitive to glutamate insults and cytosolic Ca2+ elevations [68,69,71,78,79]. In addition to these actions, calpain may actively promote ER stress via the modulation of ryanodine receptor and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [71,80,81,82]. Here, alterations in ryanodine receptors and the SERCA produce further dysregulation of the Ca2+ balance providing an additional stimulus to the production of reactive oxygen species, which, together with reactive nitrogen species that also arise due to enhanced Ca2+ levels, promote oxidative stress [64,68,69,72,77,80,83,84]. Ca2+ accumulation and nitrogen oxide disrupt the electron transport chain in mitochondria producing additional reactive oxygen species and mitochondria dysfunction [64,68,77]. Indeed, glutamate induction of both mitochondria failure and ER stress are critical factors in excitotoxic cell death [59,64,68,69,74,77]. Glutamate insult further potentiates oxidative stress by decreasing glutathione and superoxide dismutase activity, and promoting NADPH oxidase activity, eventually leading to lipid peroxidation, protein nitrosylation and cell death [72,85]. In addition, glutamate excitotoxicity also increases Ca2+ uptake by mitochondria, leading to the opening of mitochondrial permeability transition pores, and mitochondria fragmentation [66,74]. The high levels of Ca2+ uptake by mitochondria have also been related to the inhibition of mitochondrial respiration and the release of cytochrome c, thus triggering apoptosis [75,86,87]. Glutamate excitotoxicity also increases the expression of pro-apoptotic Bak protein, while also decreasing anti-apoptotic Bcl-2 protein levels [71], and altering the transcription and function of the nuclear factor Y (NF-Y) complex [45], a transcription factor closely linked to the control of apoptotic cell death [88] (see Section 8).
1.4. AMPAR in Glial Cells
The vast majority of research into glial cells has focussed on the astrocytes, oligodendrocytes and microglia. Much is therefore known regarding the expression and function of AMPAR in these CNS glia, while knowledge on this topic in other glial cells, particularly those of the autonomic system and the ependymal tissues is limited. Consequently this review will largely focus on those glia where functional AMPAR have been most thoroughly explored, although we will attempt to summarise the information that is available on other glial subtypes where possible. The following sections provide a review of each glial cell type, considering AMPAR expression, describing the known functions for these receptors in physiological and pathophysiological conditions, and highlighting emergent actions that stimulation of these AMPAR may evoke in the context of nervous system injury and disease. The major findings on AMPAR receptor expression and function in each glial cell type are summarised in Table 1, Table 2, Table 3 and Table 4.
2. Astrocytes
Astrocytes are the most numerous glial cells in the CNS. They are distributed throughout both the grey and white matter where they perform a myriad of tasks that serve to maintain neuronal function. Astrocytes make extensive contacts with synapses and nodes of Ranvier that enable them to regulate neuronal environments. They achieve this through the re-uptake of transmitters, and the buffering of extracellular ions, which together help to provide an extracellular environment that is optimised for efficient axonal and synaptic activity. In addition to their connections to neuronal compartments, astrocytes extend processes that terminate on cerebral vasculature through which they are involved in the formation of the blood brain barrier [89], and in mediating neurovascular coupling to maintain the supply of energy to the brain [90]. Astrocytes also have important functions in guiding CNS development, being involved in both the regulation of myelination [91] and synaptogenesis [92]. Astrocytes are equipped with a diverse array of neurotransmitter receptors that enable them to monitor neuronal activity, and which may provide the capacity to generate feedback signals via “glio transmitters” whose actions on adjacent neuronal synapses may involve the regulation of basal transmission and the regulation of synaptic plasticity [93] (also see [94]). Many of these neurotransmitter actions, including those stemming from glutamate, are associated with the activation of metabotropic receptors [95]. In contrast, less is known regarding the influence of AMPAR in astrocytes. The following sections will review the available literature and discuss the potential functions of astrocyte AMPAR in physiological and pathophysiological conditions. A summary of these findings is presented in Table 1.
2.1. Expression and Functional Properties of AMPAR in Astrocytes
Whole-cell voltage-clamp recordings show that astrocytes in most CNS regions exhibit functional AMPAR [96,103,104,105]. The exception to this rule appears to be the hippocampus where AMPAR-mediated currents are absent from astrocytes exhibiting high levels of GFAP-GFP transgene expression (cells expressing low GFAP-GFP transgene are now recognised as oligodendrocyte progenitors) [102]. The subunit composition of astrocyte AMPAR differs from region to region leading to variability in their permeability to Ca2+. Bergmann glia in the cerebellum exhibit strong expression of transcripts for GluA1 and GluA4, and lower levels of GluA2, leading to AMPAR with a high level of Ca2+ permeability [40,96]. Similarly, astrocytes in the olfactory bulb (OB) express significant levels of GluA1 and 4 protein, and low levels of GluA2 [103]. Although AMPA stimulation evokes a measurable influx of Ca2+ in these cells, AMPAR-mediated currents in these astrocytes are not fully blocked by specific blockade of Ca2+ permeable AMPAR, thus OB astrocytes appear to express a mix of Ca2+ permeable and impermeable receptors [103]. In other CNS regions, such as the cortex, GluA2 represents a dominant AMPAR subunit in astrocytes, with GluA1 and 4 transcripts being present at considerably lower levels [100]. As expected, AMPAR stimulation fails to elicit Ca2+ influx in cortical astrocytes unless their desensitisation is blocked by the application of cyclothiazide (CTZ) [101] indicating a low permeability to Ca2+. In the spinal cord immunohistochemical analysis reveals GluA2, 3 and 4 protein on GFAP+ astrocytes [106], while astrocytes in the thalamus exhibit a low level of Ca2+ permeability, potentially due to a dominant expression of GluA2, as revealed by the partial sensitivity of their AMPAR currents to pharmacological agents that selectively block GluA2 lacking receptors [104].
2.2. Astrocyte AMPAR Functions Under Physiological Conditions
Despite the abundance of studies exploring the expression of AMPAR in astrocytes (Section 2.2), relatively few physiological functions are ascribed to these receptors. In vitro studies using primary cultures of cortical astrocytes show that Na+ influx due to AMPAR activation produces a blockade of outward K+ currents through both A-type and delayed rectifier channels [48]. AMPAR activation in cultured Bergman glia produces similar effects on K+ currents activity [47,97]. Although this mechanism has not been confirmed in situ, it has been proposed to act as a mechanism for limiting elevations in extracellular [K+] during periods of excessive neuronal activity [48]. Astrocyte AMPAR are likely to be situated in appropriate locations to fulfil this role since astrocyte processes make extensive contact with synapses [92] and nodes of Ranvier throughout the CNS [107,108] (Figure 1A and Figure 2A). Glutamate has well documented effects on the function of peri-synaptic astrocytes [95], thus AMPAR-mediated effects on outward K+ currents seems a possibility. With regard to peri-nodal astrocytes, internodal axonal segments are known to release glutamate in an activity-dependent manner [109], yet it is unclear whether glutamate released in this way diffuses in sufficient concentrations to stimulate glial receptors located at nodes (Figure 1A). OL myelin). To do so it must cross the paranodal axoglial junctions (PAJ) that separate the internodal periaxonal and paranodal spaces. The PAJ has in fact been proposed as a route for the traffic of small molecules such as glucose [110], thus it is conceivable that glutamate may also diffuse via this structure to reach glial AMPAR located in peri-nodal spaces. Resolution of these questions could be achieved through the application of a genetically encoded glutamate sensing molecule, for example the intensity-based glutamate sensing fluorescent reporter (iGluSnFR) [111], which if targeted to astrocytes may be useful for revealing perinodal glutamate release. Alternatively, two-photon imaging may be used to analyse AMPAR-mediated Ca2+ influx at peri-nodal astrocyte process [112]. Studies of this nature could have wide-reaching impact given the important role astrocytes play in the regulation of myelination [91], and the influence that neuronal activity exerts in guiding oligodendrocyte differentiation and myelination [113]. Additionally, glutamate exerts a multitude of action in astrocyte via metabotropic glutamate receptors [95], thus evidence in support of an axonal-astrocyte glutamatergic signaling pathway could have significance for our understanding of white matter development that extends beyond the influence of AMPAR.
In contrast to the hypothetical functions of white matter astrocytes discussed above, AMPAR located on the processes of astrocytes in the molecular layer of the cerebellum perform an established function in the regulation of excitatory synapses. Here, Ca2+ influx through Ca2+-permeable AMPAR located on Bergman glial processes is linked to the regulation of glutamatergic synapses on Purkinje cell dendrites [40]. These synapses are enfolded by Bergman glia processes [114] whose expression of the glutamate transporter GLAST helps define the kinetics of AMPAR-mediated currents at Purkinje cells synapses. The function of these Ca2+-permeable AMPAR, which lack GluA2, are revealed by experiments in which the Ca2+ permeability of these AMPAR is reduced by exogenous expression of GluA2. Forced GluA2 expression leads to a retraction of the glial processes away from synapses, and an increase in the duration of Purkinje cell glutamatergic synaptic currents. These changes likely reflect alterations in the re-uptake of glutamate by Bergmann glial glutamate transporters located on the retracted processes [40]. Evidence for the significance of these synaptic effects are apparent in observations from transgenic mice with astrocyte-targeted deletions of GluA1 and 4. Here, changes in glial-neuron interactions and synaptic function are correlated with alterations in fine motor control that are consistent with the disturbances in Purkinje cell synaptic function [98].
2.3. Astrocyte AMPAR in Pathology
Astrocyte AMPAR activation has not been directly implicated in CNS injury or disease. Indeed, in contrast to cells in the OL lineage (Section 3.2), astrocytes do not appear to be vulnerable to pathological conditions associated with glutamate mediated excitotoxicity [106,115]. In fact, AMPAR mediated excitotoxicity is only observed in cultures of cortical astrocytes when AMPAR desensitisation is prevented by the application of CTZ [101]. Ca2+ imaging failed to reveal glutamate-mediated Ca2+ influx in these cultures suggesting that cortical astrocytes exhibit AMPAR with low Ca2+ permeability. These findings, and similar observations of excitotoxic resistance in hippocampal astrocytes [116], are supported by RNAseq data showing a dominance of GluA2 in both cortical and hippocampal astrocytes in situ [100]. Thus, low levels of AMPAR Ca2+ permeability may be associated with resistance to glutamate mediated injury. While excessive AMPAR activation does not appear to induce overt injury, it may trigger molecular alterations that could then trigger pathological conditions in the CNS. In this regard, prolonged stimulation of cultured Bergmann glia with AMPAR antagonists produces a downregulation in the expression of the glutamate transporter GLAST [99] (Figure 2B). Alterations in glial glutamate clearance due to a reduction in GLAST would be expected to disturb synaptic transmission, and could intensify excitotoxic conditions thus threatening more vulnerable neurons and OL. On this basis astrocyte AMPAR may represent a useful therapeutic target despite the apparent insensitivity to glutamate mediated injury of these glia.
Excitoxicity is a common feature of many CNS disease states including H-I injury, stroke, multiple sclerosis and neurodegenerative disorders including Alzheimer’s, Huntinton’s and Parkinson’s Disease [118]. Thus, CNS diseases of this type may provide valuable areas in which to search for subtle molecular alterations that could signal an involvement of astrocyte AMPAR in the initiation or propagation of the disease state. In this context, the emergence of RNAseq studies examining cell-specific gene expression in various disease models provides an opportunity to search for disease signatures that could foreshadow an involvement of glial AMPAR. The experimental autoimmune encephalomyelitis (EAE) model of inflammatory demyelination provides a promising model for this line of enquiry since AMPAR-mediated excitotoxicity has been linked to disease processes in this model [119,120,121]. In agreement with this, a recent astrocyte transcriptome analysis in the EAE model reveals a down-regulation of GluA4 in spinal cord astrocytes [122]. The consequence of this alteration in AMPAR expression are unclear. However, given the involvement of GluA4 in mediating Ca2+ influx, and the influence of astrocytes on myelin formation and viability [91], it is interesting to consider the detrimental effects that could arise due to an uncoupling of the axon-astrocytes interaction under inflammatory excitotoxic conditions. Although not direct implicating AMPAR activation, astrocytic GluA2 has also been linked to the pathogenesis of EAE. Recent work studying protein complexes containing GluA2 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a key enzyme involved in glycolytic metabolism, have identified an increase in the presence of this complex in EAE lesions [123] (Figure 1B). This is significant since excitotoxic stimulation of AMPAR induces cell death in cultured neurons via nuclear translocation of GluA2-GAPDH complexes, where it likely promotes cell death via upregulation of the p53 pathway [124,125]. Interestingly, a cell penetrating peptide designed to disrupt the formation of GluA2-GAPDH complexes ameliorates both clinical disease and the degree of astrocyte reactivity in EAE [123]. In addition, the peptide reduces inflammation-associated changes in isolated cultures of astrocytes, including increased expression of GFAP and EAAT1/2 proteins, nuclear translocation of GluR2-GAPDH, and p53 activation [126]. These findings highlight AMPAR-mediated GluA2-GAPDH nuclear translocation and p53 activation as a potential mechanism connecting glial AMPAR to inflammatory excitotoxic disease states. Importantly, GluA2-GAPDH complexes are also enriched in MS lesions [123] suggesting that this complex may represent a promising therapeutic for inflammatory demyelination. In conclusion, while evidence linking astrocyte AMPAR activation to CNS disease is limited, subtle changes in the molecular characteristics of astrocytes following pathological AMPAR activation may contribute to the initiation and propagation of CNS injury and disease. In addition, AMPAR subunits may stimulate inflammatory disease processes through participation in novel protein complexes. Consequently, further work is required to investigate the links between CNS disease states, disturbances in the astrocyte AMPAR transcriptome, and the intracellular behaviour of GluA proteins.
3. Oligodendrocytes
Discovered by Pio Hortega in 1921, OL are the myelinating cells of the CNS [127]. Along development, three different waves of OL generation have been identified in rodent forebrain: the first one, arising from OL progenitors (OPC) stemming from the medial ganglionic eminence and anterior entopeduncular area, happens around E12.5, and is almost completely replaced by early postnatal stages [128]. This wave is followed by a second that originates in the lateral and caudal ganglionic eminences around day E16.5 [128]. Finally, around birth a new wave of OPC with a cortical origin populates cortical areas [128]; although some studies have pointed out that this cortical oligodendrogenesis may occur earlier in development [129]. Concerning cerebellar OL, their origin is mainly extracerebellar and with a likely source being OPC from the ventral rhombomere 1 (r1) that populate the cerebellum by day E18.5 [130]. Two different waves of OPC have also been described in the spinal cord [131,132]. The first one around E13, that produce most of the spinal cord OL, and the second, starting on E15.5, which contributes a smaller population of OL in spinal cord [131,132].
The maturation of the OL lineage is commonly divided into four stages: OPC, late OPC (also known as preoligodendrocytes, preOL), immature OL (iOL) and mature myelinating OL (mOL) [133,134,135]. OPCs are characterized by a bipolar morphology and the expression of markers like PDGF-Receptor α and NG2 [136,137]. These cells are able to migrate to different regions after damage or during development [138,139]. Of note, OPC remain abundant in the adult CNS where they retain the potential to differentiate into myelinating OL [15]. However, a substantial number of these cells remain undifferentiated, and it is this fact, coupled with their unique physiological connection to neural circuitry (see Section 3.2), that has led to the suggestion that they represent a distinct cell type, termed ‘NG2-glia’, with functions that extend beyond the generation of myelin forming cells [15,140,141]. Upon arrival at their target sites developmental OPC remain mitotically active, elaborate their processes, and lose their migrative properties before differentiating into preOL displaying immunoreactivity to the O4 monoclonal antibody [136,142]. A further stage of differentiation sees preOLs transition into iOL, which are post-mitotic cells with a highly complex morphological structure characterized by expression of galactocerebrosidase (Galc, recognized by the O1 monoclonal antibody) [142,143,144]. Finally, these cells wrap the axon, myelinating it as mOL [145,146].
Traditionally, the main function OL has been to myelinate the axon. Myelination is associated with an improvement of action potential transmission, increasing its speed and saving energy [147]. Thus, a lower velocity of conduction has been observed after demyelination [148], or in unmyelinated axons [149]. A recent hypothesis has proposed that myelination could contribute to a type of brain plasticity. Here, activity-dependent modulation of myelination on the most active fibers could, by altering their conduction velocities, provide a mechanism for coordinating the flow of information through neural circuits [10,150,151]. Myelin also protects axons from outside metabolites that could be harmful [152,153]. Besides isolating the axon, myelin also plays a role in the energetic metabolism of the axon as reviewed elsewhere [9]. These metabolic functions include involve the provision of energy substrates such as lactate or pyruvate, whose delivery to the axon contribute to the preservation of axon function and neuron survival [153].
3.1. Expression and Functional Properties of AMPAR in Oligodendrocytes
AMPAR are expressed at all stages of the OL lineage. OPC express all four GluA subunits, although GluA1 appears to be less prominent [154,155,156,157,158]. Analysis by patch-clamp recording from OPC in primary culture and acute brain slices indicate that OPC AMPAR exhibit rapid desensitisation kinetics, and a degree of Ca2+ permeability [155,156,159,160]. In support of this electrophysiological evidence, a number of Ca2+ imaging studies of OPC in cell culture and ex vivo preparations have revealed AMPAR mediated Ca2+ influx [140,155,161]. Given the prominence of GluA2 in OPC [155], and evidence showing the dominance of the Ca2+ impermeable Q/R edited form of GluA2 in hippocampal OPC [157] it has been proposed that OPC exhibit both Ca2+ permeable (GluA2 lacking) and impermeable (containing GluA2) AMPAR simultaneously [141]. A shift in potentiation to CTZ has been observed between postnatal day 5 and 12 suggesting a developmental increase in the proportion of AMPAR containing the Flip isoform [162]. Both Flip and Flop variants have been detected in the CG-4 OL cell line [163], but the relative expression of these variants in OPC in situ has not to our knowledge been reported.
Differentiated OL (iOL/mOL) continue to express GluA mRNA and protein, although the combination of subunits present appears to differ from that observed in OPC. RNA sequencing of OPC, iOL (also known as newly formed OL) and mOL indicates that all four GluA transcipts are down-regulated upon differentiation to the iOL stage, although the decline in GluA2 and GluA4 is less marked compared to GluA3 [164]. The decrease in expression continues with the transition to the mOL stage, although as discussed below it is notable that levels of GluA4 transcript remain relatively sustained [164]. In agreement with sustained GluA4 expression, AMPA receptor activation induces intracellular Ca2+ influx in mature OL isolated from the optic nerve and cortex [65,155]. At the protein level GluA2, 3 and 4 are detected in cultured iOL and mOL [155], and immunocytochemical staining has revealed expression of GluA4 in the soma and myelin of spinal cord OL [106], and in the soma and processes of CNPase-GFP expressing OL in optic nerve [165]. In the latter study, GluA2 was not detected in OL, but GluA3 protein expression in OL soma was inferred using anti-GluA2/3. In addition, a recent study in human white matter detected localisation of GluA4 protein to MBP+ myelin sheaths [166,167]. In contrast to these data, OL lineage cells within the forebrain white matter of immature rats exhibit immunoreactivity for GluA1, 2 and 3, and a brief expression of GluA4 protein on 04+ lateOPC between postnatal days 7-9, which was not observed on GalC+ iOL [168]. The expression and functional properties of OL lineage AMPAR are summarised in Table 2.
