Telomerase and Telomeres Biology in Thyroid Cancer


Laboratory of Translational Research, Azienda Unità Sanitaria Locale-IRCCS di Reggio Emilia, 42123 Reggio Emilia, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(12), 2887; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122887
Submission received: 30 April 2019
/
Revised: 3 June 2019
/
Accepted: 11 June 2019
/
Published: 13 June 2019
(This article belongs to the Special Issue Role of Telomeres and Telomerase in Cancer and Aging 2019)
{kind=link}
Abstract
:Telomere and telomerase regulation contributes to the onset and evolution of several tumors, including highly aggressive thyroid cancers (TCs). TCs are the most common endocrine malignancies and are generally characterized by a high rate of curability. However, a small but significant percentage develops distant metastasis or progresses into undifferentiated forms associated with bad prognosis and for which poor therapeutic options are available. Mutations in telomerase reverse transcriptase (TERT) promoter are among the most credited prognostic marker of aggressiveness in TCs. Indeed, their frequency progressively increases passing from indolent lesions to aggressive and anaplastic forms. TERT promoter mutations create binding sites for transcription factors, increasing TERT expression and telomerase activity. Furthermore, aggressiveness of TCs is associated with TERT locus amplification. These data encourage investigating telomerase regulating pathways as relevant drivers of TC development and progression to foster the identification of new therapeutics targets. Here, we summarize the current knowledge about telomere regulation and TCs, exploring both canonical and less conventional pathways. We discuss the possible role of telomere homeostasis in mediating response to cancer therapies and the possibility of using epigenetic drugs to re-evaluate the use of telomerase inhibitors. Combined treatments could be of support to currently used therapies still presenting weaknesses.
Keywords:
telomeres; telomerase; thyroid cancer; TERT promoter; EMT; BRD4; epigenetic drugs; combined therapies1. Introduction
Telomere protection and/or elongation are considered among the primary mechanisms of cancer survival and aggressiveness. Telomeres, shortening at each mitosis, sense somatic cells aging and induce cell death when they become critically short. Thus, progressive reduction of telomere length is a rate-limiting step for uncontrolled and unlimited cell proliferation capacity. Telomerase is the enzyme responsible for telomere maintenance. It is a ribonucleic complex that includes the telomerase reverse transcriptase (TERT) and its RNA template, the Telomerase RNA component (TERC) [1]. Telomerase is selectively expressed in cells that undergo indefinite proliferation, such as staminal, embryonic and cancer cells. Conversely, cell commitment and differentiation requires telomerase inactivation. Still, mechanisms of telomere maintenance are required in differentiated healthy cells to avoid chromosomes crisis preventing the activation of DNA damage response (DDR) and preserving genomic stability. Thus, telomere homeostasis and telomerase access to telomeres are controlled by an intricate system of accessory protein complexes. Among these, the Shelterin complex is a group of six-proteins, including Telomere repeat factor-1 (TRF1) and 2 (TRF2), protection of telomeres-1 (POT1), TRF1 interacting protein-2 (TIN2), repressor/activator protein 1(RAP1) and the POT1-TIN2 organizing protein (TPP1), highly conserved during evolution. These proteins, positioned on the telomeric loop (t-loop), play a fundamental role in the homeostasis and stabilization of telomeres ends independently from the mechanisms involved in telomere elongation [2]. TIN2 is a bridging molecule that facilitates the assembly of the entire complex. By binding both to double (TRF1, TRF2) and single (POT1) strand DNA, Shelterins shield telomeres from inappropriate DNA repair avoiding the activation of DDR pathways and non-homologous end joining (NHEJ). In particular, RAP1 recruited by TRF2 inhibits homologous recombination. Furthermore, Shelterins protect telomeres from end-to-end fusion events and degradation [3]. Finally, TPP1 functions by regulating telomerase access to telomere, and the entire complex regulates nucleosomes distribution and contributes to the establishment of an epigenetic environment prone to telomere maintenance [4,5].
