Novel Diamide-Based Benzenesulfonamides as Selective Carbonic Anhydrase IX Inhibitors Endowed with Antitumor Activity: Synthesis, Biological Evaluation and In Silico Insights


 , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
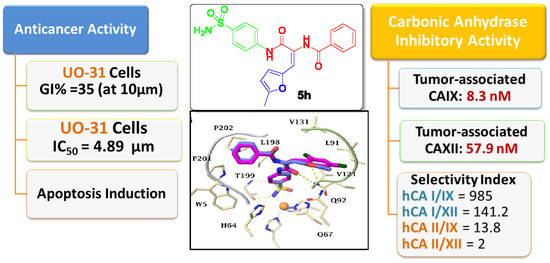
2.2. Biological Evaluation
2.2.1. Carbonic Anhydrase Inhibition
2.2.2. Antitumor Activity
Antitumor Activity towards 60 Cancer Cell Lines (NCI, USA)
Anti-Proliferative Activity towards Renal Cancer UO-31 Cell Line
Cell Cycle Analysis
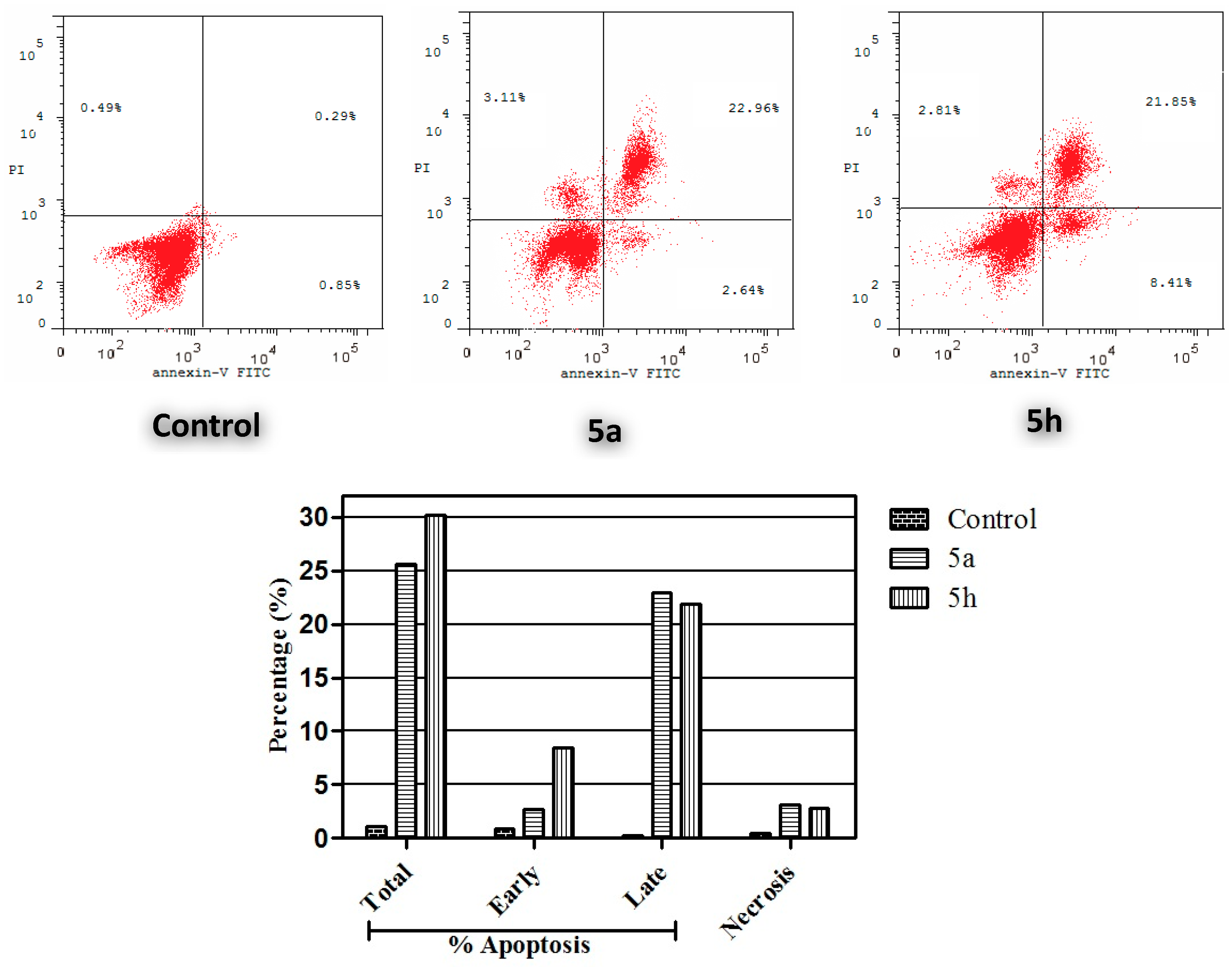
Annexin V-FITC Apoptosis Assay
2.3. Molecular Modelling Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for Preparation of Target Diamide-Based Benzenesulfonamides 5a–h
N-(1-(4-Chlorophenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5a)
N-(1-(2,4-Dichlorophenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5b)
N-(1-(4-Bromophenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5c)
N-(3-Oxo-3-((4-sulfamoylphenyl)amino)-1-(p-tolyl)prop-1-en-2-yl)benzamide (5d)
N-(1-(4-Methoxyphenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5e)
N-(1-(2,4-Dimethoxyphenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5f)
N-(1-(3,4-Dimethoxyphenyl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5g)
N-(1-(5-Methylfuran-2-yl)-3-oxo-3-((4-sulfamoylphenyl)amino)prop-1-en-2-yl)benzamide (5h)
3.2. Biological Evaluation
3.2.1. CA Inhibitory Assay
3.2.2. Anticancer Activity towards 60 Cancer Cell Lines (NCI, Bethesda, MD, USA)
3.2.3. Antiproliferative Activity towards Renal Cancer UO-31 Cell Line
3.2.4. Cell Cycle Analysis
3.2.5. Annexin V-FITC Apoptosis Assay
3.2.6. Molecular Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozensoy Guler, O.; Capasso, C.; Supuran, C.T. A magnificent enzyme superfamily: Carbonic anhydrases, their purification and characterization. J. Enzym. Inhib. Med. Chem. 2016, 31, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Vullo, D.; Durante, M.; Di Leva, F.S.; Cosconati, S.; Masini, E.; Scozzafava, A.; Novellino, E.; Supuran, C.T.; Carta, F. Monothiocarbamates Strongly Inhibit Carbonic Anhydrases in Vitro and Possess Intraocular Pressure Lowering Activity in an Animal Model of Glaucoma. J. Med. Chem. 2016, 59, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Gieling, R.G.; Babur, M.; Mamnani, L.; Burrows, N.; Telfer, B.A.; Carta, F.; Winum, J.-Y.; Scozzafava, A.; Supuran, C.T.; Williams, K.J. Antimetastatic Effect of Sulfamate Carbonic Anhydrase IX Inhibitors in Breast Carcinoma Xenografts. J. Med. Chem. 2012, 55, 5591–5600. [Google Scholar] [CrossRef]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abdel-Aziz, H.A.; Abou-Seri, S.M.; Supuran, C.T. Novel synthesized SLC-0111 thiazole and thiadiazole analogues: Determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg. Chem. 2019, 87, 794–802. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Berrino, E.; Vullo, D.; Ghabbour, H.A.; Al-Rashood, S.T.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Alharbi, A.; et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorganic Chem. 2019, 83, 549–558. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Allam, H.A.; Mahmoud, W.R.; Bonardi, A.; Nocentini, A.; Gratteri, P.; Ibrahim, E.S.; Abdel-Aziz, H.A.; Supuran, C.T. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: Selective inhibitors for the tumor-associated hCA IX isoform. Eur. J. Med. Chem. 2018, 152, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Fares, M.; Ceruso, M.; Ghabbour, H.A.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; El Ella, D.A.A.; Supuran, C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016, 110, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; El-Haggar, R.S.; Bua, S.; Bonardi, A.; Al-Rashood, S.T.; Hassan, G.S.; Gratteri, P.; Abdel-Aziz, H.A.; et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: Design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 162, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Nocentini, A.; Al-Rashood, S.T.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Reda, A.M.; Abdel-Aziz, H.A.; Supuran, C.T. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro anticancer activity towards colon cancer, Bioorg. Med. Chem. 2018, 81, 425–432. [Google Scholar]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, E.E. Ueber die Condensation der Hippursäure mit Phtalsäureanhydrid und mit Benzaldehyd. Justus Liebigs Ann. Chem. 1893, 275, 1–8. [Google Scholar] [CrossRef]
- Acheson, R.M.; Booth, D.A.; Brettle, R.; Harris, A.M. 694. The synthesis of some acylglycines and related oxazolones. J. Chem. Soc. 1960, 3457. [Google Scholar] [CrossRef] [Green Version]
- Cleary, T.; Rawalpally, T.; Kennedy, N.; Chavez, F. Catalyzing the Erlenmeyer Plöchl reaction: Organic bases versus sodium acetate. Tetrahedron Lett. 2010, 51, 1533–1536. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Boil. Chem. 1971, 246, 2561–2573. [Google Scholar]
- Skehan, P.; Scudiero, D.; Vistica, D.; Bokesch, H.; Kenney, S.; Storeng, R.; Monks, A.; McMahon, J.; Warren, J.T.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Kerdawy, A.M.; Al-Ansary, G.H.; Al-Rashood, S.T.; Ali, M.M.; Mahmoud, A.E. Type IIA-Type IIB protein tyrosine kinase inhibitors hybridization as an efficient approach for potent multikinase inhibitor development: Design, synthesis, anti-proliferative activity, multikinase inhibitory activity and molecular modeling of novel indolinone-based ureides and amides. Eur. J. Med. Chem. 2019, 163, 37–53. [Google Scholar]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Alsherbiny, M.A.; Aly, M.H.; Ghabbour, H.A.; Abdel-Aziz, H.A.; Eynde, J.J.V.; Mayence, A. Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties. Molecules 2016, 21, 762. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Nocentini, A.; Ferraroni, M.; Carta, F.; Ceruso, M.; Gratteri, P.; Lanzi, C.; Masini, E.; Supuran, C.T. Benzenesulfonamides Incorporating Flexible Triazole Moieties Are Highly Effective Carbonic Anhydrase Inhibitors: Synthesis and Kinetic, Crystallographic, Computational, and Intraocular Pressure Lowering Investigations. J. Med. Chem. 2016, 59, 10692–10704. [Google Scholar] [CrossRef]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Parveen, M.; Ali, A.; Ahmed, S.; Malla, A.M.; Alam, M.; Pereira Silva, P.S.; Silva, M.R.; Lee, D.U. Synthesis, bioassay, crystal structure and ab initio studies of Erlenmeyer azlactones, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 104, 538–545. [Google Scholar] [CrossRef]
- Conway, P.A.; Devine, K.; Paradisi, F. A simple and efficient method for the synthesis of Erlenmeyer azlactones. Tetrahedron 2009, 65, 2935–2938. [Google Scholar] [CrossRef]
- Alafeefy, A.M.; Ahmad, R.; Abdulla, M.; Eldehna, W.M.; Al-Tamimi, A.M.S.; Abdel-Aziz, H.A.; Al-Obaid, O.; Carta, F.; Al-Kahtani, A.A.; Supuran, C.T. Development of certain new 2-substituted-quinazolin-4-yl-aminobenzenesulfonamide as potential antitumor agents. Eur. J. Med. Chem. 2016, 109, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Fares, M.; Eladwy, R.A.; Nocentini, A.; El Hadi, S.R.A.; Ghabbour, H.A.; Abdel-Megeed, A.; Eldehna, W.M.; Abdel-Aziz, H.A.; Supuran, C.T. Synthesis of bulky-tailed sulfonamides incorporating pyrido [2, 3-d][1,2,4] triazolo [4, 3-a] pyrimidin-1 (5H)-yl) moieties and evaluation of their carbonic anhydrases I, II, IV and IX inhibitory effects. Bioorg. Med. Chem. 2017, 25, 2210–2217. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abou-Seri, S.M.; Supuran, C.T. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur. J. Med. Chem. 2018, 157, 28–36. [Google Scholar] [CrossRef]
- Melis, C.; Meleddu, R.; Angeli, A.; Distinto, S.; Bianco, G.; Capasso, C.; Cottiglia, F.; Angius, R.; Supuran, C.T.; Maccioni, E. Isatin: A privileged scaffold for the design of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 68–73. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Beverly, A.T. Cancer Drug Discovery and Development. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials and Approval, 2nd ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 41–62, (Chapter 1). [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Al-Warhi, T.; Altyar, A.E.; Alkahtani, H.M.; Almehizia, A.A.; Abdel-Aziz, H.A. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Abo-Ashour, M.F.; Eldehna, W.M.; George, R.F.; Abdel-Aziz, M.M.; Elaasser, M.M.; Gawad, N.M.A.; Gupta, A.; Bhakta, S.; Abou-Seri, S.M. Novel indole-thiazolidinone conjugates: Design, synthesis and whole-cell phenotypic evaluation as a novel class of antimicrobial agents. Eur. J. Med. Chem. 2018, 160, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Almahli, H.; Hadchity, E.; Jaballah, M.Y.; Daher, R.; Ghabbour, H.A.; Kabil, M.M.; Al-Shakliah, N.S.; Eldehna, W.M. Development of novel synthesized phthalazinone-based PARP-1 inhibitors with apoptosis inducing mechanism in lung cancer. Bioorg. Chem. 2018, 77, 443–456. [Google Scholar]
- Ismail, R.S.; Abou-Seri, S.M.; Eldehna, W.M.; Ismail, N.S.; Elgazwi, S.M.; Ghabbour, H.A.; Ahmed, M.S.; Halaweish, F.T.; El Ella, D.A.A. Novel series of 6-(2-substitutedacetamido)-4-anilinoquinazolines as EGFR-ERK signal transduction inhibitors in MCF-7 breast cancer cells. Eur. J. Med. Chem. 2018, 155, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Sabt, A.; Abdelhafez, O.M.; El-Haggar, R.S.; Madkour, H.M.F.; Eldehna, W.M.; El-Khrisy, E.E.-D.A.M.; Abdel-Rahman, M.A.; Rashed, L.A. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and QSAR studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; El-Naggar, D.H.; Hamed, A.R.; Ibrahim, H.S.; Ghabbour, H.A.; Abdel-Aziz, H.A.; Ghabbour, H.A. Abdel-Aziz One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDAMB-231 cells. J. Enzym. Inhib. Med. Chem. 2018, 33, 309–318. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Almahli, H.; Al-Ansary, G.H.; Ghabbour, H.A.; Aly, M.H.; Ismael, O.E.; Al-Dhfyan, A.; Abdel-Aziz, H.A. Synthesis and in vitro anti-proliferative activity of some novel isatins conjugated with quinazoline/phthalazine hydrazines against triple-negative breast cancer MDA-MB-231 cells as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2017, 32, 600–613. [Google Scholar] [CrossRef] [Green Version]
- Eldehna, W.M.; Abo-Ashour, M.F.; Ibrahim, H.S.; Al-Ansary, G.H.; Ghabbour, H.A.; Elaasser, M.M.; Ahmed, H.Y.A.; Safwat, N.A. Novel [(3-indolylmethylene)hydrazono]indolin-2-ones as apoptotic anti-proliferative agents: Design, synthesis and in vitro biological evaluation. J. Enzym. Inhib. Med. Chem. 2018, 33, 686–700. [Google Scholar] [CrossRef]
- Schrödinger Suite Release 2018-2: (a) Maestrov.11.6; (b) Epik, v.4.4; (c) Impact, v.7.9; (d) Prime, v.5.2; (e) Macromodel v.12.0. (f) Glide, v.7.9; Schrödinger, L.L.C.: New York, NY, USA, 2018.