3.2. Oligodendrocyte AMPAR Functions Under Physiological Conditions
It is tempting to hypothesize that the pronounced alterations of AMPAR expression observed during OL differentiation [155,164] are related with the receptors role in OL maturation and myelination. Indeed, in line with evidence from other neural cell types, AMPAR have been implicated in developmental OL maturation and myelination [39,41,43,46,180] (Figure 1). AMPAR activation seems to have an inhibitory effect on OPC proliferation. This is observed by the reduction of the proliferative ratio after AMPAR stimulation in cell cultures [42], as well as its increase after AMPAR antagonism in organotypic cerebellar slice [41,43]. In agreement with this, a promotion of OPC proliferation has been studied after suppressing axonal release of glutamate [148]. By contrast, AMPAR activation appears to play a role in the recruitment of OPCs to target axons [148], perhaps via effects on OPC migration speed [37]. AMPAR also play a key role in OL differentiation, promoting the elongation and branching of OPC processes [41]. Indeed, this receptor seems to participate in the early stages of OPC lineage maturation rather than in developmental myelination [46,148], with alterations of this receptors inducing delays of myelination by reducing the number of mature oligodendrocytes [46].
The involvement of OPC AMPAR in developmental myelination has been challenged by recent in vivo work involving mice carrying a conditional deletion of OPC GluA2, 3, and 4 [46]. OPC AMPAR currents are completely abolished under these conditions, yet analysis of OPC in tissues from these mice failed to detect a change in OPC proliferation or differentiation [46]. Instead, the genetic ablation of OPC AMPAR resulted in a decrease in OPC survival. Importantly, parameters of myelination considered to be senstive to neuronal acitvity such as internodal length [181] and number [182] also remained unaltered. These findings are in contrast with data from larval zebrafish and rodent optic nerve where suppression of neuronal activity and attenuation of vesicular glutamate induce a measurable decrease in internode number and length respectively [182,183]. Technical differences in the approaches used to modulate glutamatergic actions on OPC could account for these contrasting results. Kougioumtzidou et al. [46] used a non-inducible Cre strategy to delete Gria2 and Gria4 in OPC on a constitute Gria3−/− background. These tripple transgenic OPC lacked functional AMPAR through development, thus they would have been insensitive to all sources of glutamate-mediated AMPAR stimulation, be that vesicular or otherwise. One consideration is whether complete starvation of AMPAR-mediated signaling through the life-span may induce compensatory changes in OPC that mask the function of AMPAR in OPC maturation and myelination. In support of this view, recent work using OPC-specific retroviral-mediated modulations that alter OPC AMPAR only during postnatal development have identified an involvement of these receptors in OPC proliferation and differentiation, but not survival [180] (discussed in more detail below). With regard to the study by Kougioumtzidou et al. [46], it should also be noted that Gria2, 3, and 4 lacking OPC would continue to receive glutamatergic stimulation from NMDAR [165,184,185], thus actvitity-dependent glutamate signaling could still influence OL maturation and myelination [185] (but see [186]). In contrast, Mensch et al. [183] and Etxeberria et al. [182] targeted glutamate release, rather than AMPAR expression, thus OPC in these studies could continute to receive stimulation from glutamate released by non-vesicular sources, which may act on both AMPAR and NMDAR. The use of an inducible-conditional Gria 2, 3, 4 deletion, perhaps via a multiplex CRISPR-based knockout strategy, could help to bring further clarity to the role of AMPAR signaling in OPC maturation and myelination.
Notably, OPC AMPAR are activated by vesicular release of glutamate from unmyelinated axons in white and grey matter [141,187,188,189] (Figure 1A and Figure 2A). The function of these neuro-glial synapses is unknown, but it is hypothesised that they may signal levels of activity within neural circuits, perhaps allowing OPC to regulate their proliferation or differentiation at sites of increased activity [141,190]. In agreement with this idea, AMPAR-mediated input declines upon differentiation of OPC [191], and synaptic activity can induce Ca2+ influx into OPC via AMPAR [159,160], thus the synaptic activation of pro-differentiation Ca2+-dependent intracellular signals seems a possibility. However, recent evidence suggests a role for axon-OPC synapses in regulating proliferation but not differentiation [180]. In this work increases in the Ca2+ permeability of OPC AMPR via OPC specific expression of either non Q/R edited GluA2 subunits, or a “pore dead” GluA2 construct, promoted OPC proliferation without affecting differentiation or survival. Thus neuronal activity may influence OPC proliferation via the activation of OPC AMPAR and the subsequent activation of Ca2+-dependent signaling pathways. Interestingly, an additional strategy that reduced the proportion of Ca2+ permeable AMPAR in OPC without affecting GluA2 channel properties caused an increase in the size of the OPC population without altering proliferation or survival [180] suggesting further complexities in the influence of AMPAR on OPC development. Contrasts between these findings, and those indicating an enhancement of OPC proliferation following AMPAR antagonism in cerebellar slice cultures [41,43] may be explained if bath applied AMPAR blockers, as used on ex vivo slices, affect additional mechanisms that impinge on OPC functions. One possibility, as highlighted previously [41], would be an effect on neuronal synapses whose inhibition would be expected to produce similar effects to that seen when neuronal activity is blocked pharmacologically. Of note, both TTX and the AMPAR antagonist GYKI induce a similar stimulation of OPC proliferation in cerebellar slice cultures [41]. Taken together there is considerable evidence that OPC AMPAR, including those recruited via neuron-OPC synapses, exert influences on OPC migration, proliferation and survival during CNS development (Figure 1A).
Interestingly, a large numbers of OPC, or NG2-glia, persist in the adult CNS where they continue to receive synaptic input from neuronal circuits [reviewed by 182]. These NG2+ cells seem able to respond to this activity since, like their developmental counterparts [161], they exhibit activity-dependent and neurotransmitter receptor dependent Ca2+ transients [192]. These observations, and morphological data showing that their processes make intimate contact with multiple neuronal and astrocyte elements, are suggestive of specialized functions within the CNS [192]. Indeed, it has been proposed that NG2+ cells might regulate glutamatergic synapses by modulating postsynaptic AMPA [193], although this idea remains controversial at this time [194]. Aside from a role in remyelination (Section 3.2) other functions for OPC/NG2-glia in the adult CNS remains an open question.
Regarding differentiated OL, both iOL and mOL continue to express AMPAR (Section 3.1), and GluA4 has been detected in rodent spinal cord myelin [106] and human cortical white matter [167]. Ca2+-permeable AMPAR containing GluA4 may contribute to activity-dependent signaling between axons and myelin. Electrical stimulation of an ex vivo optic nerve preparation induces Ca2+ signaling in myelin that is blocked by NMDAR and AMPAR antagonists [109]. These data suggest that AMPAR activation may provide a depolarizing stimuli that relieves Mg2+ blockade from NMDAR, and indeed imaging in zero Mg2+ revealed a subtle NDMAR-dependent elevation in Ca2+ [109]. This form of activity-dependent signaling involves vesicular release of glutamate: Ca2+ signals were blocked by bafilomycin and tetanus toxin, both of which disrupt vesicular release; and enhanced by hypertonic sucrose stimulation, which encourages vesicle release. Whether or not this form of axon-myelin signaling occurs in other regions of the CNS, and what function it may play in regulating axon-OL interactions remains unclear. However, it has been proposed that an ‘axo-myelinic’ synapse may enable mOL to sense activity in target axons, allowing them to adjust the provision of metabolic support [195], perhaps via the provision of lactate [196], or to fine tune myelin internode parameters such as length and thickness in response to changes in demand [195].
3.3. Oligodendrocyte AMPAR in Pathology
The prominent expression of Ca2+ permeable AMPAR in the OL lineage places these cells at threat of injury from excitotoxic conditions (Figure 2B). Indeed, AMPAR-mediated excitotoxicity has been described in numerous in vitro studies using cultures of both OPC/preOL [45,170,171] and differentiated OL [65,163,197]. Importantly, excitotoxicity in these cells is associated with excessive Ca2+ influx [65,171], thus expression of Ca2+ permeable AMPAR, as described in Section 2.2, appears to render these cells vulnerable to high levels of extracellular glutamate. In support of this idea, specific antagonists of Ca2+ permeable AMPAR reduce injury in OL subjected to oxygen glucose deprivation [171], while forced expression of Q/R edited GluA2 subunits protected OPC from an AMPAR-induced excitotoxic injury [173]. Given the links between Ca2+ influx and cellular injury responses in OPC, the degree of Ca2+ permeable AMPAR expression in OPC would be expected to regulate vulnerability to excitotoxic injury. In this respect, it is interesting that group 1 mGluR activation leads to an increased surface expression of Ca2+ permeable AMPAR in OPC [198]. This mechanism, which involves specific TARP-dependent trafficking of the Ca2+ permeable GluA4 subunit, may act to amplify the sensitivity of OPC to elevated levels of extracellular glutamate. However, at this time the links between mGluR activation and OPC excitotoxicity remain unclear.
AMPAR-mediated injury has been observed in a number of in vivo models involving OL and myelin injury. For example, the AMPA antagonists NBQX reduces OL and myelin loss in rats subjected to a weight-drop spinal cord contusion injury [179], and as discussed below, ameliorates inflammatory demyelination in the EAE model of MS [119,120]. These observations are significant since excitotoxicity is implicated in a number of pathologies (described in Section 1.3) including Cerebral White Matter Injury (WMI) and MS [199,200]. In premature infants WMI includes different neuropathological cerebral white matter [201], the survivors of which eventually develop long-term neurological disabilities [199,202]. Within the framework of pre-term conditions, H-I damage has been specifically correlated to oligodendrocyte maturation, with preOL being identified as being particularly vulnerable in several species [115,203,204,205] including human [206]. For a detailed discussion of perinatal WMI see the recent review by Back et al. [199]. Importantly, within the OL lineage, preOL in developing white matter exhibit the greatest abundance of GluA4 [168,207], a Ca2+ permeable subunit highly expressed in neural cells exhibiting vulnerability to excitotoxic death [208]. Importantly, topiramate and NBQX reduce OL death in rodent models of neonatal H-I injury [172,178]. Thus, the protection of OL via the targeting of Ca2+ permeable AMPAR, or relevant downstream pathways, represents a potential strategy for the prevention of WMI in pre-term infants. However, despite these promising pre-clnical findings, it should be noted that other pathogenic events are likely to be relevant since preOLs are also very sensitive to increased levels of TNFα and oxidative stress, both of which are characteristic of WMI [209,210,211].
Related results are obtained after stroke in neonatal and adult rodent models. In both cases, cerebral ischemia is followed by a decrease in the mOL population [212,213] as well as OPC proliferation and migration into the damage areas [213,214]. mOL death is reproduced in vitro by AMPA but not NMDA administration, and is reduced by AMPAR antagonist after oxygen and glucose deprivation [197,215]. The administration of AMPAR competitive antagonist, SPD 502, mimics this oligoprotection after cerebral stroke induced in adult rats [216]. Interestingly, this effect is regionally restricted since the oligoprotection is specific to cortical OL. As mentioned above, brain OL are heterogenous both in respect of their developmental origins, and their differentiation rates in grey and white matter [217,218]. Therefore, deeper studies to analyze how AMPAR modulation affects specific OL populations are warranted if AMPAR-targeting therapeutics are to be developed further.
A number of observations link glutamate dysregulation and OL AMPAR to pathology in MS. First, MS is characterized by high levels of glutamate within CSF [5,62]. Second, an increase in levels of the Ca2+ permeable subunit GluA1, but not GluA2, in oligodendrocytes located next to MS plaques has been observed in post-mortem CNS samples of MS patients [219]. Third, a greater level of GluR2–GAPDH complexes are detected in MS plaques [123], and disruption of the GluA2-GAPDH complex has been shown to produce a therapeutic/protective action in EAE [123] (see Section 2.3 for a related discussion on this topic). Fourth, Ca2+ permeable AMPAR are implicated in CNS pathology in EAE where OL death, axonal damage and demyelination are reduced in mice lacking Gria3, the gene encoding the Ca2+ permeable GluA3 subunit [220]. In agreement with these latter findings, other work in the EAE model encourages the use of AMPAR as therapeutic targets in MS since blockade of these receptors via subcutaneous injection of NBQX reduces clinical disease and protects OL [119,120]. These promising preclinical results from models of MS [221], and also stroke [216,222], must be weighed against the negative findings from clinical studies that have observed worsened outcomes following the use of AMPAR-targeting drugs in patients with acute ischemic stroke, [223,224]. These disappointing results may be explained by the low specificity of the drug used, ZK200775, and the wide expression of these receptors in CNS circuitry, where their inhibition may be expected to produce numerous unwanted side effects that may interfere with any potential therapeutic benefits. As discussed above (Section 3.2) AMPAR influence the development and survival of OL. Consequently, protection of OL from excitotoxic conditions associated with CNS inflammation may not be achieved without compromising the regenerative capacity of OPC (but see [172]). Nonetheless, glutamate dysregulation plays wider roles in MS pathogenesis via actions on T cell function such as migration and cytokine secretion, and even glutamate release [5], thus therapies focused on controlling glutamate levels may provide significant benefits that extend beyond oligodendrocyte protection.
The activation of OPC AMPAR by axonal synaptic input may play a role in regulating OPC-mediated remyelination (Figure 1). New OPC recruited to lesions express AMPAR, which may be involved in both migration and the early stages of myelination [39,148]. Within the caudal cerebellar peduncle, axons demyelinated by infusion of ethidium bromide establish de novo glutamatergic synapses with recruited OPCs, while infusion of antagonists of specific voltage-gated Ca2+ channels, selected to specifically act on axonal channels, reduce the degree of remyelination [148,176]. Thus, synaptic activation of OPC AMPAR appears to be important for remyelination. Another study tracked the occurrence of synaptic inputs on OPC following lysolecithin lesions in the corpus callosum [176]. Here the authors report a disruption of synaptic input shortly after demyelination that correlates with a reduction in immunohistologically identified axonal synaptic connections on proliferating OPC in the lesion. Interestingly, a recovery in synaptic innervation coincides with a reduction in OPC proliferation, suggesting that AMPAR activation may inhibit cell division [176]. This observation agrees with recent studies indicating a role for synaptic Ca2+ permeable AMPAR in stimulating OPC proliferation [180], and with data from brain slice cultures and in vivo studies that show that blockade of axonal activity increases OPC proliferation [41,148,225,226]. However, blockade of activity in ex vivo brain slices also produces an increae in OL differentiation [41] that was not observed under in vivo remyelinating conditions [148]. These contrasting outcomes may be explained by differences in experimental models (ex vivo vs. in vivo) and the developmental status of the tissues examined (developing white vs adult remyelinating). Despite these differences the data from these studies, and many others from a variety of different CNS systems, clearly indicate an important role for axonal activity and AMPAR in the regulation of OL myelination [113,181,182,183,225,227] (Figure 1).
In conclusion, the excessive activation of OL AMPAR poses numerous threats to the viability of OL and the myelin they support. This loss of myelin compromises neurological function in a wide range of CNS disease states, thus therapies that protect OL from excitotoxic AMPAR-mediated injury are an important clinical goal. Nevertheless, AMPAR are powerful regulators of OL development and survival, and may also play unidentified functions in adult-NG2-glia. Therefore, AMPAR-targeting therapies capable of protection OL may induce unwanted side effects, and potentially hinder other aspects of OL regeneration and myelin repair. Further basic and pre-clinical research will be necessary to fully identify the functions of OL AMPAR, and determine the benefits and feasibility of targeting these receptors in a therapeutic context.
4. Microglia
Microglia are highly abundant CNS glia, whose origin within the embryonic yolk sac [6], and macrophage-like functions, distinguish them from all other parenchymal glia in the nervous system. In the healthy brain microglia exhibit highly motile processes that scan the local tissue landscape, seemingly searching for signs of disease or injury whose detection may trigger them into acquiring an activated amoeboid phenotype capable of launching inflammatory responses and engaging in phagocytic activity [228]. Microglia are considered to serve a number of protective roles that collectively help to maintain a healthy environment with the CNS. These functions include the removal of dead and dying cells, the regulation of synapse number via pruning of excess connections, and the production of various neurotrophic factors capable of influencing the development and survival of other neural cells [22]. In addition, microglia express major histocompatibility complex (MHC) I and II molecules, particularly under pathological conditions including perinatal hypoxia, thus a role in the detection of pathological conditions and the stimulation of adaptive immune responses are ascribed to these cells [229]. In addition to protecting the healthy CNS, microglia contribute to the generation of pathological conditions via the synthesis and release of cytotoxic molecules such as nitric oxide, reactive oxygen species and proinflammatory cytokines [230]. They also contribute to CNS repair processes by releasing neuroprotective factors, reducing inflammation, and encouraging regenerative processes such as OPC differentiation and remyelination [230]. These contrasting roles in CNS protection and injury have frequently been cast within a model where microglia exhibit polarised states, the so-called M1 (pro-inflammatory)/M2 (anti-inflammatory pro-repair) phenotypes [231]. However, a number of observations, such as the co-existence of cardinal M1/M2 molecules within microglia in vivo, have lead this dichotomy to be described as unsuitable and unhelpful for the understanding of microglial biology within the intact CNS [232]. Nevertheless, microglia certainly exert profound and varied influences in the context of CNS protection, immune activation, injury and repair, whose regulation by AMPAR may contribute to the induction and progression of a number of disease states.