Being the sensor of cell proliferative capacity, aberrant telomere elongation occurs in the majority of cancers due to telomerase reactivation (in 90% of malignancies) and/or alternative lengthening of telomeres (ALT) [6]. Alteration in TERT expression and/or activity has been reported in many cancers and associated with increased aggressiveness. Among the mechanisms leading to aberrant telomerase activation, TERT promoter mutations C228T and C250T (−124 bp and −146 bp upstream of TERT’s transcription starting site, respectively) are frequent in cancer and associated with increased aggressiveness and metastatic potential in several tumor settings. These alterations cause the formation of ectopic binding sites for specific transcription factors (TFs) within the TERT promoter inducing its overexpression [7]. Similarly, aberrant expression and/or mutations of Shelterin proteins can favor tumorigenesis either by induction of chromosomal rearrangements and genome instability and by contributing to alteration of telomere length [8]. ALT occurs often in cancer that do not express telomerase, as the consequence of recombination-dependent replication pathways of telomere extension. ALT activation mainly correlates with the presence of mutations in ATRX chromatin remodeler (ATRX) and death domain associated protein (DAXX) genes in both tumors and cell lines [9,10,11,12]. In several tumors, mutations in the TERT promoter and ATRX have been reported to be mutually exclusive. However, melanomas with loss-of-function mutations in ATRX have also been found to have TERT promoter mutations [11,13].
Thyroid cancers (TCs) are the most common endocrine malignancy. Metastasis and aggressive clinical behavior in these tumors are generally limited to a small but significant group of high-grade lesions (about 10% of all diagnosed thyroid tumors) with poor biological characterization and very limited therapeutic options [14]. These tumors, associated with poor prognosis and low rate of survival, comprise a range of highly heterogeneous diseases that includes well differentiated TCs that developed Distant Metastases (DMs), Poorly Differentiated Thyroid Cancers (PDTCs) and Anaplastic Thyroid Cancers (ATCs). Clinical management of TC patients encompasses a spectrum of options, ranging from sonographic surveillance to total thyroidectomy plus lymph node neck dissection and/or radioiodine ablation (RAI). The lack of reliable markers for predicting the aggressive behavior of these tumors is a severe limitation to a precise risk-based stratification of this disease, thus contributing to the issue of patients’ overtreatment [15]. Furthermore, over 75% of high-grade TCs are radioiodine refractory and no effective therapies are available. These patients represent a relevant clinical challenge [16]. As for other tumors, TCs heavily rely on aberrant telomere regulation. In this review, we aim to summarize the current knowledge about telomere regulation in TC, with a particular focus on highly aggressive lesions and with the intent to discuss the potential application of these observations for defining both new prognostic tools and new potential targets to improve patient management.
2. TERT Promoter Mutations Are a Hallmark of TC Aggressiveness
Mutations in the TERT promoter are strongly over-represented in highly aggressive TCs, with the C228T usually being more frequent than C250T [17,18,19,20]. Their presence strongly correlates with disease stage III/IV, metastatic behavior, cancer recurrence and reduced survival probability [15,17,21,22,23]. A recently published study, conducted on data from The Cancer Genome Atlas (TCGA) consortium, revealed that TCs harboring TERT promoter mutations consistently present distinct transcriptomic profiles associated with enhanced cell cycle progression and metabolic activities as compared to wild-type patients [24]. As in other settings, also in thyroid, TERT promoter mutations are markers of malignant transformation, since no evidence of their occurrence in benign thyroid lesions has been reported so far. Furthermore, TERT promoter mutation frequency increases dramatically from low-grade microcarcinomas (where they were reported in fewer than 5% of lesions [25]) to highly aggressive poorly differentiated and undifferentiated TC (48.8% and 41.8%, respectively) independently of the ethnic and geographical backgrounds of the studies. In well-differentiated papillary thyroid cancers (PTCs), TERT promoter mutations are markers of metastatic behavior. Indeed, while in non-metastatic lesions TERT mutations