- Nocentini, A.; Carta, F.; Tanc, M.; Selleri, S.; Supuran, C.T.; Bazzicalupi, C.; Gratteri, P. Deciphering the Mechanism of Human Carbonic Anhydrases Inhibition with Sulfocoumarins: Computational and Experimental Studies. Chemistry 2018, 24, 7840–7844. [Google Scholar] [CrossRef]
- Nocentini, A.; Gratteri, P.; Supuran, C.T. Phosphorus versus Sulfur: Discovery of Benzenephosphonamidates as Versatile Sulfonamide-Mimic Chemotypes Acting as Carbonic Anhydrase Inhibitors. Chemistry 2019, 25, 1188–1192. [Google Scholar] [CrossRef]
- Nocentini, A.; Bonardi, A.; Gratteri, P.; Cerra, B.; Gioiello, A.; Supuran, C.T. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J. Enzym. Inhib. Med. Chem. 2018, 33, 1453–1459. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R | R1 | R2 | KI (nM)* | |||
|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | ||||
| 5a | H | H | Cl | 3955.7 | 68.3 | 8.8 | 16.1 |
| 5b | Cl | H | Cl | 5977.6 | 223.9 | 18.3 | 10.5 |
| 5c | H | H | Br | 2397.8 | 251.9 | 33.5 | 55.4 |
| 5d | H | H | CH3 | 796.1 | 94.4 | 62.1 | 60.2 |
| 5e | H | H | OCH3 | 1006.4 | 127.7 | 78.0 | 42.8 |
| 5f | OCH3 | H | OCH3 | 5132.7 | 294.2 | 73.7 | 134.5 |
| 5g | H | OCH3 | OCH3 | 2207.7 | 448.0 | 123.3 | 9.8 |
| 5h | - | - | - | 8175.4 | 114.8 | 8.3 | 57.9 |
| AAZ | - | - | - | 250.0 | 12.0 | 25.0 | 5.7 |
 | |||||||
| Cmpd | I/IX | II/IX | I/XII | II/XII |
|---|---|---|---|---|
| 5a | 449.5 | 7.8 | 245.7 | 4.2 |
| 5b | 326.6 | 12.2 | 569.2 | 21.3 |
| 5c | 71.6 | 7.5 | 43.3 | 4.5 |
| 5d | 12.8 | 1.5 | 13.2 | 1.7 |
| 5e | 12.9 | 1.4 | 23.5 | 3 |
| 5f | 69.9 | 4 | 38.2 | 2.2 |
| 5g | 17.9 | 3.6 | 225.3 | 45.7 |
| 5h | 985 | 13.8 | 141.2 | 2 |
| AAZ | 10.0 | 0.5 | 43.9 | 2.2 |
| Compound | IC50 (μM) a |
|---|---|
| UO-31 | |
| 5a | 6.53 ± 0.38 |
| 5b | 16.68 ± 0.92 |
| 5h | 4.89 ± 0.22 |
| Staurosporine | 7.25 ± 0.43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelrahman, M.A.; Eldehna, W.M.; Nocentini, A.; Bua, S.; Al-Rashood, S.T.; Hassan, G.S.; Bonardi, A.; Almehizia, A.A.; Alkahtani, H.M.; Alharbi, A.; et al. Novel Diamide-Based Benzenesulfonamides as Selective Carbonic Anhydrase IX Inhibitors Endowed with Antitumor Activity: Synthesis, Biological Evaluation and In Silico Insights. Int. J. Mol. Sci. 2019, 20, 2484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102484
Abdelrahman MA, Eldehna WM, Nocentini A, Bua S, Al-Rashood ST, Hassan GS, Bonardi A, Almehizia AA, Alkahtani HM, Alharbi A, et al. Novel Diamide-Based Benzenesulfonamides as Selective Carbonic Anhydrase IX Inhibitors Endowed with Antitumor Activity: Synthesis, Biological Evaluation and In Silico Insights. International Journal of Molecular Sciences. 2019; 20(10):2484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102484
Chicago/Turabian StyleAbdelrahman, Mohamed A., Wagdy M. Eldehna, Alessio Nocentini, Silvia Bua, Sara T. Al-Rashood, Ghada S. Hassan, Alessandro Bonardi, Abdulrahman A. Almehizia, Hamad M. Alkahtani, Amal Alharbi, and et al. 2019. "Novel Diamide-Based Benzenesulfonamides as Selective Carbonic Anhydrase IX Inhibitors Endowed with Antitumor Activity: Synthesis, Biological Evaluation and In Silico Insights" International Journal of Molecular Sciences 20, no. 10: 2484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102484