4.1. Expression and Functional Properties of AMPAR in Microglia
Consistent evidence for function AMPAR expression in microglia has been reported in experiments on cultures of cortical rat microglia [233]. Reverse transcription PCR indicates that these cells express GluA transcripts 2, 3 and 4 [234], although a subsequent study using more sensitive quantitative RT-PCR method detected GluA, 1, 2 and 3 mRNA [235]. Expression of GluA1 has also been shown at the protein level in rat cortical microglia via immunocytochemistry [236]. Functionality for these AMPAR has been confirmed by patch-clamp analysis where CNQX sensitive glutamate currents are detected [233]. These currents were potentiated by CTZ, and showed little Ca2+ permeability [233], consistent with the presence of GluA2 transcripts. Interestingly, experiments with AMPAR modulators show that microglial glutamate currents exhibit a greater degree of potentiation to CTZ compared to PEPA [234]. CTZ is selective for Flip variants, while PEPA exhibits a greater affinity for Flop subunits, thus these data suggest a dominance of Flip splice variants in microglia [234]. In line with this finding transcript analysis in the same study revealed a predominance of GluA1-3 Flip variants, a configuration that may render cells more sensitive to excessive glutamate levels (Section 1.2). In contrast to the in vitro studies described above, evidence for microglial AMPAR expression in vivo is more uncertain. AMPAR expression has been detected by immunostaining for GluA2/3 and 4 in OX-42 cells in rat forebrain sections [237], although this work was done under permeabalising conditions hence it is not clear if the immunosignals represent surface expressed receptors. Importantly, immunostaining did not identify receptor subunits on the surface of retinal microglia, and patch-clamp recordings from microglia have failed to detect glutamate-mediated currents in a range of CNS tissues including spinal cord, hippocampus and retina (reviewed in [238]). Interestingly, hypoxia causes a marked increase in microglial GluA2-4 protein expression within forebrain periventricular white matter [237], and hippocampal microglia express GluA4 protein only after exposure to an ischemic injury [239]. These in situ observations suggest that microglial AMPAR expression may be low or absent under healthy conditions, but may be induced in microglia activated under pathological conditions [238] (Figure 2). Furthermore, the contrast between these findings, and those from in vitro studies, suggest that the physiological properties of microglia may differ significantly under cell culture conditions. Indeed, differences in the expression of voltage-gated K+ channels are observed when patch-clamp recordings are made from microglia in actute vs cultured hippocampal brain slices [240]. A summary of findings on the expression and functional properties of microglial AMPAR are summarised in Table 3.
4.2. Microglial AMPAR in Pathology
Evidence linking AMPAR activation to microglial functions under physiological conditions appear to be limited to effects on chemotaxis, where glutamate stimulates a directed-migration of microglia in cell cultures and spinal cord slices via AMPAR receptors [117]. In contrast, several interesting actions are associated with the activation of microglial AMPAR under pathological conditions that are linked with excessive glutamate signaling. Studies using a rat model of periventricular white matter (PWM) injury, and microglial cell cultures exposed to hypoxia, have identified links between hypoxia-induced elevations in glutamate, and the regulation of protective and inflammatory microglial-derived substances [237] (Figure 2B). This work shows that hypoxia causes increases in PWM concentrations for both glutamate and IGF-1, a neurotrophic factor associated with the protection of neurons [241] and oligodendrocytes [169,170] following H-I injury. Importantly, elevated IGF-1 protein is localised to microglial with an activated amoeboid morphology that also exhibit enhanced expression of GluA2, 3 and 4 subunits [237] (Figure 1B). While increased expression of IGF-1 may play a protective role following this injury, the increase in AMPAR expression in these cells appears to prime them for a more pathological response since a subsequent exposure to glutamate simultaneously attenuates IGF-1 production, while promoting TNF-α release and IL1-beta [237]. This inflammatory response is mechanistically linked to the reduced levels of IGF-1 seen after glutamate stimulation since knock-down of IGF-1 gene expression promotes TNF- α and IL-1 beta gene expression. Overall, the study by Sivakumar et al. [237] suggests that increased expression of microglial AMPAR in hypoxic PWM tissue sensitises microglia to glutamate leading to a regulatory response that reduces protective IGF-1 release while increasing the release of inflammatory mediators.
Other evidence suggests that microglial AMPAR activation, and a subsequent production and release of TNF-α, may contribute to the propagation of CNS disease states (Figure 2B). First, cell culture and in vivo experiments show that the activation of microglia AMPAR stimulates the production and release of TNF-α [234,237]. Second, TNF-α signaling is heavily associated with a number of glial-dependent actions, including the emission of glutamate via gap-junction hemichannels from microglia [242] and astrocytes, Fas ligand release from microglia, and the downregulation of EAAT2/GLT1 glutamate transporters in astrocytes, which trigger neurotoxicity in the CNS [2]. Third, TNF-α release from astrocytes stimulates the trafficking of AMPAR to the surface of hippocampal and cortical neurons, rendering them more sensitive to excitotoxic conditions involving excessive levels of extra cellular glutamate [243]. Fourth, conditioned medium containing TNF-α released from kainate-stimulated microglia induces the upregulation of voltage-gated Ca2+ channels and NMDA-type glutamate receptors in hippocampal neurons, and is associated with neuronal apoptosis [244]. In summary, microglial AMPAR-mediated TNF-α release, acting through a range of autocrine and paracrine routes, are positioned to elevate extracellular glutamate concentrations, release apoptosis inducing signals, and increase the sensitivity of neurons to excitotoxicity. As outlined in Section 1.3, excitotoxic injury has a link to a wide range of CNS injury and disease states [1,4], thus further investigation of the influence of the microglial AMPAR-TNF-α axis, and its actions in propagating excitotoxic conditions, are warranted. In this regard, a promising area for research can be found in the context of MS, where impaired cognitive function may be linked to pathological alterations in synaptic structures that arise early in the course of the disease [245]. Research using the EAE model of inflammatory demyelination shows that TNF-α contribute to the mechanisms driving these synaptic dysfunctions, at least within the striatum, where these pathophysiological actions are correlated with the activation of microglia and astrocytes [246,247]. While the involvement of microglial AMPAR is in these actions is unknown, the established links between their activation and the release of TNF-α [234,237] indicates that they could be capable of amplifying an initial inflammation-induced elevation in extracellular glutamate into additional excitotoxic insults.
Microglial AMPAR have also been implicated in neurodegenerative disease processes through a series of cell culture studies examining the regulation of GluA subunit surface expression in activated microglia [248]. Here, the authors examined the hypothesis that down-regulation of GluA2, as is observed in a number of neurodegenerative conditions including MS [249], drives neurotoxicity via increased release of pro-inflammatory cytokines from microglia. Analysis of cultured microglia generated from a global GluA2 null line exhibited a greater degree of Ca2+ permeability, and enhanced TNF-α production following AMPAR activation. In line with this data, conditioned medium from GluA2 -/- microglia exerted a greater neurotoxic effect on cultured neurons. Thus, as proposed by Noda and Beppu [250], the loss of microglial GluA2 under pathological conditions may serve to accelerate neuronal injury and loss via alterations in the subunit composition of their AMPAR that favour Ca2+ entry and inflammatory cytokine release. While this hypothesis provides an attractive link between microglial AMPAR function and disease, further work should be performed using a conditional deletion of GluA2 in microglia to refine the loci of action and confirm the relevance of this mechanism in relevant in vivo disease models.
In summary, microglial are equipped with AMPAR whose activation may help to synchronise the production and release of inflammatory molecules to inflammatory excitotoxic conditions within the tissue. These actions could trigger de novo excitotoxicity or may contribute to the propagation of an ongoing disease state. Based on these actions, therapies targeting microglial AMPAR represent a valuable ambition, the achievement of which may provide a means to modulate neurotoxicity in a number of neurodegenerative CNS conditions.
5. AMPAR in Other Glial Cells
AMPAR are expressed in glial cells beyond the principle types described in Section 2, Section 3 and Section 4, yet their functions in these ‘other glia’ are less characterized. Nonetheless, these glia, which in this review include Radial Glia, Schwann Cells, Satellite Glia and Enteric Glia, have important functions in both CNS and PNS. Therefore, AMPAR might yet play significant roles in their behavior under physiological and pathophysiological conditions. Here we present a brief overview of AMPAR in these other glial cells. See Table 4 for a summary of the expression and functional properties of AMPAR in these glial cells.
5.1. Radial Glia
Radial glia (RG) gives rise to astrocytes, neurons, and oligodendrocytes during embryonic development [16,262,263]. In primary cell cultures of RG-like neural progenitor cells (NPC) qPCR reveals a high level of GluA1 mRNA, and lower levels of GluA2, 3 and 4, while immunostaining detects GluA1, 2 and 3 protein expression on RG/NPC expressing the glial marker GLAST [251]. The functionality of these AMPAR is evident in Ca2+ imaging data where transient AMPA induced elevations in intracellular Ca2+ are enhacned by CTZ and reduced by philantotoxin, a specific antagonist of Ca2+ permeable AMPAR [251]. In contrast, another study revealed high levels of GluA3 and 4 mRNA, and an absence of GluA1 and 2, in NPC [252]. In agrement with this data, immunohistochemical staining reveals strong GluA3 and 4 localisation to RG in the developing white matter of rodents [168], while GluA4 is localised to RG in fetal human RG [207]. Differentiation of RG/NPC into astrocytes or neurons involves an increase of GluA2 and a decrease of GluA3/4, thus suggesting an increase in the proportion of Ca2+ impermeable AMPAR [168,207,252,253,264]. These findings suggests a role for glutamate acting via AMPAR in RG proliferation and differentiation. In agreement with this idea, AMPAR are involved in the regulation of RG processes length and RG motility [265].
RG-like cells are also present in the adult subgranular zone of the hippocampus [166,253]. In contrast to developmental RG, these adult RG-like cells possess AMPAR that are mainly composed of GluA2 subunits, and which are expressed in the cell’s processes but not in the soma [166]. The administration of kainate promotes RG-like cell proliferation and survival, both in vitro, and in vivo, in an animal model of status epilepticus [253].
Very little is known about AMPAR expression in ependymal cells derived from radial glia, although GluA2/3 expression has been found in cell bodies and proximal thick processes of tanycytes [254].
5.2. Schwann Cells
As mentioned in the introduction (Section 1.1), Schwann cells are the myelinating cells of the PNS. As in the CNS, PNS axons extend significant distances, thus in addition to the benefits associated with OL myelination, such as enhanced conduction velocity and axonal isolation (Section 3), trophic support arising from myelinating Schwann cells is likely to be important for sustaining the viability of axons located long distances from the cell body [152]. Distinct from the myelinating forms, the perisynaptic Schwann cells (PSCs) are non-myelinating cells that are part of the neuromuscular junction (NMJ) formed between the presynaptic nerve terminal and the postsynaptic specialization [264]. These cells plays an important role in the growth and maintenance of NMJs as well as in the modulation of its synaptic properties [264,266,267]. In comparison to OL, the very little less is known regarding the expression and function of AMPAR in SCs.
During development mRNA expression of all AMPAR subunits is detected in mouse sciatic nerve, although it is not clear if this expression arises from the axon or glial cells [256,268]. While GluA2/3 and GluA4 are detected by electron microscopy in Schwann cells of the vestibular system of rats and guinea pigs; only GluA1 and GluA4, but no GluA2 or GluA3, protein expression are found in primary SC culture [255,258]. Patch-clamp recording from developing SC in peripheral nerve tissue and cell cultures exhibit functional Ca2+ permeable AMPAR [256,257]. A detailed study performed in sciatic nerve preparation of mouse pups at embryonic day 16th–18th or postnatal day 0–2nd has proved that AMPAR are modulated along development, with functional AMPAR only expressed in developing SCs while more mature myelinating SCs are unresponsiveness to glutamate [256]. Furthermore, the amount of AMPAR might also be modulated in developing SCs [256].
However, the AMPAR role in SCs is still not clear. Metabotropic glutamate receptors activation induces SCs proliferation, whereas migration induced by glutamate are also NMDAR-dependent [268,269,270]. AMPAR activation on PNS myelinated axons induced an increase of axoplasmic Ca2+, although myelin abnormalities are only observed after prolonged activation of NMDA, but not AMPAR [271]. AMPAR activation has also be found to control ATP release after glutamate stimulation in SCs culture [258].
5.3. Satellite Glia
Among satellite Glia cells (SGCs), those of the sensory ganglia has been the most studied. [25]. SGCs are in close contact with neurons, ensheathing them and playing a role in neurotransmission and glutamate homeostasis [25,259,260,272]. To that end, SGCs express several glutamate transporters, metabotropic glutamate receptors, NMDAR, KR and AMPAR [259,260]. Regarding AMPAR, SGCs express mainly GluA4 and in lesser quantities GluA2/3 but no GluA1 [259,273]. AMPAR activation induces a rapid Ca2+ influx in SGCs inline with their expression of GluA4 [259]. In addition, a role for SGC NMDAR has been described in pathological situations like hyperalgesia [274]. It is therefore tempting to hypothesize that AMPAR could also play a role in SGCs response after damage.
5.4. Enteric Glia
Enteric glia are non-myelinating glial cells of the enteric nervous system, localized within the wall of intestines they are critical in the control of gut motility [275,276]. Despite showing some similarities to astrocytes, enteric glia display a unique transcription profile [275,276]. The connection between enteric neurons and enteric glia is not well characterized, although ionotropic glutamate receptors have been found in both cell types [261,275]. Concerning AMPAR only GluA1 and GluA3 subunits have been found in enteric glia, but the Ca2+ permeability of the functional AMPAR has not been fully elucidated yet [261]. AMPAR are involved in excitotoxic damage to enteric neurons in studies performed in myenteric ganglia [277,278]. Although it is unclear whether these pathological actions involve AMPAR located on enteric glia, their expression of GluA proteins suggests they may also be targets during ischemic injury in these enteric tissues.
6. Cannabinoids and AMPA Receptor
Several studies have pointed out that cannabinoids and the Endocannabinoid System (ECS) might modulate glutamatergic transmission, including AMPAR. This modulation has been principally studied in neurons, although glial receptor involvement has also been described [279,280,281].
The medical use of Cannabis sativa has been explored for millennia worldwide [282]. Modern research in the cannabinoids field started during the last century with the isolation of Δ9- tetrahydrocannabinol or THC [283], a discovery that eventually lead to the recognition of the ECS. The ECS is an endogenous system that plays important physiological roles, modulating neuronal synapses, the immune response or energy and metabolism regulation, among others [284,285,286,287,288].
As has been recently reviewed, the ECS modulates OL lineage cells and myelination [289]. Different elements of the ECS are expressed in all OL stages, and observations that the ECS is modulated during OL maturation, suggest that they may play a role in OL differentiation [290,291]. The endocannabinoid 2-arachidonoylglycerol (2-AG) and cannabinoid receptors CB1 and CB2 activation promote OPC proliferation, migration and maturation, and oligodendrocyte myelination [290,291,292,293,294,295]. Indeed, different pharmacological studies have proved that the reduction of 2-AG levels, or blocking either CB1 or CB2 reduces myelination, both in vitro and in vivo [291,296].
Based on the links between the ECS and OL lineage functions described above, the modulation of the ECS has been studied as a possible treatment for demyelinating diseases like MS [289]. A cannabinoid derived drug, Sativex, is approved to treat MS spasticity and pain [297,298], and in different animal models of MS, cannabinoid treatment based on cannabidiol solely, or cannabidiol plus THC (Sativex), reduces the myelin injury, inflammation and functional impairment [299,300,301]. The modulation of 2-AG levels effectively reduces motor symptoms, along with inflammation and demyelination [302,303]. Some of this protective effect is mediated by cytosolic Ca2+ modulation. Either the inhibition of 2-AG degrading enzyme, or the direct administration of 2-AG preventing intracellular Ca2+ increase via AMPARs, and the subsequent pattern of mitochondrial dysfunction and ROS increase [302]. Similar results are obtained in cultured OL when CB1 receptor agonist are administered after AMPAR activation, or K+- induced depolarization, with both producing a reduction in intracellular Ca2+ [302,304] (Figure 1B). These processes might involve the activation of Gi/0 proteins, the blockade of Kir or voltage-gated Ca2+ channels (VGCCs) [304,305]. The administration of the phytocannabinoid cannabidiol (CBD), a promising therapeutic molecule, also modulates intracellular Ca2+ via mechanisms involving mitochondria, and can, under specific conditions, induce OL cell death [306]. In spite of the controversy concerning the deleterious effect of CBD on OL cell cultures, some authors have identified a dose-dependent response [306,307]. Furthermore, CBD has been reported to be oligoprotective after an inflammatory damage [307], and to reduce excitotoxicity in several pathologies such as stroke or neonatal hypoxia-ischemia [58,308,309,310]. Results of our group have proved this dual effect. While the exposure of mouse organotypic cerebellar slices to CBD resulted in a reduction of the OL population; CBD applied before and during an excitotoxic insult was able to reduce OL cell death (Ceprian M, Ng J, Fulton D, unpublished). Promising oligoprotective findings from CBD treatments suggest that more research in this area are warranted. An important area for this work relates to the mechanism of CBD action on glia, which at this time remains unclear.
After an excitotoxic insult, there is an increase of endocannabinoids and endocannabinoids-like molecules, probably released by neurons [308,311]. Interestingly, the administration of the endocannabinoids 2-AG or anandamide (AEA) or synthetic cannabinoid agonist, HU-210, reduces cell death after AMPA administration in a mixed culture of astrocytes and neurons [221,311]. HU-210 neuroprotective mechanism are also seen in vivo and requires astrocytes and CB1 and CB2 activation [221]. AEA also exerts its neuroprotection via CB1 and CB2 receptors, and by preventing the AMPAR-induced downregulation of astrocyte glutamate transporters GLAST and GLT-1, as evidenced in both primary cultures of astrocytes, and an in vivo model of MS [311] (Figure 1B). In agreement with these results, the agonism of both cannabinoid receptors by WIN55,212-2 increases GLAST and GLT-1 expression in the spinal cord of EAE induced animals [312], while another study in the EAE model has found a decrease of AEA and 2-AG, which due to the actions on GLAST and GLT-1 described above, would be expected contribute to glutamate excitotoxicity [313] (Figure 1B).
Finally, the administration of the phytocannabinoids THC and CBD modulate the expression of neuronal GluA2-AMPAR in rodent models of drug abuse [314,315]. Further research to analyze if a similar AMPAR subunit modulation occurs after cannabinoid administration in excitotoxic models could help to elucidate cannabinoid’s protective action on glial cells.
7. AMPAR-Stimulated Gene Expression in Glial Cells: Contributions to Injury and Disease?
Activity-dependent receptor-mediated gene expression, particularly immediate early genes (IEGs), regulates key functions in the CNS including development and synaptic plasticity [316,317]. Gene expression in glial cells can also regulated by neuronal activity, for example in astrocytes where neuronally derived notch signals regulates a broad range of genes leading to alterations in astrocyte development, metabolism and neurotransmitter uptake functions [318]. As described in Section 2.1 astrocytes express functional AMPAR that allow them to respond to glutamate released from neuronal synapses. Given the broad range of astrocyte genes regulated by neuronal activity [318] it is tempting to speculate on the potential of AMPAR to contribute to these actions. For example, AMPAR activation on Bergmann glial cells leads to a downregulation in transcription of the glutamate transporter GLAST via a mechanism involving Ca2+ influx, PKC signaling and the activation of the IEG c-jun [99]. GLAST downregulation was observed with prolonged exposures to glutamate. Triggering of this regulatory response during periods of elevated extracellular glutamate, as occurs in pathological conditions such as MS [121], stroke and hypoxic-ischemia [4], could therefore lead to an amplification of the pathophysiological levels of glutamate (Figure 2).