are detected in about 10% of cases, 33% of PTCs that develop distant metastasis harbor mutations in the promoter of TERT gene [23], highlighting a role of these alterations in conferring aggressiveness to PTCs. Furthermore, De Biase et al., with the intent of exploring potential commonalities, compared the genetic background of 29 DMs and 18 ATCs. They showed that these highly aggressive thyroid lesions are characterized by different genetic backgrounds and likely evolve through different molecular mechanisms with the sole exception of TERT promoter mutations, which were the only high-frequency genetic events shared between these two separate groups of TCs. Together, these observations define the presence of TERT promoter mutations as a unique and reliable feature of aggressive TCs and suggest these genetic alterations as candidates for being a potential prognostic tool for identifying aggressive forms of TC at diagnosis [26]. In line with this, TERT expression and telomerase activity are detected in thyroid carcinoma, but not in normal thyrocytes or in benign thyroid tumors [17]. Interestingly, in the vast majority of published studies, TERT promoter mutations correlate with B-Raf proto-oncogene, serine/threonine kinase (BRAF) V600E mutation, considered a major oncogenic determinant of TC. Patients harboring both BRAF V600E and TERT promoter mutations display more severe clinical factors, suggesting that these mutations contribute to different molecular mechanisms resulting in synergic effects [17,18,27]. Additionally, a significant association was also observed between TERT promoter mutations and RAS mutations, which are frequent in PDTCs and ATCs [17,19,21]. To further strengthen the role of TERT in high-grade TC, our group identified the first genetic signature distinctive of DMs and significantly associated with increased mortality. This signature, called THYT1, includes, in addition to Ch1q duplication, not only TERT promoter mutations, but also duplication of TERT locus on chromosome 5 [15]. These data highlight the importance of TERT locus in the progression of TC. Furthermore, THYT1 may represent a prognostic marker to improve risk-based stratification and management of pre-operative PTCs.
Differently from the vast majority of cancer driver mutations that fall within the coding region of oncogenes and/or tumor suppressors altering the resulting proteins, TERT promoter mutations occur within non-coding elements, and their mechanism of action has only partially been elucidated. Both of the most frequent mutations (C228T and C250T) introduce within the TERT promoter new binding sites for members of the E-Twenty-Six (ETS) family of transcription factors. The increased activity of these TFs enhances TERT expression, leading to an augmented telomerase activity [28]. Experiments of site-direct mutagenesis recently conducted in glioblastoma cell lines confirmed the high specificity of the ETS consensus sequence necessary for TERT transcription activation. Different substitutions in the same position like G228C, G250C or G250T showed no effect on TERT transcription. In contrast, the G228T mutation partially induced promoter activity, being collocated in a position of ETS motif frequently degenerated for A/T. To identify which among the ETS transcription factors was responsible for TERT activation, a small interfering RNA screening of 13 molecules was conducted. The GA-binding protein alpha subunit (GABPA) KD resulted in a substantial decrease in TERT expression. Consistently, only GABPA KD impaired mutant promoter activity, despite having no effect on the wild type sequence in Luciferase assay indicating that GABPA is the only ETS factor able to affect TERT expression in presence of cancer-associated mutations [28]. The selective binding of GABPA to mutant promoter was confirmed in vivo and extended to different tumor settings by the analysis of ChIP-seq data from ENCODE project [29]. In contrast, Yuan et al. observed an inverse correlation between GABPA and TERT expression in primary TCs, where lower GABPA expression was associated with aggressive disease and reduced overall survival. These observations propose a potential tumor suppressor activity of GABPA. According to the model suggested by these authors, GABPA exerts a potent anti-metastatic function by directly stimulating the expression of Dicer1, a master suppressor of cancer initiation and progression. This discrepancy with regard to the role of GABPA as both promoter and suppressor of cancer aggressiveness warrants further investigation [30].