AMPAR have also been shown to regulate IEG in cortical cultures of OPC [44] where stimulation of AMPAR induced a Ca2+-dependent upregulation in ngfi-a and other IEGs (Figure 1A). Ngfi-a (also known as zif-268, egr-1 and krox-20) is implicated in regulation of the cell cycle [319], and is induced dramatically in the brain in a depolarisation-dependent manner [320]. Thus Ngfi-a activation by AMPAR may contribute to the regulation of OPC proliferation observed following modulation of neuronal activity and AMPAR [41,148,210]. AMPAR stimulation inhibits OPC proliferation [210], thus pathological elevations in glutamate concentrations could act to reduce the supply of remyelinating OPC. As discussed in Section 3.3, excessive AMPAR activation injures OL lineage cells via Ca2+-dependent mechanisms involving mitochondrial stress and the induction of pro-apoptotic Bcl-2 molecules [65,169,321]. Apoptosis is closely regulated by the induction of pro- and anti-apoptotic Bcl-2 genes [322,323], so the pathways connecting pathophysiological AMPAR stimulation may provide an interesting range of targets for therapeutic research. Related to this idea, we recently examined the transcriptional events induced in OPC following pathophysiological AMPAR stimulation [45]. To focus our work we searched for potential regulators of Gria4, the gene encoding GluA4, via an in silico analysis. Gria4 was an attractive target since it has prominent expression in OPC, and has been linked to excitotoxic injury in other cell types [208]. From among the set of candidate regulators we selected NF-Y subunit b (NF-Yb), a member of the NF-Y complex whose activity is closely associated with the regulation of apoptototic cell death [88,324]. Experiments using an OPC cell line revealed that excitotoxic AMPAR stimulation altered NF-Yb expression, modulated its binding to regulatory regions within Gria4, and altered the expression of Gria4 transcripts. Thus NF-Yb is regulated by pathophysiological stimulation of AMPAR leading to alterations in the expression of target genes. Further experiments using primary OPC showed that excitotoxic stimulation produced a parallel increase in the expression of NF-Yb and its target gene Gria4. Interestingly GluA4 protein is upregulated in OL in an in vivo hypoxia model [237], thus it is tempting to speculate on the involvement of NF-Yb in these actions. NF-Yb is linked to the control of apoptosis [88,324] thus we performed a transcriptomic analysis to identify genes that were differentially regulated by NF-Yb modulation and excitotoxicity. For this analysis we compared the effects of a treatment with Garcinol, a compound that both blocked NF-Yb binding to Gria4 and reduced cellular viability, with an excitotoxic AMPAR stimulation proven to injure OPC. Both treatments induced transcriptional regulation in the same set of apoptotic genes underscoring the link between NF-Y function, excitotoxic injury, and apoptosis in OPC (Figure 1B). As described in this review, AMPAR are ubiquitous in neurons and glial cells so the clinical use of systemic AMPAR blockade is likely to produce numerous side effects that may complicate the evaluation of therapeutic outcomes. Cell-specific targeting could reduce these problems, but the technologies available are not translatable, and in any case, AMPAR influence numerous physiological functions in glial cells whose modulation may worsen disease conditions. Consequently, molecular targets downstream of pathological AMPAR activation, for example those contained within the NF-Y transcriptome, represent an attractive proposition for research aiming to provide protection against pathological conditions involving AMPAR-mediated injury.
8. Summary
AMPAR are expressed widely by glial cells throughout the nervous system. Their activation modulates numerous cellular actions in glia including ion channel function, gene expression, migration, various aspects of growth and differentiation, and even the expression and subunit composition of AMPAR themselves. Additionally, at the tissue level glial AMPAR influence homeostatic functions and shape neuronal function by regulating myelin production and modulating synaptic function. Numerous pathological conditions, including several notable neurological and neurodegenerative conditions, involve dysregulation of extracellular glutamate levels. These conditions can lead to excessive AMPAR stimulation triggering injury responses within glia and releasing additional excitotoxic signals in the form of glutamate and cytokines. Glia are a key feature of the nervous system whose normal activities maintain an environment that is optimal for healthy development and neuronal circuit activity, thus the identification of therapeutic approaches capable of protecting glial functions and viability under excitotoxic conditions are a critical target for future research.
Acknowledgments
M.C. was a predoctoral fellow supported by the Ayudas para la formación de profesorado universitario (FPU) program (Ministerio de Ciencia, Innovación y Universidades, Madrid, Spain). DNA vector graphic in Figure 1 PixLoger from Pixabay.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Butt, A.M.; Fern, R.F.; Matute, C. Neurotransmitter signaling in white matter. Glia 2014, 62, 1762–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediators Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Fern, R.F.; Matute, C.; Stys, P.K. White matter injury: Ischemic and nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef]
- Levite, M. Glutamate, T cells and multiple sclerosis. J. Neural Transm. 2017, 124, 775–798. [Google Scholar] [CrossRef]
- Yamane, T. Mouse Yolk Sac Hematopoiesis. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Nedergaard, M. Functions of Astrocytes and their Potential As Therapeutic Targets. Neurotherapeutics 2010, 7, 338–353. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D. A new mechanism of nervous system plasticity: Activity-dependent myelination. Nat. Rev. Neurosci. 2015, 16, 756–767. [Google Scholar] [CrossRef] [PubMed]
- de Faria, O.; Pama, E.A.C.; Evans, K.; Luzhynskaya, A.; Káradóttir, R.T. Neuroglial interactions underpinning myelin plasticity. Dev. Neurobiol. 2018, 78, 93–107. [Google Scholar] [CrossRef]
- Richardson, W.D.; Young, K.M.; Tripathi, R.B.; McKenzie, I. NG2-glia as Multipotent Neural Stem Cells: Fact or Fantasy? Neuron 2011, 70, 661–673. [Google Scholar] [CrossRef]
- Nishiyama, A.; Boshans, L.; Goncalves, C.M.; Wegrzyn, J.; Patel, K.D. Lineage, fate, and fate potential of NG2-glia. Brain Res. 2015, 1638, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Wigley, R.; Hamilton, N.; Nishiyama, A.; Kirchhoff, F.; Butt, A.M. Morphological and physiological interactions of NG2-glia with astrocytes and neurons. Proc. J. Anat. 2007, 210, 661–670. [Google Scholar] [Green Version]
- Hill, R.A.; Nishiyama, A. NG2 cells (polydendrocytes): Listeners to the neural network with diverse properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spassky, N. Adult Ependymal Cells Are Postmitotic and Are Derived from Radial Glial Cells during Embryogenesis. J. Neurosci. 2005, 25, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Götz, M.; Barde, Y.-A. Radial Glial Cells. Neuron 2005, 46, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Parnavelas, J.G.; Nadarajah, B. Radial glial cells: Are they really glia? Neuron 2001, 31, 881–884. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Choroid plexus in the central nervous system: Biology and physiopathology. J. Neuropathol. Exp. Neurol. 2000, 59, 561–574. [Google Scholar] [CrossRef]
- Bolborea, M.; Dale, N. Hypothalamic tanycytes: Potential roles in the control of feeding and energy balance. Trends Neurosci. 2013, 36, 91–100. [Google Scholar] [CrossRef]
- Kaur, C.; Rathnasamy, G.; Ling, E.A. Biology of microglia in the developing brain. J. Neuropathol. Exp. Neurol. 2017, 76, 736–753. [Google Scholar] [CrossRef]
- Kidd, G.J.; Ohno, N.; Trapp, B.D. Biology of Schwann cells. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 55–79. ISBN 9780444529022. [Google Scholar]
- Kim, J.; Lee, H.; Park, H. Two faces of Schwann cell dedifferentiation in peripheral neurodegenerative diseases: Pro-demyelinating and axon-preservative functions. Neural Regen. Res. 2014, 9, 1952. [Google Scholar] [CrossRef]
- Hanani, M. Satellite glial cells in sympathetic and parasympathetic ganglia: In search of function. Brain Res. Rev. 2010, 64, 304–327. [Google Scholar] [CrossRef]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res. 2015, 1621, 5–16. [Google Scholar] [CrossRef]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Kirchhoff, F. NMDA receptors in glia. Neuroscientist 2007, 13, 28–37. [Google Scholar] [CrossRef]
- Dzamba, D.; Honsa, P.; Anderova, M. NMDA Receptors in Glial Cells: Pending Questions. Curr. Neuropharmacol. 2013, 11, 250–262. [Google Scholar] [CrossRef]
- Swanson, G.T.; Kamboj, S.K.; Cull-Candy, S.G. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J. Neurosci. 1997, 17, 58–69. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Huganir, R.L. The Cell Biology of Synaptic Plasticity: AMPA Receptor Trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613–643. [Google Scholar] [CrossRef]
- Geiger, J.R.P.; Melcher, T.; Koh, D.S.; Sakmann, B.; Seeburg, P.H.; Jonas, P.; Monyer, H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 1995, 15, 193–204. [Google Scholar] [CrossRef]
- Hollmann, M.; Hartley, M.; Heinemann, S. Ca2+ permeability of KA-AMPA - gated glutamate receptor channels depends on subunit composition. Science 1991, 252, 851–853. [Google Scholar] [CrossRef]
- Palacios, J.M.; Pazos, A.; Tomiyama, M.; Cortés, R.; Mengod, G.; Rodríguez-Puertas, R. Flip and flop splice variants of AMPA receptor subunits in the spinal cord of amyotrophic lateral sclerosis. Synapse 2002, 45, 245–249. [Google Scholar] [Green Version]
- Haering, S.C.; Tapken, D.; Pahl, S.; Hollmann, M. Auxiliary subunits: Shepherding AMPA receptors to the plasma membrane. Membranes 2014, 4, 469–490. [Google Scholar] [CrossRef]
- Bowie, D. Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J. Biol. Chem. 2018, 293, 18789–18802. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Sasaki, T.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Harlow, D.E.; Saul, K.E.; Komuro, H.; Macklin, W.B. Myelin Proteolipid Protein Complexes with v Integrin and AMPA Receptors In Vivo and Regulates AMPA-Dependent Oligodendrocyte Progenitor Cell Migration through the Modulation of Cell-Surface GluR2 Expression. J. Neurosci. 2015, 35, 12018–12032. [Google Scholar] [CrossRef]
- Iino, M.; Goto, K.; Kakegawa, W.; Okado, H.; Sudo, M.; Ishiuchi, S.; Miwa, A.; Takayasu, Y.; Saito, I.; Tsuzuki, K.; et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 2001, 292, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fannon, J.; Tarmier, W.; Fulton, D. Neuronal activity and AMPA-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia 2015, 63, 1021–1035. [Google Scholar] [CrossRef]
- Gallo, V.; Zhou, J.; McBain, C.; Wright, P.; Knutson, P.; Armstrong, R. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 1998, 125, 2901–2914. [Google Scholar]
- Pende, M.; Holtzclaw, L.A.; Curtis, J.L.; Russell, J.T.; Gallo, V. Glutamate regulates intracellular calcium and gene expression in oligodendrocyte progenitors through the activation of DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 3215–3219. [Google Scholar] [CrossRef]
- Begum, G.; Otsu, M.; Ahmed, U.; Ahmed, Z.; Stevens, A.; Fulton, D. NF-Y-dependent regulation of glutamate receptor 4 expression and cell survival in cells of the oligodendrocyte lineage. Glia 2018, 66, 1896–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kougioumtzidou, E.; Shimizu, T.; Hamilton, N.B.; Tohyama, K.; Sprengel, R.; Monyer, H.; Attwell, D.; Richardson, W.D. Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. Elife 2017, 6, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.; Kettenmann, H. Blockade of K+ channels induced by AMPA/kainate receptor activation in mouse oligodendrocyte precursor cells is mediated by NA+ entry. J. Neurosci. Res. 1995, 42, 579–593. [Google Scholar] [CrossRef]
- Robert, A.; Magistretti, P.J. AMPA/kainate receptor activation blocks K+ currents via internal Na+ increase in mouse cultured stellate astrocytes. Glia 1997, 20, 38–50. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, N.; Guo, D.-Z.; Pan, S.-Y.; Li, H.; Yang, C. High Plasma Glutamate Levels are Associated with Poor Functional Outcome in Acute Ischemic Stroke. Cell. Mol. Neurobiol. 2014, 35, 159–165. [Google Scholar] [CrossRef]
- Castellanos, M.; Sobrino, T.; Pedraza, S.; Moldes, O.; Pumar, J.M.; Silva, Y.; Serena, J.; Garcia-Gil, M.; Castillo, J.; Davalos, A. High plasma glutamate concentrations are associated with infarct growth in acute ischemic stroke. Neurology 2008, 71, 1862–1868. [Google Scholar] [CrossRef]
- Martínez-Sánchez, P.; Gutiérrez-Fernández, M.; Fuentes, B.; Masjuán, J.; de Leciñana Cases, M.A.; Novillo-López, M.E.; Díez-Tejedor, E. Biochemical and inflammatory biomarkers in ischemic stroke: translational study between humans and two experimental rat models. J. Transl. Med. 2014, 12, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Zhang, T.; Liren, C. Excitatory amino acids in cerebrospinal fluid of patients with acute head injuries. Clin. Chem. 2001, 47, 1458–1462. [Google Scholar]
- Kierans, A.S.; Kirov, I.I.; Gonen, O.; Haemer, G.; Nisenbaum, E.; Babb, J.S.; Grossman, R.I.; Lui, Y.W. Myoinositol and glutamate complex neurometabolite abnormality after mild traumatic brain injury. Neurology 2014, 82, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, M.; Jander, S. T-Cell Cytokines in Injury-Induced Neural Damage Michael Schroeter and Sebastian Jander *. NeuroMolecular Med. 2005, 7, 183–195. [Google Scholar] [CrossRef]
- Pazos, M.R.; Cinquina, V.; Gómez, A.; Layunta, R.; Santos, M.; Fernández-Ruiz, J.; Martínez-Orgado, J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. [Google Scholar] [CrossRef]
- Dang, Y.X.; Shi, K.N.; Wang, X.M. Early changes in glutamate metabolism and perfusion in basal ganglia following hypoxia-ischemia in neonatal piglets: A multi-sequence 3.0t MR study. Front. Physiol. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Ceprián, M.; Jimenez-Sanchez, L.; Vargas, C.; Barata, L.; Hind, W.; Martínez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 2017, 116, 151–159. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural Transm. 2014, 121, 849–859. [Google Scholar] [CrossRef]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [Green Version]
- Al Gawwam, G.; Sharquie, I.K. Serum Glutamate Is a Predictor for the Diagnosis of Multiple Sclerosis. Sci. World J. 2017, 2017. [Google Scholar] [CrossRef]
- Foran, E.; Trotti, D. Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2009, 11, 1587–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.; Patel, V. Hypoxic ischaemic encephalopathy. Paediatr. Child Heal. (United Kingdom) 2014, 24, 385–389. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Marino, A.; Matute, C. Ca2+ influx through AMPA or kainate receptors alone is sufficient to initiate excitotoxicity in cultured oligodendrocytes. Neurobiol. Dis. 2002, 9, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, S.; Wang, P.; Wang, W. Transient mitochondrial permeability transition mediates excitotoxicity in glutamate-sensitive NSC34D motor neuron-like cells. Exp. Neurol. 2015, 271, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Randall, R.D.; Thayer, S.A. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 1992, 12, 1882–1895. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.; Baburamani, A.A.; Kichev, A.; Hagberg, H. Oxidative stress and endoplasmic reticulum (ER) stress in the development of neonatal hypoxic–ischaemic brain injury. Biochem. Soc. Trans. 2017, 45, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentice, H.; Modi, J.P.; Wu, J.-Y. Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Estrada-Sánchez, A.M.; Montiel, T.; Páramo, B.; Massieu, L.; Morán, J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef]
- Blanco, S.; Hernández, R.; Franchelli, G.; Ramos-Álvarez, M.M.; Peinado, M.Á. Melatonin influences NO/NOS pathway and reduces oxidative and nitrosative stress in a model of hypoxic-ischemic brain damage. Nitric Oxide - Biol. Chem. 2017, 62, 32–43. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef]
- Andreyev, A.; Tamrakar, P.; Rosenthal, R.E.; Fiskum, G. Calcium uptake and cytochrome c release from normal and ischemic brain mitochondria. Neurochem. Int. 2018, 117, 15–22. [Google Scholar] [CrossRef]
- Pottorf, W.J., II; Johanns, T.M.; Derrington, S.M.; Strehler, E.E.; Eneyde, A.; Thayer, S. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J. Neurochem. 2006, 98, 1646–1656. [Google Scholar] [CrossRef]
- Thornton, C.; Jones, A.; Nair, S.; Aabdien, A.; Mallard, C.; Hagberg, H. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. 2017, 1–19. [Google Scholar] [CrossRef]
- Brostrom, M.A.; Brostrom, C.O. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: Implications for cell growth and adaptability. Cell Calcium 2003, 34, 345–363. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic reticulum stress and oxidative stress: A vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernández, H.; Fernández-Torrón, R.; López de Munain, A.; Vallejo-Illarramendi, A. Calpain 3 deficiency affects SERCA expression and function in the skeletal muscle. Expert Rev. Mol. Med. 2016, 18, e7. [Google Scholar] [CrossRef]
- French, J.P.; Quindry, J.C.; Falk, D.J.; Staib, J.L.; Lee, Y.; Wang, K.K.W.; Powers, S.K. Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am. J. Physiol. Circ. Physiol. 2005, 290, H128–H136. [Google Scholar] [CrossRef]
- Pedrozo, Z.; Sánchez, G.; Torrealba, N.; Valenzuela, R.; Fernández, C.; Hidalgo, C.; Lavandero, S.; Donoso, P. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta - Mol. Basis Dis. 2010, 1802, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Neginskaya, M.; Berezhnaya, E.; Uzdensky, A.B.; Abramov, A.Y. Reactive oxygen species produced by a photodynamic effect induced calcium signal in neurons and astrocytes. Mol. Neurobiol. 2018, 55, 96–102. [Google Scholar] [CrossRef]
- Ruiz, A.; Matute, C.; Alberdi, E. Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium 2009, 46, 273–281. [Google Scholar] [CrossRef]
- Mei, B.; Sun, Z.-W.; Zhang, L.; Zhu, S.-J.; Chen, W.-C. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar] [Green Version]
- Martínez-Fábregas, J.; Díaz-Moreno, I.; González-Arzola, K.; Janocha, S.; Navarro, J.A.; Hervás, M.; Bernhardt, R.; Velázquez-Campoy, A.; Díaz-Quintana, A.; De la Rosa, M.A. Structural and Functional Analysis of Novel Human Cytochrome c Targets in Apoptosis. Mol. Cell. Proteomics 2014, 13, 1439–1456. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAf-1 · cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Imbriano, C.; Gnesutta, N.; Mantovani, R. The NF-Y/p53 liaison: Well beyond repression. Biochim. Biophys. Acta - Rev. Cancer 2012, 1825, 131–139. [Google Scholar] [CrossRef]
- Filous, A.R.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef]
- Mishra, A. Binaural blood flow control by astrocytes: Listening to synapses and the vasculature. J. Physiol. 2017, 595, 1885–1902. [Google Scholar] [CrossRef] [PubMed]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Nedergaard, M.; Verkhratsky, A. Artifact versus reality-How astrocytes contribute to synaptic events. Glia 2012, 60, 1013–1023. [Google Scholar] [CrossRef]
- Bradley, S.J.; Challiss, R.A.J. G protein-coupled receptor signalling in astrocytes in health and disease: A focus on metabotropic glutamate receptors. Biochem. Pharmacol. 2012, 84, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Burnashev, N.; Khodorova, A.; Jonas, P.; Helm, P.J.; Wisden, W.; Monyer, H.; Seeburg, P.H.; Sakmann, B. Calcium-permeable AMPA-kainate receptors in fusiform cerebellar glial cells. Science 1992, 256, 1566–1570. [Google Scholar] [CrossRef]
- Müller, T.; Möller, T.; Berger, T.; Schnitzer, J.; Kettenmann, H. Calcium entry through kainate receptors and resulting potassium-channel blockade in Bergmann glial cells. Science 1992, 256, 1563–1566. [Google Scholar] [CrossRef]
- Saab, A.S.; Neumeyer, A.; Jahn, H.M.; Cupido, A.; Le Meur, K.; Deitmer, J.W.; Monyer, H.; Boele, H.-J.; Scheller, A.; Gotz, M.; et al. Bergmann Glial AMPA Receptors Are Required for Fine Motor Coordination. Science 2012, 337, 749–753. [Google Scholar] [CrossRef]
- López-Bayghen, E.; Espinoza-Rojo, M.; Ortega, A. Glutamate down-regulates GLAST expression through AMPA receptors in Bergmann glial cells. Mol. Brain Res. 2003, 115, 1–9. [Google Scholar] [CrossRef]
- Mölders, A.; Koch, A.; Menke, R.; Klöcker, N. Heterogeneity of the astrocytic AMPA-receptor transcriptome. Glia 2018, 66, 2604–2616. [Google Scholar] [CrossRef]
- David, J.C.; Yamada, K.A.; Bagwe, M.R.; Goldberg, M.P. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J. Neurosci. 1996, 16, 200–209. [Google Scholar] [CrossRef]
- Matthias, K.; Kirchhoff, F.; Seifert, G.; Hüttmann, K.; Matyash, M.; Kettenmann, H.; Steinhäuser, C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J. Neurosci. 2003, 23, 1750–1758. [Google Scholar] [CrossRef]
- Droste, D.; Seifert, G.; Seddar, L.; Jädtke, O.; Steinhaüser, C.; Lohr, C. Ca2+-permeable AMPA receptors in mouse olfactory bulb astrocytes. Sci. Rep. 2017, 7, 44817. [Google Scholar] [CrossRef]
- Höft, S.; Griemsmann, S.; Seifert, G.; Steinhäuser, C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: Insights from the thalamus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Lalo, U. NMDA Receptors Mediate Neuron-to-Glia Signaling in Mouse Cortical Astrocytes. J. Neurosci. 2006, 26, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Stys, P.K. Mechanisms of Ionotropic Glutamate Receptor-Mediated Excitotoxicity in Isolated Spinal Cord White Matter. J. Neurosci. 2000, 20, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Colquhoun, K.; Tutton, M.; Berry, M. Three-dimensional morphology of astrocytes and oligodendrocytes in the intact mouse optic nerve. J. Neurocytol. 1994, 23, 469–485. [Google Scholar] [CrossRef]
- Serwanski, D.R.; Jukkola, P.; Nishiyama, A. Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J. Comp. Neurol. 2017, 525, 535–552. [Google Scholar] [CrossRef]
- Micu, I.; Plemel, J.R.; Lachance, C.; Proft, J.; Jansen, A.J.; Cummins, K.; van Minnen, J.; Stys, P.K. The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 2016, 276, 41–50. [Google Scholar] [CrossRef]
- Rosenbluth, J. Multiple functions of the paranodal junction of myelinated nerve fibers. J. Neurosci. Res. 2009, 87, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Savtchouk, I.; Carriero, G.; Volterra, A. Studying Axon-Astrocyte Functional Interactions by 3D Two-Photon Ca2+ Imaging: A Practical Guide to Experiments and “Big Data” Analysis. Front. Cell. Neurosci. 2018, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.G.; Lyons, D.A. On Myelinated Axon Plasticity and Neuronal Circuit Formation and Function. J. Neurosci. 2017, 37, 10023–10034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosche, J.; Kettenmann, H.; Verkhratsky, A.; Matyash, V.; Möller, T.; Reichenbach, A. Microdomains for neuron–glia interaction: Parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2002, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J. Neurosci. 1984, 4, 1884–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.J.; Nagarajah, R.; Banati, R.B.; Bennett, M.R. Glutamate induces directed chemotaxis of microglia. Eur. J. Neurosci. 2009, 29, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Salińska, E.; Danysz, W.; Łazarewicz, J.W. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005, 43, 322–339. [Google Scholar] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Kanwar, J.R.; Kanwar, R.K.; Krissansen, G.W. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain 2004, 127, 1313–1331. [Google Scholar] [CrossRef] [Green Version]
- Groom, A.J.; Smith, T.; Turski, L. Multiple sclerosis and glutamate. Proc. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar]
- Kaito, M.; Ren, E.; Voskuhl, R.R.; Johnsonbaugh, H.; Tassoni, A.; Itoh, N.; Burda, J.; Ao, Y.; Farkhondeh, V.; Sofroniew, M.V.; et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 2017, 115, E302–E309. [Google Scholar] [Green Version]
- Lee, F.H.F.; Jia, Z.; Zhang, S.; Wong, A.H.C.; D’Souza, C.; Zhang, L.; Su, P.; Zhai, D.; Liu, F. Blocking GluR2-GAPDH ameliorates experimental autoimmune encephalomyelitis. Ann. Clin. Transl. Neurol. 2015, 2, 388–400. [Google Scholar]
- Wang, M.; Li, S.; Zhang, H.; Pei, L.; Zou, S.; Lee, F.J.S.; Wang, Y.T.; Liu, F. Direct interaction between GluR2 and GAPDH regulates AMPAR-mediated excitotoxicity. Mol. Brain 2012, 5, 13. [Google Scholar] [CrossRef]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 2014, 7, 20. [Google Scholar] [CrossRef]
- Lee, F.H.F.; Zhang, H.; Jiang, A.; Zai, C.C.; Liu, F. Specific Alterations in Astrocyte Properties via the GluA2-GAPDH Complex Associated with Multiple Sclerosis. Sci. Rep. 2018, 8, 12856. [Google Scholar] [CrossRef]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. Pío del Río Hortega and the discovery of the oligodendrocytes. Front. Neuroanat. 2015, 9, 7–12. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Winkler, C.C.; Yabut, O.R.; Fregoso, S.P.; Gomez, H.G.; Dwyer, B.E.; Pleasure, S.J.; Franco, S.J. The Dorsal Wave of Neocortical Oligodendrogenesis Begins Embryonically and Requires Multiple Sources of Sonic Hedgehog. J. Neurosci. 2018, 38, 5237–5250. [Google Scholar] [CrossRef]
- Hashimoto, R.; Sakai, K.; Masuyama, N.; Akiyama, H.; Inoue, Y.U.; Hoshino, M.; Inoue, T.; Hayase, Y.; Kawaguchi, Y.; Koizumi, S.; et al. Origins of oligodendrocytes in the cerebellum, whose development is controlled by the transcription factor, Sox9. Mech. Dev. 2016, 140, 25–40. [Google Scholar] [CrossRef]
- Fogarty, M.; Richardson, W.D.; Kessaris, N. A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development 2005, 132, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Pringle, N.P.; Yu, W.P.; Guthrie, S.; Roelink, H.; Lumsden, A.; Peterson, A.C.; Richardson, W.D. Determination of neuroepithelial cell fate: Induction of the oligodendrocyte lineage by ventral midline cells and Sonic hedgehog. Dev. Biol. 1996, 177, 30–42. [Google Scholar] [CrossRef]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta - Mol. Cell Res. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Zusso, M.; Marinelli, C.; Giusti, P.; Bertalot, T. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Front. Cell. Neurosci. 2016, 10, 1–19. [Google Scholar]
- Anna Cunningham, L.; Newville, J.; Jantzie, L.L. Embracing oligodendrocyte diversity in the context of perinatal injury. Neural Regen Res 2017, 12, 1575–1585. [Google Scholar] [CrossRef]
- Roy, N.S.; Wang, S.; Harrison-Restelli, C.; Benraiss, A.; Fraser, R.A.R.; Gravel, M.; Braun, P.E.; Goldman, S.A. Identification, Isolation, and Promoter-Defined Separation of Mitotic Oligodendrocyte Progenitor Cells from the Adult Human Subcortical White Matter. J. Neurosci. 1999, 19, 9986–9995. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Nogawa, S.; Suzuki, S.; Dembo, T.; Kosakai, A. Upregulation of oligodendrocyte progenitor cells associated with restoration of mature oligodendrocytes and myelination in peri-infarct area in the rat brain. Brain Res. 2003, 989, 172–179. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-Activated ERK Signaling Mediated FAK-Paxillin Interaction in Oligodendrocyte Progenitor Cell Migration. J. Mol. Neurosci. 2019, 1, 10. [Google Scholar] [CrossRef]
- Biname, F.; Sakry, D.; Dimou, L.; Jolivel, V.; Trotter, J. NG2 Regulates Directional Migration of Oligodendrocyte Precursor Cells via Rho GTPases and Polarity Complex Proteins. J. Neurosci. 2013, 33, 10858–10874. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, N.; Vayro, S.; Wigley, R.; Butt, A.M. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia 2010, 58, 66–79. [Google Scholar] [CrossRef]
- Larson, V.A.; Zhang, Y.; Bergles, D.E. Electrophysiological properties of NG2+cells: Matching physiological studies with gene expression profiles. Brain Res. 2016, 1638, 138–160. [Google Scholar] [CrossRef]
- Gard, A.L.; Williams, C., II; Burrell, M.R. Oligodendroblasts Distinguished from O-2A Glial Progenitors by Surface Phenotype (O4+GalC-) and Response to Cytokines Using Signal Transducer LIFRB. Dev. Biol. 1995, 167, 596–608. [Google Scholar] [CrossRef]
- Niu, J.; Mei, F.; Wang, L.; Tian, Y.; Mo, W.; Li, H.; Lu, Q.R.; Xiao, L. Phosphorylated Olig1 Localizes to the Cytosol of Oligodendrocytes and Promotes Membrane Expansion and Maturation. Glia 2012, 60, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Rivkin, M.J.; Flax, J.; Mozell, R.; Osathanondh, R.; Volpe, J.J.; Villa-Komaroff, L. Oligodendroglial development in human fetal cerebrum. Ann. Neurol. 2005, 38, 92–101. [Google Scholar] [CrossRef]
- Liu, S.; Qu, Y.; Stewart, T.J.; Howard, M.J.; Chakrabortty, S.; Holekamp, T.F.; McDonald, J.W. Embryonic stem cells differentiate into oligodendrocytes and myelinate in culture and after spinal cord transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 6126–6131. [Google Scholar] [CrossRef] [Green Version]
- Ziemka-Nalecz, M.; Janowska, J.; Strojek, L.; Jaworska, J. Impact of neonatal hypoxia-ischaemia on oligodendrocyte survival, maturation and myelinating potential. J. Cell. Mol. Med. 2018, 22, 207–222. [Google Scholar] [CrossRef]
- Stiefel, K.M.; Torben-Nielsen, B.; Coggan, J.S. Proposed evolutionary changes in the role of myelin. Front. Neurosci. 2013, 7, 1–9. [Google Scholar] [CrossRef]
- Gautier, H.O.B.; Franklin, R.J.M.; Reynolds, R.; Lao-Peregrin, C.; Lundgaard, I.; Evans, K.A.; Sitnikov, S.; James, R.; Volbracht, K.; James, F.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef] [Green Version]
- Drobyshevsky, A.; Jiang, R.; Lin, L.; Derrick, M.; Luo, K.; Back, S.A.; Tan, S. Unmyelinated axon loss with postnatal hypertonia after fetal hypoxia. Ann. Neurol. 2014, 75, 533–541. [Google Scholar] [CrossRef]
- Mitew, S.; Lulu, Y.; Merson, T.D. Axonal activity-dependent myelination in development: Insights for myelin repair. J. Chem. Neuroanat. 2016, 76, 2–8. [Google Scholar] [CrossRef]
- Stedehouder, J.; Brizee, D.; Shpak, G.; Kushner, S.A. Activity-Dependent Myelination of Parvalbumin Interneurons Mediated by Axonal Morphological Plasticity. J. Neurosci. 2018, 38, 3631–3642. [Google Scholar] [CrossRef]
- Nave, K.-A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Lee, K.Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Hossain, S.; Liu, H.N.; Fragoso, G.; Almazan, G. Agonist-induced down-regulation of AMPA receptors in oligodendrocyte progenitors. Neuropharmacology 2014, 79, 506–514. [Google Scholar] [CrossRef]
- Itoh, T.; Beesley, J.; Itoh, A.; Cohen, A.S.; Kavanaugh, B.; Coulter, D.A.; Grinspan, J.B.; Pleasure, D. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J. Neurochem. 2002, 81, 390–402. [Google Scholar] [CrossRef] [Green Version]
- Gallo, V.; Patneau, D.K.; Mayer, M.L.; Vaccarino, F.M. Excitatory amino acid receptors in glial progenitor cells: Molecular and functional properties. Glia 1994, 11, 94–101. [Google Scholar] [CrossRef]
- Seifert, G.; Rehn, L.; Weber, M.; Steinhäuser, C. AMPA receptor subunits expressed by single astrocytes in the juvenile mouse hippocampus. Mol. Brain Res. 1997, 47, 286–294. [Google Scholar] [CrossRef]
- Wright, P.; Chew, L.-J.; Fleck, M.W.; Gallo, V.; Scherer, S.E.; Mayer, M.L. Growth Factor-Induced Transcription of GluR1 Increases Functional AMPA Receptor Density in Glial Progenitor Cells. J. Neurosci. 2018, 17, 227–240. [Google Scholar]
- Bergles, D.E.; Roberts, J.D.B.; Somogyl, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef]
- Zhou, W.; Jan, Y.N.; Luo, Q.; Ge, W.-P.; Jan, L.Y. Dividing glial cells maintain differentiated properties including complex morphology and functional synapses. Proc. Natl. Acad. Sci. USA 2008, 106, 328–333. [Google Scholar] [Green Version]
- Haberlandt, C.; Derouiche, A.; Wyczynski, A.; Haseleu, J.; Pohle, J.; Karram, K.; Trotter, J.; Seifert, G.; Frotscher, M.; Steinhäuser, C.; et al. Gray Matter NG2 Cells Display Multiple Ca2+-Signaling Pathways and Highly Motile Processes. PLoS ONE 2011, 6, e17575. [Google Scholar] [CrossRef]
- Seifert, G.; Zhou, M.; Steinhäuser, C. Analysis of AMPA Receptor Properties During Postnatal Development of Mouse Hippocampal Astrocytes. J. Neurophysiol. 1997, 78, 2916–2923. [Google Scholar] [CrossRef]
- Yoshioka, A.; Bacskai, B.; Pleasure, D. Pathophysiology of oligodendroglial excitotoxicity. J. Neurosci. Res. 1996, 46, 427–437. [Google Scholar] [CrossRef]
- Zhang, Y.; Bennett, M.L.; Maniatis, T.; Wu, J.Q.; Deng, S.; O’Keeffe, S.; Guarnieri, P.; Scholze, A.R.; Chen, K.; Zhang, C.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.G.; Fern, R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Renzel, R.; Sadek, A.R.; Chang, C.H.; Gray, W.P.; Seifert, G.; Steinhäuser, C. Polarized distribution of AMPA, but not GABAA, receptors in radial glia-like cells of the adult dentate gyrus. Glia 2013, 61, 1146–1154. [Google Scholar] [CrossRef]
- Christensen, P.C.; Samadi-Bahrami, Z.; Pavlov, V.; Stys, P.K.; Moore, G.R.W. Ionotropic glutamate receptor expression in human white matter. Neurosci. Lett. 2016, 630, 1–8. [Google Scholar] [CrossRef]
- Talos, D.M.; Fishman, R.E.; Park, H.; Folkerth, R.D.; Follett, P.L.; Volpe, J.J.; Jensen, F.E. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J. Comp. Neurol. 2006, 497, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Ness, J.K.; Scaduto, R.C.; Wood, T.L. IGF-I Prevents Glutamate-Mediated Bax Translocation and Cytochrome C Release in O4+ Oligodendrocyte Progenitors. Glia 2004, 46, 183–194. [Google Scholar] [CrossRef]
- Ness, J.K.; Wood, T.L. Insulin-like growth factor I, but not neurotrophin-3, sustains Akt activation and provides long-term protection of immature oligodendrocytes from glutamate-mediated apoptosis. Mol. Cell. Neurosci. 2002, 20, 476–488. [Google Scholar] [CrossRef]
- Deng, W.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 6801–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follett, P.L.; Deng, W.; Dai, W.; Talos, D.M.; Massillon, L.J.; Rosenberg, P.A.; Volpe, J.J.; Jensen, J.B. Glutamate Receptor-Mediated Oligodendrocyte Toxicity in Periventricular Leukomalacia: A Protective Role for Topiramate. J. Neurosci. 2004, 24, 4412–4420. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Neve, R.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. α-Amino-3-hydroxy-5-methyl-4-isoxazole Propionate Receptor Subunit Composition and cAMP-response Element-binding Protein Regulate Oligodendrocyte Excitotoxicity. J. Biol. Chem. 2006, 281, 36004–36011. [Google Scholar] [CrossRef] [Green Version]
- Jabs, R.; Kirchhoff, F.; Kettenmann, H.; Steinhäuser, C. Kainate activates Ca2+-permeable glutamate receptors and blocks voltage-gated K+ currents in glial cells of mouse hippocampal slices. Pflügers Arch. Eur. J. Physiol. 1994, 426, 310–319. [Google Scholar] [CrossRef]
- Ge, W.P.; Yang, X.J.; Zhang, Z.; Wang, H.K.; Shen, W.; Deng, Q.D.; Duan, S. Long-term potentiation of neuron-glia synapses mediated by Ca 2+-permeable AMPA receptors. Science 2006, 312, 1533–1537. [Google Scholar] [CrossRef]
- Sahel, A.; Ortiz, F.C.; Kerninon, C.; Maldonado, P.P.; Angulo, M.C.; Nait-Oumesmar, B. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell. Neurosci. 2015, 9, 1–12. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Sánchez-Gómez, M.V.; Pérez-Samartín, A.; Rodríguez-Antigüedad, A.; Pérez-Cerdá, F. Excitotoxic damage to white matter. Proc. J. Anat. 2007, 210, 693–702. [Google Scholar] [Green Version]
- Follett, P.L.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 2000, 20, 9235–9241. [Google Scholar] [CrossRef]
- Rosenberg, L.J.; Teng, Y.D.; Wrathall, J.R. 2,3-Dihydroxy-6-Nitro-7-Sulfamoyl-Benzo(f)Quinoxaline Reduces Glial Loss and Acute White Matter Pathology after Experimental Spinal Cord Contusion. J. Neurosci. 1999, 19, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861.e7. [Google Scholar] [CrossRef]
- Hines, J.H.; Ravanelli, A.M.; Schwindt, R.; Scott, E.K.; Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 2015, 18, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Etxeberria, A.; Mei, F.; Ullian, E.M.; Dao, D.Q.; Redmond, S.A.; Mayoral, S.R.; Hokanson, K.C.; Chan, J.R. Dynamic Modulation of Myelination in Response to Visual Stimuli Alters Optic Nerve Conduction Velocity. J. Neurosci. 2016, 36, 6937–6948. [Google Scholar] [CrossRef] [Green Version]
- Mensch, S.; Baraban, M.; Almeida, R.; Czopka, T.; Ausborn, J.; El Manira, A.; Lyons, D.A. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 2015, 18, 628–630. [Google Scholar] [CrossRef] [Green Version]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- Lundgaard, I.; Luzhynskaya, A.; Stockley, J.H.; Wang, Z.; Evans, K.A.; Swire, M.; Volbracht, K.; Gautier, H.O.B.; Franklin, R.J.M.; Ffrench-Constant, C.; et al. Neuregulin and BDNF Induce a Switch to NMDA Receptor-Dependent Myelination by Oligodendrocytes. PLoS Biol. 2013, 11, e1001743. [Google Scholar] [CrossRef]
- De Biase, L.M.; Pucak, M.L.; Mishina, M.; Kang, S.H.; Calabresi, P.A.; Baxi, E.G.; Fukaya, M.; Bergles, D.E. NMDA Receptor Signaling in Oligodendrocyte Progenitors Is Not Required for Oligodendrogenesis and Myelination. J. Neurosci. 2011, 31, 12650–12662. [Google Scholar] [CrossRef]
- Kukley, M.; Capetillo-Zarate, E.; Dietrich, D. Vesicular glutamate release from axons in white matter. Nat. Neurosci. 2007, 10, 311–320. [Google Scholar] [CrossRef]
- Ziskin, J.L.; Nishiyama, A.; Rubio, M.; Fukaya, M.; Bergles, D.E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 2007, 10, 321–330. [Google Scholar] [CrossRef] [Green Version]
- De Biase, L.M.; Nishiyama, A.; Bergles, D.E. Excitability and Synaptic Communication within the Oligodendrocyte Lineage. J. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef] [Green Version]
- Gallo, V.; Mangin, J.M.; Kukley, M.; Dietrich, D. Synapses on NG2-expressing progenitors in the brain: Multiple functions? J. Physiol. 2008, 586, 3767–3781. [Google Scholar] [CrossRef]
- Kukley, M.; Nishiyama, A.; Dietrich, D. The Fate of Synaptic Input to NG2 Glial Cells: Neurons Specifically Downregulate Transmitter Release onto Differentiating Oligodendroglial Cells. J. Neurosci. 2010, 30, 8320–8331. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Hamilton, N.; Hubbard, P.; Pugh, M.; Ibrahim, M. Synantocytes: The fifth element. J. Anat. 2005, 207, 695–706. [Google Scholar] [CrossRef]
- Sakry, D.; Neitz, A.; Singh, J.; Frischknecht, R.; Marongiu, D.; Binamé, F.; Perera, S.S.; Endres, K.; Lutz, B.; Radyushkin, K.; et al. Oligodendrocyte Precursor Cells Modulate the Neuronal Network by Activity-Dependent Ectodomain Cleavage of Glial NG2. PLoS Biol. 2014, 12, e1001993. [Google Scholar] [CrossRef]
- Passlick, S.; Trotter, J.; Seifert, G.; Steinhäuser, C.; Jabs, R. The NG2 Protein Is Not Required for Glutamatergic Neuron-NG2 Cell Synaptic Signaling. Cereb. Cortex 2016, 26, 51–57. [Google Scholar] [CrossRef]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–57. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat. Med. 1998, 4, 291–297. [Google Scholar] [CrossRef]
- Zonouzi, M.; Renzi, M.; Farrant, M.; Cull-Candy, S.G. Bidirectional plasticity of calcium-permeable AMPA receptors in oligodendrocyte lineage cells. Nat. Neurosci. 2011, 14, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A.; Rosenberg, P.A. Pathophysiology of Glia in Perinatal White Matter Injury. Glia 2014, 62, 1790–1815. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- Volpe, J.J. Confusions in Nomenclature: “Periventricular Leukomalacia” and “White Matter Injury”—Identical, Distinct, or Overlapping? Pediatr. Neurol. 2017, 73, 3–6. [Google Scholar] [CrossRef]
- Back, S.A.; Riddle, A.; McClure, M.M. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 2007, 38, 724–730. [Google Scholar] [CrossRef]
- Buser, J.R.; Segovia, K.N.; Dean, J.M.; Nelson, K.; Beardsley, D.; Gong, X.; Luo, N.L.; Ren, J.; Wan, Y.; Riddle, A.; et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1053–1065. [Google Scholar] [CrossRef] [Green Version]
- Riddle, A.; Luo, N.L.; Manese, M.; Beardsley, D.; Green, L.; Rorvik, D.A.; Kelly, K.A.; Barlow, C.H.; Kelly, J.J.; Hohimer, A.R.; et al. Spatial Heterogeneity in Oligodendrocyte Lineage Maturation and Not Cerebral Blood Flow Predicts Fetal Ovine Periventricular White Matter Injury. J. Neurosci. 2006, 26, 3045–3055. [Google Scholar] [CrossRef] [Green Version]
- Segovia, K.N.; Mcclure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested Oligodendrocyte Lineage Maturation in Chronic Perinatal White Matter Injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J. Neuropathol. Exp. Neurol. 2002, 61, 197–211. [Google Scholar] [CrossRef]
- Talos, D.M.; Follett, P.L.; Folkerth, R.D.; Fishman, R.E.; Trachtenberg, F.L.; Volpe, J.J.; Jensen, F.E. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J. Comp. Neurol. 2006, 497, 61–77. [Google Scholar] [CrossRef]
- Page, K.J.; Everitt, B.J. The Distribution of Neurons Coexpressing Immunoreactivity to AMPA-sensitive Glutamate Receptor Subtypes (GluR1-4) and Nerve Growth Factor Receptor in the Rat Basal Forebrain. Eur. J. Neurosci. 1995, 7, 1022–1033. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef]
- Su, Z.; Yuan, Y.; Chen, J.; Zhu, Y.; Qiu, Y.; Zhu, F.; Huang, A.; He, C. Reactive astrocytes inhibit the survival and differentiation of oligodendrocyte precursor cells by secreted TNF-α. J. Neurotrauma 2011, 28, 1089–1100. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Ling, E.; Kaur, C. Iron and Iron Regulatory Proteins in Amoeboid Microglial Cells Are Linked to Oligodendrocyte Death in Hypoxic Neonatal Rat Periventricular White Matter through Production of Proinflammatory Cytokines and Reactive Oxygen/Nitrogen Species. J. Neurosci. 2011, 31, 17982–17995. [Google Scholar] [CrossRef] [Green Version]
- Villapol, S.; Fau, S.; Renolleau, S.; Biran, V.; Charriaut-Marlangue, C.; Baud, O. Melatonin promotes myelination by decreasing white matter inflammation after neonatal stroke. Pediatr. Res. 2011, 69, 51–55. [Google Scholar] [CrossRef]
- Wada, T.; Sawano, T.; Tanaka, T.; Furuyama, T.; Fukumoto, M.; Yamaguchi, W.; Saino, O.; Takeda, Y.; Kogo, M.; Matsuyama, T.; et al. Absence of Sema4D improves oligodendrocyte recovery after cerebral ischemia/reperfusion injury in mice. Neurosci. Res. 2016, 108, 6–11. [Google Scholar] [CrossRef]
- Bonfanti, E.; Gelosa, P.; Fumagalli, M.; Dimou, L.; Viganò, F.; Tremoli, E.; Cimino, M.; Sironi, L.; Abbracchio, M.P. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 2017, 8, e2871. [Google Scholar] [CrossRef] [Green Version]
- Rebai, O.; Amri, M. Chlorogenic Acid Prevents AMPA-Mediated Excitotoxicity in Optic Nerve Oligodendrocytes Through a PKC and Caspase-Dependent Pathways. Neurotox. Res. 2018, 34, 559–573. [Google Scholar] [CrossRef]
- McCracken, E.; Fowler, J.H.; Dewar, D.; Morrison, S.; McCulloch, J. Grey matter and white matter ischemic damage is reduced by the competitive AMPA receptor antagonist, SPD 502. J. Cereb. Blood Flow Metab. 2002, 22, 1090–1097. [Google Scholar] [CrossRef]
- Viganò, F.; Möbius, W.; Götz, M.; Dimou, L. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci. 2013, 16, 1370–1372. [Google Scholar] [CrossRef]
- Lentferink, D.H.; Jongsma, J.M.; Werkman, I.; Baron, W. Grey matter OPCs are less mature and less sensitive to IFNγ than white matter OPCs: Consequences for remyelination. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Newcombe, J.; Uddin, A.; Dove, R.; Patel, B.; Turski, L.; Nishizawa, Y.; Smith, T. Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol. 2008, 18, 52–61. [Google Scholar] [CrossRef]
- Bannerman, P.; Horiuchi, M.; Feldman, D.; Hahn, A.; Itoh, A.; See, J.; Jia, Z.P.; Itoh, T.; Pleasure, D. GluR2-free alfa-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors intensify demyelination in experimental autoimmune encephalomyelitis. J. Neurochem. 2007, 102, 1064–1070. [Google Scholar] [CrossRef]
- Docagne, F.; Muñetón, V.; Clemente, D.; Ali, C.; Loría, F.; Correa, F.; Hernangómez, M.; Mestre, L.; Vivien, D.; Guaza, C. Excitotoxicity in a chronic model of multiple sclerosis: Neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol. Cell. Neurosci. 2007, 34, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Suda, S.; Sowa, K.; Sakamoto, Y.; Nito, C.; Nishiyama, Y.; Aoki, J.; Ueda, M.; Yokobori, S.; Yamada, M.; et al. AMPA Receptor Antagonist Perampanel Ameliorates Post-Stroke Functional and Cognitive Impairments. Neuroscience 2018, 386, 256–264. [Google Scholar] [CrossRef]
- Elting, J.-W.; Diener, H.C.; Teelken, A.W.; Lees, K.R.; Sulter, G.A.; Hommel, M.; Kaste, M.; Versavel, M.; De Keyser, J. AMPA Antagonist ZK200775 in Patients With Acute Ischemic Stroke. Stroke 2002, 33, 2813–2818. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.R.; Kaste, M.; Lees, K.R.; Diener, H.C.; Hommel, M.; De Keyser, J.; Steiner, H.; Versavel, M. The AMPA antagonist ZK 200775 in patients with acute ischaemic stroke: A double-blind, multicentre, placebo-controlled safety and tolerability study. Cerebrovasc. Dis. 2005, 20, 304–309. [Google Scholar] [CrossRef]
- Gibson, E.M.; Barres, B.A.; Bieri, G.; Lin, G.L.; Woo, P.J.; Wood, L.S.; Vogel, H.; Zuchero, J.B.; Mount, C.W.; Purger, D.; et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef]
- Barres, B.A.; Hart, I.K.; Coles, H.S.R.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell 1992, 70, 31–46. [Google Scholar] [CrossRef]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of local protein synthesis and initial events in myelination by action potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Gan, W.B. Microglia dynamics and function in the CNS. Curr. Opin. Neurobiol. 2010, 20, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef]
- Miron, V.E.; Franklin, R.J.M. Macrophages and CNS remyelination. J. Neurochem. 2014, 130, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci. 2000, 20, 251. [Google Scholar] [CrossRef]
- Hagino, Y.; Kariura, Y.; Manago, Y.; Amano, T.; Wang, B.; Sekiguchi, M.; Nishikawa, K.; Aoki, S.; Wada, K.; Noda, M. Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 2004, 47, 68–77. [Google Scholar] [CrossRef]
- Christensen, R.N.; Ha, B.K.; Sun, F.; Bresnahan, J.C.; Beattie, M.S. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. J. Neurosci. Res. 2006, 84, 170–181. [Google Scholar] [CrossRef]
- Sivakumar, V.; Ling, E.A.; Lu, J.; Kaur, C. Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia 2010, 58, 507–523. [Google Scholar] [CrossRef]
- Wong, W.T.; Wang, M.; Li, W. Regulation of microglia by ionotropic glutamatergic and GABAergic neurotransmission. Neuron Glia Biol. 2012, 7, 41–46. [Google Scholar] [CrossRef]
- Gottlieb, M.; Matute, C. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 290–300. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007, 1186, 21–28. [Google Scholar] [CrossRef]
- Guan, J.; Bennet, L.; Gluckman, P.D.; Gunn, A.J. Insulin-like growth factor-1 and post-ischemic brain injury. Prog. Neurobiol. 2003, 70, 443–462. [Google Scholar] [CrossRef]
- Takeuchi, H.; Suzumura, A. Gap junctions and hemichannels composed of connexins: Potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Braithwaite, S.P.; Beattie, M.S.; Beattie, E.C.; Leonoudakis, D.D. TNFα-induced AMPA-receptor trafficking in CNS neurons; relevance to excitotoxicity? Neuron Glia Biol. 2004, 1, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Zheng, H.; Li, Z.; Wang, W.; Shao, X.; Yao, Q. Excitotoxicity of TNFα derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J. Neurochem. 2010, 114, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Mandolesi, G.; De Vito, F.; Fresegna, D.; Gentile, A.; Marfia, G.A.; Bullitta, S.; Sepman, H.; Musella, A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Centonze, D.; Ecconi, F.; Battistini, L.; Bernardi, G.; Bergamaschi, A.; Furlan, R.; Muzio, L.; Cencioni, M.T.; Cavallucci, V.; De Chiara, V.; et al. Inflammation Triggers Synaptic Alteration and Degeneration in Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G.; et al. Tumor Necrosis Factor and Interleukin-1β Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. 2018, 2018, 8430123. [Google Scholar] [CrossRef]
- Beppu, K.; Kosai, Y.; Kido, M.A.; Akimoto, N.; Mori, Y.; Kojima, Y.; Fujita, K.; Okuno, Y.; Yamakawa, Y.; Ifuku, M.; et al. Expression, subunit composition, and function of AMPA-type glutamate receptors are changed in activated microglia; possible contribution of GluA2 (GluR-B)-deficiency under pathological conditions. Glia 2013, 61, 881–891. [Google Scholar] [CrossRef]
- Carter, T.L.; Rissman, R.A.; Mishizen-Eberz, A.J.; Wolfe, B.B.; Hamilton, R.L.; Gandy, S.; Armstrong, D.M. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer’s disease patients according to Braak stage. Exp. Neurol. 2004, 187, 299–309. [Google Scholar] [CrossRef]
- Noda, M.; Beppu, K. Possible Contribution of Microglial Glutamate Receptors to Inflammatory Response upon Neurodegenerative Diseases. J. Neurol. Disord. 2013, 1. [Google Scholar] [CrossRef]
- Jansson, L.C.; Wigren, H.K.; Nordström, T.; Åkerman, K.E. Functional α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in differentiating embryonic neural progenitor cells. Neuroreport 2011, 22, 282–287. [Google Scholar] [CrossRef]
- Muth-Köhne, E.; Pachernegg, S.; Karus, M.; Faissner, A.; Hollmann, M. Expression of NMDA receptors and Ca-impermeable AMPA receptors requires neuronal differentiation and allows discrimination between two different types of neural stem cells. Cell. Physiol. Biochem. 2010, 26, 935–946. [Google Scholar] [CrossRef]
- Shtaya, A.; Sadek, A.R.; Zaben, M.; Seifert, G.; Pringle, A.; Steinhäuser, C.; Gray, W.P. AMPA receptors and seizures mediate hippocampal radial glia-like stem cell proliferation. Glia 2018, 66, 2397–2413. [Google Scholar] [CrossRef]
- Kawakami, S.I. Glial and neuronal localization of ionotropic glutamate receptor subunit-immunoreactivities in the median eminence of female rats: GluR2/3 and GluR6/7 colocalize with vimentin, not with glial fibrillary acidic protein (GFAP). Brain Res. 2000, 858, 198–204. [Google Scholar] [CrossRef]
- Demêmes, D.; Lleixa, A.; Dechesne, C.J. Cellular and subcellular localization of AMPA-selective glutamate receptors in the mammalian peripheral vestibular system. Brain Res. 1995, 671, 83–94. [Google Scholar] [CrossRef]
- Chen, T.-J.; Fröhlich, N.; Kula, B.; Barzan, R.; Kukley, M. Glutamate Activates AMPA Receptor Conductance in the Developing Schwann Cells of the Mammalian Peripheral Nerves. J. Neurosci. 2017, 37, 11818–11834. [Google Scholar] [CrossRef]
- Fink, T.; Davey, D.F.; Ansselin, A.D. Glutaminergic and adrenergic receptors expressed on adult guinea pig Schwann cells in vitro. Can. J. Physiol. Pharmacol. 1999, 77, 204–210. [Google Scholar] [CrossRef]
- Liu, G.J.; Bennett, M.R. ATP secretion from nerve trunks and Schwann cells mediated by glutamate. Neuroreport 2006, 14, 2079. [Google Scholar] [CrossRef]
- Kung, L.H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for Glutamate as a Neuroglial Transmitter within Sensory Ganglia. PLoS ONE 2013, 8, e68312. [Google Scholar] [CrossRef]
- Fernández-Montoya, J.; Avendaño, C.; Negredo, P. The glutamatergic system in primary somatosensory neurons and its involvement in sensory input-dependent plasticity. Int. J. Mol. Sci. 2018, 19, 69. [Google Scholar] [CrossRef]
- Von Boyen, G.; Steinkamp, M.; Adler, G.; Kirsch, J. Glutamate receptor subunit expression in primary enteric glia cultures. J. Recept. Signal Transduct. 2006, 26, 329–336. [Google Scholar] [CrossRef]
- Goffredo, D.; Conti, L.; Di Febo, F.; Biella, G.; Tosoni, A.; Vago, G.; Biunno, I.; Moiana, A.; Bolognini, D.; Toselli, M.; et al. Setting the conditions for efficient, robust and reproducible generation of functionally active neurons from adult subventricular zone-derived neural stem cells. Cell Death Differ. 2008, 15, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Buffo, A.; Rossi, F. Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 2013, 109, 1–22. [Google Scholar] [CrossRef]
- Ko, C.; Robitaille, R. Perisynaptic Schwann Cells at the Neuromuscular Synapse: Adaptable, Multitasking Glial Cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a020503. [Google Scholar] [CrossRef] [Green Version]
- Jansson, L.C.; Louhivuori, L.; Wigren, H.K.; Nordström, T.; Louhivuori, V.; Castrén, M.L.; Åkerman, K.E. Effect of glutamate receptor antagonists on migrating neural progenitor cells. Eur. J. Neurosci. 2013, 37, 1369–1382. [Google Scholar] [CrossRef]
- Herrera, A.A.; Qiang, H.; Ko, C.P. The role of perisynaptic Schwann cells in development of neuromuscular junctions in the frog (Xenopus laevis). J. Neurobiol. 2000, 45, 237–254. [Google Scholar] [CrossRef]
- Todd, K.J.; Darabid, H.; Robitaille, R. Perisynaptic Glia Discriminate Patterns of Motor Nerve Activity and Influence Plasticity at the Neuromuscular Junction. J. Neurosci. 2010, 30, 11870–11882. [Google Scholar] [CrossRef] [Green Version]
- Campana, W.M.; Mantuano, E.; Azmoon, P.; Henry, K.; Banki, M.A.; Kim, J.H.; Pizzo, D.P.; Gonias, S.L. Ionotropic glutamate receptors activate cell signaling in response to glutamate in Schwann cells. FASEB J. 2017, 31, 1744–1755. [Google Scholar] [CrossRef]
- Mantuano, E.; Lam, M.S.; Shibayama, M.; Campana, W.M.; Gonias, S.L. The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J. Cell Sci. 2015, 128, 3478–3488. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, F.; Wakatsuki, S.; Tokunaga, S.; Fujieda, H.; Araki, T. Glutamate signals through mGluR2 to control Schwann cell differentiation and proliferation. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Christensen, P.C.; Welch, N.C.; Brideau, C.; Stys, P.K. Functional ionotropic glutamate receptors on peripheral axons and myelin. Muscle&Nerve 2016, 54, 451–459. [Google Scholar]
- Rebillard, G.; Ruel, J.; Nouvian, R.; Saleh, H.; Pujol, R.; Dehnes, Y.; Raymond, J.; Puel, J.L.; Devau, G. Glutamate transporters in the guinea-pig cochlea: Partial mRNA sequences, cellular expression and functional implications. Eur. J. Neurosci. 2003, 17, 83–92. [Google Scholar] [CrossRef]
- Tachibana, M.; Wenthold, R.J.; Morioka, H.; Petralia, R.S. Light and electron microscopic immunocytochemical localization of AMPA-selective glutamate receptors in the rat spinal cord. J. Comp. Neurol. 1994, 344, 431–454. [Google Scholar] [CrossRef]
- Ferrari, L.F.; Lotufo, C.M.; Araldi, D.; Rodrigues, M.A.; Macedo, L.P.; Ferreira, S.H.; Parada, C.A. Inflammatory sensitization of nociceptors depends on activation of NMDA receptors in DRG satellite cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18363–18368. [Google Scholar] [CrossRef] [Green Version]
- Grubišić, V.; Gulbransen, B.D. Enteric glia: The most alimentary of all glia. J. Physiol. 2017, 595, 557–570. [Google Scholar] [CrossRef]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express Proteolipid Protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef]
- Carpanese, E.; Moretto, P.; Filpa, V.; Marchet, S.; Moro, E.; Crema, F.; Frigo, G.; Giaroni, C. Antagonism of ionotropic glutamate receptors attenuates chemical ischemia-induced injury in rat primary cultured myenteric ganglia. PLoS ONE 2014, 9, 1–17. [Google Scholar] [CrossRef]
- Kirchgessner, A.L.; Liu, M.-T.; Alcantara, F. Excitotoxicity in the Enteric Nervous System. J. Neurosci. 1997, 17, 8804–8816. [Google Scholar] [CrossRef] [PubMed]
- Shouman, B.; Fontaine, R.H.; Baud, O.; Schwendimann, L.; Keller, M.; Spedding, M.; Lelièvre, V.; Gressens, P. Endocannabinoids potently protect the newborn brain against AMPA-kainate receptor-mediated excitotoxic damage. Br. J. Pharmacol. 2006, 148, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Islas, C.; Garcia-Bereguiain, M.A.; Wenner, P. Tonic and Transient Endocannabinoid Regulation of AMPAergic Miniature Postsynaptic Currents and Homeostatic Plasticity in Embryonic Motor Networks. J. Neurosci. 2012, 32, 13597–13607. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, M.; McGuire, P.; Pertwee, R.G.; Bhattacharyya, S. Effect of cannabis on glutamate signalling in the brain: A systematic review of human and animal evidence. Neurosci. Biobehav. Rev. 2016, 64, 359–381. [Google Scholar] [CrossRef] [Green Version]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [Green Version]
- Gaoni, Y.; Mechoulam, R. Isolation, structure and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Nature Publishing Group: London, UK, 2018; Volume 43, ISBN 4149558493. [Google Scholar]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-gómez, E.; Matias, I.; Delamarre, A.; Metna-laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Shosaku, T.; Kano, M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol. 2014, 29, 1–898. [Google Scholar] [CrossRef]
- Augustin, S.M.; Lovinger, D.M. Functional Relevance of Endocannabinoid-Dependent Synaptic Plasticity in the Central Nervous System. ACS Chem. Neurosci. 2018, 9, 2146–2161. [Google Scholar] [CrossRef]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Tanimura, A.; Kita, Y.; Sano, Y.; Shimizu, T.; Di Marzo, V.; Kano, M. Acute inhibition of diacylglycerol lipase blocks endocannabinoid-mediated retrograde signalling: Evidence for on-demand biosynthesis of 2-arachidonoylglycerol. J. Physiol. 2013, 591, 4765–4776. [Google Scholar] [CrossRef]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-holgado, F.; Borrel, J.; Guaza, C. Cannabinoids Promote Oligodendrocyte Progenitor Survival: Involvement of Cannabinoid Receptors and Phosphatidylinositol-3 Kinase/Akt Signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef] [Green Version]
- Gomez, O.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Ortega-Gutierrez, S.; Cisneros, J.A.; Almazan, G.; Sánnchez-Rodriguez, M.A.; Molina-Holgado, F.; Molina-Holgado, E. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010, 58, 1913–1927. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, M.A.; Ortega-Gutierrez, S.; Vazquez-Villa, H.; Guaza, C.; Molina-Holgado, F.; Molina-Holgado, E. A Basal Tone of 2-Arachidonoylglycerol Contributes to Early Oligodendrocyte Progenitor Proliferation by Activating Phosphatidylinositol 3-Kinase (PI3K)/AKT and the Mammalian Target of Rapamycin (MTOR) Pathways. J. Neuroimmune Pharmacol. 2015, 10, 309–317. [Google Scholar] [CrossRef]
- Tomas-Roig, J.; Wirths, O.; Salinas-Riester, G.; Havemann-Reinecke, U. The Cannabinoid CB1/CB2 Agonist WIN55212.2 Promotes Oligodendrocyte Differentiation In Vitro and Neuroprotection During the Cuprizone-Induced Central Nervous System Demyelination. CNS Neurosci. Ther. 2016, 22, 387–395. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, A.; Le, M.Q.U.; Sanchez-Caro, C.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 2011, 163, 1520–1532. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Rodriguez, M.A.; Gomez, O.; Esteban, P.F.; Garcia-Ovejero, D.; Molina-Holgado, E. The endocannabinoid 2-arachidonoylglycerol regulates oligodendrocyte progenitor cell migration. Biochem. Pharmacol. 2018, 157, 180–188. [Google Scholar] [CrossRef]
- Arevalo-Martin, A.; Garcia-Ovejero, D.; Rubio-Araiz, A.; Gomez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 2007, 26, 1548–1559. [Google Scholar] [CrossRef] [Green Version]
- Giacoppo, S.; Bramanti, P.; Mazzon, E. Sativex in the management of multiple sclerosis-related spasticity: An overview of the last decade of clinical evaluation. Mult. Scler. Relat. Disord. 2017, 17, 22–31. [Google Scholar] [CrossRef]
- Russo, M.; Naro, A.; Leo, A.; Sessa, E.; D’Aleo, G.; Bramanti, P.; Calabrò, R.S. Evaluating sativex® in neuropathic pain management: A clinical and neurophysiological assessment in multiple sclerosis. Pain Med. (United States) 2016, 17, 1145–1154. [Google Scholar] [CrossRef]
- Feliú, A.; Moreno-Martet, M.; Mecha, M.; Carrillo-Salinas, F.J.; De Lago, E.; Fernández-Ruiz, J.; Guaza, C. A Sativex®-like combination of phytocannabinoids as a disease-modifying therapy in a viral model of multiple sclerosis. Br. J. Pharmacol. 2015, 172, 3579–3595. [Google Scholar] [CrossRef]
- Giacoppo, S.; Rajan, T.S.; Galuppo, M.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Purified Cannabidiol, the main non-psychotropic component of Cannabis sativa, alone, counteracts neuronal apoptosis in experimental multiple sclerosis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4906–4919. [Google Scholar]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sánchez-Gómez, M.; Pérez-Samartín, A.; Rodríguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef]
- Manterola, A.; Bernal-Chico, A.; Cipriani, R.; Canedo-Antelo, M.; Moreno-García, Á.; Martín-Fontecha, M.; Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Ortega-Gutiérrez, S.; Brown, J.M.; et al. Deregulation of the endocannabinoid system and therapeutic potential of ABHD6 blockade in the cuprizone model of demyelination. Biochem. Pharmacol. 2018, 157, 189–201. [Google Scholar] [CrossRef]
- Mato, S.; Alberdi, E.; Ledent, C.; Watanabe, M.; Matute, C. CB1 cannabinoid receptor-dependent and -independent inhibition of depolarization-induced calcium influx in oligodendrocytes. Glia 2009, 57, 295–306. [Google Scholar] [CrossRef]
- Lozovaya, N.; Min, R.; Tsintsadze, V.; Burnashev, N. Dual modulation of CNS voltage-gated calcium channels by cannabinoids: Focus on CB1 receptor-independent effects. Cell Calcium 2009, 46, 154–162. [Google Scholar] [CrossRef]
- Mato, S.; Victoria Sánchez-Gómez, M.; Matute, C. Cannabidiol induces intracellular calcium elevation and cytotoxicity in oligodendrocytes. Glia 2010, 58, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Torrao, A.S.; Mestre, L.; Carrillo-Salinas, F.J.; Mechoulam, R.; Guaza, C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012, 3, e331. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT1A and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Khaksar, S.; Bigdeli, M.R. Anti-excitotoxic effects of cannabidiol are partly mediated by enhancement of NCX2 and NCX3 expression in animal model of cerebral ischemia. Eur. J. Pharmacol. 2016, 794, 270–279. [Google Scholar] [CrossRef]
- El-Remessy, A.B.; Khalil, I.E.; Matragoon, S.; Abou-Mohamed, G.; Tsai, N.J.; Roon, P.; Caldwell, R.B.; Caldwell, R.W.; Green, K.; Liou, G.I. Neuroprotective Effect of(-)Δ9-Tetrahydrocannabinol and Cannabidiol in N-Methyl-D-Aspartate-Induced Retinal Neurotoxicity: Involvement of Peroxynitrite. Am. J. Pathol. 2003, 163, 1997–2008. [Google Scholar] [CrossRef]
- Loría, F.; Petrosino, S.; Hernangómez, M.; Mestre, L.; Spagnolo, A.; Correa, F.; Di Marzo, V.; Docagne, F.; Guaza, C. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol. Dis. 2010, 37, 166–176. [Google Scholar] [CrossRef] [Green Version]
- De Lago, E.; Moreno-Martet, M.; Cabranes, A.; Ramos, J.A.; Fernández-Ruiz, J. Cannabinoids ameliorate disease progression in a model of multiple sclerosis in mice, acting preferentially through CB1 receptor-mediated anti-inflammatory effects. Neuropharmacology 2012, 62, 2299–2308. [Google Scholar] [CrossRef]
- Cabranes, A.; Venderova, K.; De Lago, E.; Fezza, F.; Sánchez, A.; Mestre, L.; Valenti, M.; García-Merino, A.; Ramos, J.A.; Di Marzo, V.; et al. Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis. Neurobiol. Dis. 2005, 20, 207–217. [Google Scholar] [CrossRef]
- Good, C.H.; Lupica, C.R. Afferent-specific AMPA receptor subunit composition and regulation of synaptic plasticity in midbrain dopamine neurons by abused drugs. J. Neurosci. 2010, 30, 7900–7909. [Google Scholar] [CrossRef] [Green Version]
- Luján, M.Á.; Castro-Zavala, A.; Alegre-Zurano, L.; Valverde, O. Repeated Cannabidiol treatment reduces cocaine intake and modulates neural proliferation and CB1R expression in the mouse hippocampus. Neuropharmacology 2018, 143, 163–175. [Google Scholar] [CrossRef]
- Pérez-Cadahía, B.; Drobic, B.; Davie, J.R. Activation and function of immediate-early genes in the nervous system. Biochem. Cell Biol. 2011, 89, 61–73. [Google Scholar] [CrossRef]
- Minatohara, K.; Akiyoshi, M.; Okuno, H. Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front. Mol. Neurosci. 2016, 8, 78. [Google Scholar] [CrossRef]
- Hasel, P.; Dando, O.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef] [Green Version]
- Molnar, G.; Crozat, A.; Pardee, A.B. The immediate-early gene Egr-1 regulates the activity of the thymidine kinase promoter at the G0-to-G1 transition of the cell cycle. Mol. Cell. Biol. 2015, 14, 5242–5248. [Google Scholar] [CrossRef]
- Sukhatme, V.P.; Cao, X.; Chang, L.C.; Tsai-Morris, C.H.; Stamenkovich, D.; Ferreira, P.C.P.; Cohen, D.R.; Edwards, S.A.; Shows, T.B.; Curran, T.; et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 1988, 53, 37–43. [Google Scholar] [CrossRef]
- Sanchez-Gomez, M.V.; Perez-Navarro, E.; Alberdi, E.; Alberch, J.; Matute, C. Bax and Calpain Mediate Excitotoxic Oligodendrocyte Death Induced by Activation of Both AMPA and Kainate Receptors. J. Neurosci. 2011, 31, 2996–3006. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Cakouros, D. Transcriptional control of the core cell-death machinery. Trends Biochem. Sci. 2004, 29, 193–199. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Benatti, P.; Basile, V.; Merico, D.; Fantoni, L.I.; Tagliafico, E.; Imbriano, C. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 2008, 36, 1415–1428. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
AMPAR functions for CNS white matter glia in health and disease. (A) Illustration depicting the interaction of glial elements in CNS white matter. Astrocytes (AST) and oligodendrocyte progenitors (OPC) extend processes that make contact with unmyelinated axons and the nodes of Ranvier on myelinated axons. These glial processes contain functional AMPAR since white matter astrocytes exhibit AMPAR-mediated Ca2+ signals, and OPC display AMPAR-mediated synaptic currents. Both unmyelinated and myelinated axons release glutamate via vesicular mechanisms. Glutamate released at unmyelinated axons drives synaptic input on OPC, but it is unclear whether glutamate released at internodal sites diffuses in concentrations sufficient to activate glial AMPAR at nodes of Ranvier. The functional consequences of astrocyte AMPAR in white matter remains unknown but may include a role in the regulation of outward K+ currents. AMPAR activation influences multiple functions in OPC including migration, proliferation, differentiation and survival. In addition, AMPAR activation in OPC influences events in the nucleus including the induction of immediate early genes involved in cellular growth. Note, OPC actions depicted also occur in CNS grey matter. (B) Depiction of excitotoxic events and glial cell injury in CNS white matter. Excitotoxic and in inflammatory conditions involve an increase in extracellular glutamate levels that damage OL and myelin internodes. Glutamate is released from damaged axons and from gap-junction hemichannels on activated microglia. Excessive astrocyte AMPAR activation may aggravate excitotoxic conditions via the downregulation of astrocyte GLAST. Myelin can be restored by OPC recruited to demyelinated axons. GluA2-GAPDH complexes formed in astrocytes at inflammatory demyelinating lesions may undergo nuclear translocation leading to the initiation of disease processes. Myelin repair involves the recruitment of OPC to demyelinated axons where they establish AMPAR-mediated synaptic connections. Axon-OPC synapses may play a role in guiding OPC to target axons, and in controlling their differentiation into myelinating OL. The modulation of the Endocannabinoid System is able to prevent several of these pathogenic pathways. Either the increase of endocannabinoid tone, or the direct agonism of CB1 receptors, reduces cytosolic Ca2+ influx in the oligodendrocyte after an AMPA stimulus. Similarly, increased AEA tone prevents GLAST and GLT-1 downregulation in a mechanism that involves at least CB1 receptor, and AEA tone increase or CB1/ CB2 agonism potentiates GLAST and GLT-1 expression in mouse models of MS. Note, other cytotoxic mediators involved in inflammatory demyelination, such as cytokines and complement cascade components are not shown for clarity.
Figure 1.
AMPAR functions for CNS white matter glia in health and disease. (A) Illustration depicting the interaction of glial elements in CNS white matter. Astrocytes (AST) and oligodendrocyte progenitors (OPC) extend processes that make contact with unmyelinated axons and the nodes of Ranvier on myelinated axons. These glial processes contain functional AMPAR since white matter astrocytes exhibit AMPAR-mediated Ca2+ signals, and OPC display AMPAR-mediated synaptic currents. Both unmyelinated and myelinated axons release glutamate via vesicular mechanisms. Glutamate released at unmyelinated axons drives synaptic input on OPC, but it is unclear whether glutamate released at internodal sites diffuses in concentrations sufficient to activate glial AMPAR at nodes of Ranvier. The functional consequences of astrocyte AMPAR in white matter remains unknown but may include a role in the regulation of outward K+ currents. AMPAR activation influences multiple functions in OPC including migration, proliferation, differentiation and survival. In addition, AMPAR activation in OPC influences events in the nucleus including the induction of immediate early genes involved in cellular growth. Note, OPC actions depicted also occur in CNS grey matter. (B) Depiction of excitotoxic events and glial cell injury in CNS white matter. Excitotoxic and in inflammatory conditions involve an increase in extracellular glutamate levels that damage OL and myelin internodes. Glutamate is released from damaged axons and from gap-junction hemichannels on activated microglia. Excessive astrocyte AMPAR activation may aggravate excitotoxic conditions via the downregulation of astrocyte GLAST. Myelin can be restored by OPC recruited to demyelinated axons. GluA2-GAPDH complexes formed in astrocytes at inflammatory demyelinating lesions may undergo nuclear translocation leading to the initiation of disease processes. Myelin repair involves the recruitment of OPC to demyelinated axons where they establish AMPAR-mediated synaptic connections. Axon-OPC synapses may play a role in guiding OPC to target axons, and in controlling their differentiation into myelinating OL. The modulation of the Endocannabinoid System is able to prevent several of these pathogenic pathways. Either the increase of endocannabinoid tone, or the direct agonism of CB1 receptors, reduces cytosolic Ca2+ influx in the oligodendrocyte after an AMPA stimulus. Similarly, increased AEA tone prevents GLAST and GLT-1 downregulation in a mechanism that involves at least CB1 receptor, and AEA tone increase or CB1/ CB2 agonism potentiates GLAST and GLT-1 expression in mouse models of MS. Note, other cytotoxic mediators involved in inflammatory demyelination, such as cytokines and complement cascade components are not shown for clarity.

Figure 2.
AMPAR functions for peri-synaptic CNS glia in health and disease. (A) In the healthy CNS astrocyte (AST) and OPC/NG2-glia processes exhibit physical and functional contacts with neuronal synapses. Ca2+ permeable AMPAR on Bergmann glia sustain the physical interaction between glial processes and neuronal synapses allowing efficient clearance of glutamate from Purkinje cell synapses. Activation of astrocyte AMPAR also induces a blockade of outward K+ currents that may support neuronal function during sustained periods of neuronal activity. OPC/NG2-glia in developing and adult CNS tissues exhibit Ca2+ permeable AMPAR and receive AMPAR-mediated synaptic input that may regulate their migration, maturation and survival. In the adult CNS NG2-glia continue to receive synaptic input, the functions of which remain unclear. The role of microglial AMPAR in the healthy CNS remain unknown although in vitro data suggest a role in chemotaxis [117]. (B) Glial cell AMPAR amplify pathological conditions and mediate glial cell injury in the CNS. Stimulation of AMPAR under excitotoxic conditions induces the release of glutamate from activated microglia via gap-junction hemichannels. AMPAR activation under hypoxic conditions also stimulates the upregulation of microglial AMPAR leading to an imbalance in anti- and pro-inflammatory cytokine release characterised by enhanced release of TNF-α. TNF-α released from activated microglia may intensify excitotoxic conditions by inducing the downregulation of astrocytic GLT1. Similarly, direct stimulation of astrocyte AMPAR may worsen excitotoxic conditions by inducing the downregulation of GLAST. TNF-α also increases the vulnerability of neurons to excitotoxicity by stimulating increased surface trafficking of AMPAR. Excessive AMPAR activation induces direct injury to OPC via numerous mechanisms including oxidative and ER stress and mitochondrial dysfunction (depicted by red organelles). In addition, excitotoxic stimulation of OPC AMPAR alters the function of transcription factor complex NF-Y leading to alterations in the expression of Ca2+ permeable GluA4 subunits and the regulation of genes involved in apoptosis.
Figure 2.
AMPAR functions for peri-synaptic CNS glia in health and disease. (A) In the healthy CNS astrocyte (AST) and OPC/NG2-glia processes exhibit physical and functional contacts with neuronal synapses. Ca2+ permeable AMPAR on Bergmann glia sustain the physical interaction between glial processes and neuronal synapses allowing efficient clearance of glutamate from Purkinje cell synapses. Activation of astrocyte AMPAR also induces a blockade of outward K+ currents that may support neuronal function during sustained periods of neuronal activity. OPC/NG2-glia in developing and adult CNS tissues exhibit Ca2+ permeable AMPAR and receive AMPAR-mediated synaptic input that may regulate their migration, maturation and survival. In the adult CNS NG2-glia continue to receive synaptic input, the functions of which remain unclear. The role of microglial AMPAR in the healthy CNS remain unknown although in vitro data suggest a role in chemotaxis [117]. (B) Glial cell AMPAR amplify pathological conditions and mediate glial cell injury in the CNS. Stimulation of AMPAR under excitotoxic conditions induces the release of glutamate from activated microglia via gap-junction hemichannels. AMPAR activation under hypoxic conditions also stimulates the upregulation of microglial AMPAR leading to an imbalance in anti- and pro-inflammatory cytokine release characterised by enhanced release of TNF-α. TNF-α released from activated microglia may intensify excitotoxic conditions by inducing the downregulation of astrocytic GLT1. Similarly, direct stimulation of astrocyte AMPAR may worsen excitotoxic conditions by inducing the downregulation of GLAST. TNF-α also increases the vulnerability of neurons to excitotoxicity by stimulating increased surface trafficking of AMPAR. Excessive AMPAR activation induces direct injury to OPC via numerous mechanisms including oxidative and ER stress and mitochondrial dysfunction (depicted by red organelles). In addition, excitotoxic stimulation of OPC AMPAR alters the function of transcription factor complex NF-Y leading to alterations in the expression of Ca2+ permeable GluA4 subunits and the regulation of genes involved in apoptosis.


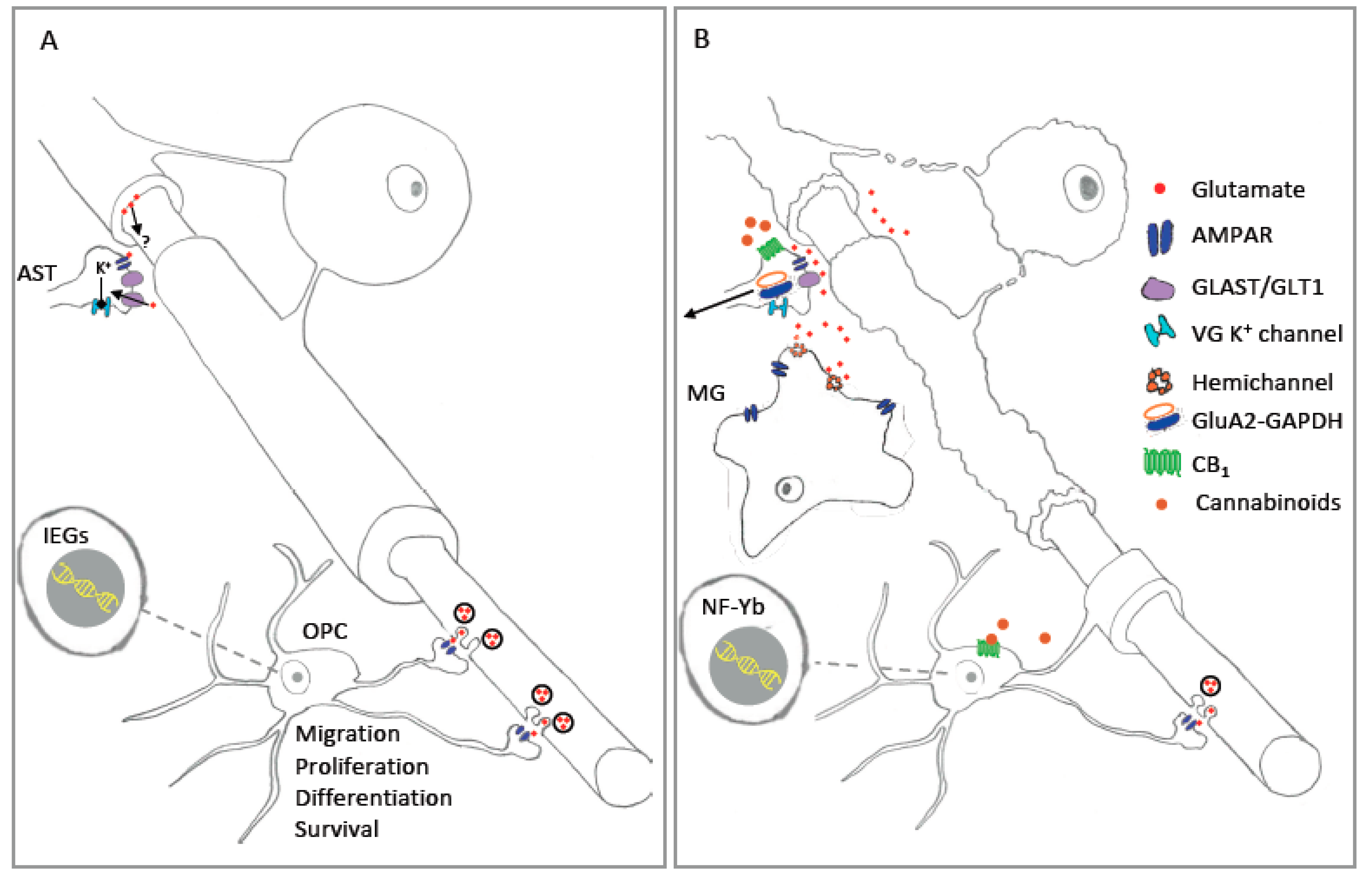{kind=link}
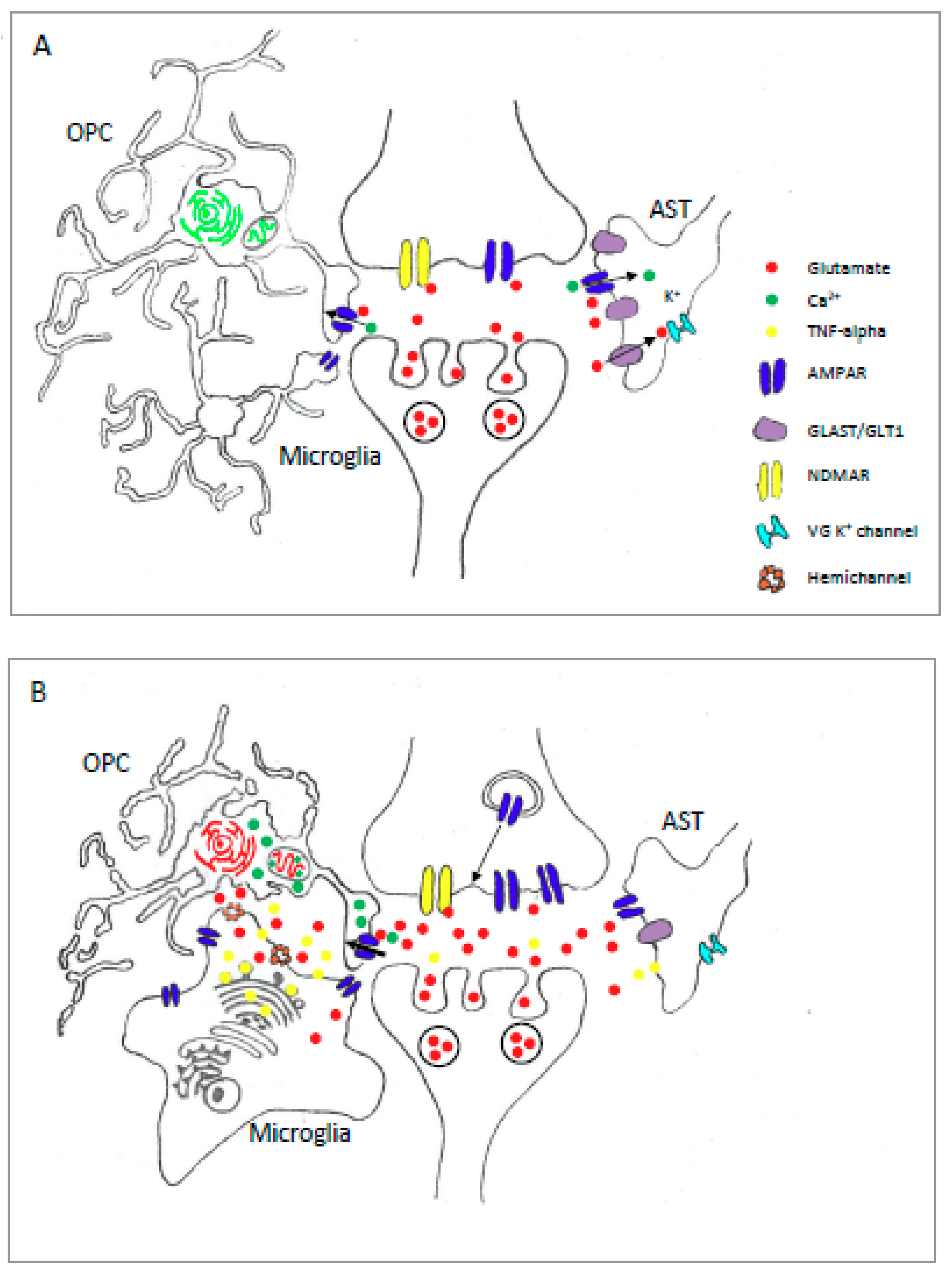{kind=link}
Table 1.
Summary of AMPAR expression and functions in astrocytes.
| Region | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| Cb | GluA1, GluA4 [96] | ++ | ePhys [96] | Synapse modulation [40] Blockade of K+ currents [97] | Altered motor control [98] GLAST downregulation [99] |
| Ctx | GluA2 dominant (qPCR) [100] | +/− | Ci [101] | Regulate K+ currents [48] | Excitotoxicity [101] |
| Hp | GluA2 dominant (qPCR) [100] No expression (ephys) [102] | No AMPA current | ePhys [102] | N.D. | N.D. |
| OB | GluA1, 2, 4 (IHC) [103] | + | ci, ePhys [103] | N.D. | N.D. |
| Th | GluA1-4 (RT-PCR) [104] | +/− | ePhys [104] | N.D. | N.D. |
+/− low Ca2+ permeability; + intermediate Ca2+ permeability; ++ high Ca2+ permeability Cb: cerebellum; Ci: calcium imaging analysis; ePhys: electrophysiological analysis; Ctx: cortex; Hp: hippocampus; IHC: immunohistochemistry; N.D.: not determined; OB: olfactory bulb; qPCR: quantitative real-time PCR; RT-PCR: reverse transcription PCR; Th: Thalamus.
Table 2.
Summary of AMPAR expression and functions in oligodendrocytes.
| Preparation | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| OPC in vitro | GluA2, 3, 4 (qPCR, WB) [154,155] | ++ | Ci [155,169] ePhys [155] | IEG expression [44] | Excitotoxicity [170] OGD injury [171,172,173] NF-Y-dependent apoptosis [45] |
| OPC ex vivo | N.D. | + | Ci, ePhys [161] | Proliferation, Differentiation [41,43] Migration [39] Blockade of K+ currents [43,174] OPC LTP [175] | OGD injury [165] |
| OPC in vivo | GluA2-4 (FISH) [46] | N.D. | N.D. | Survival [165] | H-I injury [172] Remyelination [148,176] |
| OL in vitro | GluA2, 3, 4 (qPCR, WB) [155] | + | Ci [155,177] ePhys [155] | N.D. | Excitotoxicity [65] |
| OL ex vivo | GluA4 (IHC) [106] | + | Ci [109] | Activate NMDAR [109] | Excitotoxicity [106] |
| OL in vivo | GluA2/3 (IHC) [177] GluA4 IHC [167] | N.D. | N.D. | N.D. | Demyelination [119] H-I injury [178] Spinal cord injury [179] |
+ intermediate Ca2+ permeability; ++ high Ca2+ permeability Cb: cerebellum; Ci: calcium imaging analysis; ePhys: electrophysiological analysis; IEG: immediate early gene; IHC: immunohistochemistry; H-I: hypoxic ischemia; FISH: fluorescent in situ hybridization; N.D.: not determined; OGD: oxygen glucose deprivation; qPCR: quantitative real-time PCR; WB: Western blot.
Table 3.
Summary of AMPAR expression and functions in microglia.
| Preparation | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| In vitro | GluA2,3,4 (RT-PCR) [233] GluA1,2,3 (qPCR) [234] GluA1 (ICC) [236] | - | ePhys [225,226] | Chemotaxis [117] | Hypoxia induced increase in GluA2-4 [237] Release of IL-1b and TNF-a [234,237] TNF-a release [234] |
| In vivo | GluA2/3, 4 (IHC) [237] No AMPAR (ePhys) [238] | No AMPAR | ePhys [238] | N.D. | Hypoxia/Ischemia increased GluA2-4 [237,239] |
–: negligible Ca2+ permeability; ePhys: electrophysiological analysis; ICC: immunocytochemistry; IHC: immunohistochemistry; qPCR: quantitative real-time PCR; RT-PCR: reverse transcription PCR; N.D.: not determined.
Table 4.
Summary of AMPAR expression and functions in other glia.
| Tissue Age | GluA Expression | Ca2+ Permeability | Functions | ||
|---|---|---|---|---|---|
| Degree | Evidence | Physiological | Pathophysiological | ||
| RG Developing Tissue | GluA1, 2, 3 (qPCR, ICC) [251] GluA3, 4 (qPCR) [252] GluA3,4 [252] | + | Ci [251] | N.D. | N.D. |
| RG Adult tissue | GluA2, 3 and 4 (qPCR) [166] | N.D. | N.D. | Proliferation, survival [253] | N.D. |
| Tanycte | GluA2/3 (IHC) [254] | N.D. | N.D. | N.D. | N.D. |
| Schwann cell | GluA2/3, 4 (IHC, I-EM) [255] Functional AMPAR in developing cells [256] | + | ePhys [256,257] | ATP release [258] | N.D. |
| Satellite glia | GluA4 (IHC) [259,260] | + | Ci [259,260] | N.D. | N.D. |
| Enteric glia | GluA1, 3 (qPCR, WB) [261] | N.D. | N.D. | N.D. | N.D. |
+: intermediate Ca2+ permeability; ePhys: electrophysiological analysis; ICC: immunocytochemistry; I-EM: immunogold electron microscopy; IHC: immunohistochemistry; qPCR: quantitative real-time PCR; RG: Radial Glia; N.D.: not determined; Western blot: WB.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20, 2450. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102450
AMA Style
Ceprian M, Fulton D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. International Journal of Molecular Sciences. 2019; 20(10):2450. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102450
Chicago/Turabian StyleCeprian, Maria, and Daniel Fulton. 2019. "Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease" International Journal of Molecular Sciences 20, no. 10: 2450. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102450
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.