The methylation status of the chromatin surrounding TERT promoter is also affected by the presence of cancer-associated mutations reflecting the transcriptional activity of this region. The mutant TERT promoter exhibits high levels of H3K4me2/3, markers of opened chromatin and low CpG methylation upstream of the transcription starting site. On the contrary, high levels of H3K27me3 and CpG methylation outline wild-type TERT promoter, indicating a more silenced transcriptional state [31]. Recently, Liu R et al. proposed a model to explain the synergistic effects of TERT promoter mutations and BRAF V600E that perfectly fits in this context. It is known that binding of GABP to mutant TERT promoter, necessary for TERT activation, is influenced by the presence of native ETS motif spanning the two mutations and allowing the binding of tetramers of GABPA and GABPB. These authors demonstrated that BRAF V600E enhances the activation of Mitogen-activated protein kinase (MAPK) pathway, leading to the FOS-mediated expression of GABPB, which, in turn, binding to TERT mutated promoters in tetrameric form, induces TERT overexpression [32]. In agreement with this, a recent report showed that TERT expression in TC patients is positively correlated with both TERT promoter and BRAF V600E mutations. Mechanistically, this was due to the BRAF-induced upregulation of the PEA3 subfamily of ETS transcription factors (ETV1, ETV4 and ETV5), which in turn bind and selectively activate the mutant promoter driving TERT expression [33].
Ankicilar et al. investigated how TERT promoter mutations drive TERT activation and, by circular chromosome conformation capture (4C) assay, they demonstrated that GABP mediates a long-range chromatin interaction that along with an enrichment of active histone marks consequently drives TERT transcription. Indeed CRISPR/CAS9-mediated reversal of TERT promoter mutations to the wild type residue abrogates this long-range chromatin interaction and RNA Polymerase II recruitment, and reduces TERT transcription and telomerase activity. Furthermore, they found that a major player of this complex assembly is Bromodomain-containing protein 4 (BRD4), which is able to interact with the long-range region 300kb upstream of mutant TERT promoter enhancing TERT expression. Accordingly, BRD4 KD led to significant loss of active histone marks and impaired long-range chromatin interaction to an extent similar to GABPA KD. Furthermore, BRD4 regulates GABPA transcription by directly binding its promoter [34]. BRD4 belongs to the BET family of proteins, it is able to bind acetylated lysines, and it has both acetyl-transferase and kinase activities. It promotes cell cycle progression and cell growth in cancer. In particular, a recent study demonstrated a direct role of BRD4 in ATCs, where its silencing significantly inhibited tumor growth both in vitro and in vivo [35]. Furthermore, it has been already demonstrated in mouse models that the employment of BET inhibitors (BETi), a new class of anticancer drugs design to block the activity of BRD4 and the other BET proteins, is able to suppress ATC growth and to improve survival [36]. In line with this, Wang et al. showed that treatment with different kinds of BETi blocks telomere elongation and Telomerase complex activity [37]. Thus, this evidence suggests that telomere alterations in aggressive TCs may rely on BRD4 activity, prompting the use of BETi already employed in clinical trials for several tumor settings. Thus, the use of BETi may represent an alternative way to counteract telomeres deregulation avoiding side effects of direct telomerase inhibitors. Furthermore, the employment of BETi may present a selective strategy for targeting cancer cells presenting TERT promoter mutations sparing the ones that are not affected by these mutations, including normal cells.
3. Additional Mechanisms of Aberrant TERT Expression Regulation in Cancer
TERT promoter contains binding sites for several known regulatory factors, which include GC-motif and E-boxes. It has been widely demonstrated that MYC proto-oncogene, bHLH transcription factor (c-Myc) directly regulates TERT expression by binding to E-boxes within the TERT promoter [38]. Several studies have described c-Myc activation in TCs and its association with features of aggressiveness [39,40]. Still, whether and to which extent telomere deregulation relies on c-Myc activity is not well defined. Interestingly, the BRAF V600E - MAPK pathway axis may activate TERT expression through c-Myc in a TERT promoter mutation-independent manner, even if this mechanism seems to be less robust than that previous cited in TERT promoter mutated lesions [32]. c-Myc alone is likely not sufficient to drive TERT expression and the cooperation with Specificity Protein 1 (Sp1), a transcription factor that binds GC-motifs next to E-boxes, seems to be required for an efficient TERT promoter activation. Intriguingly, BETi, which we previously described as a potential therapeutic options for aggressive TCs harboring TERT promoter mutations, have been shown to drastically suppress c-Myc expression, and this effect has been widely documented in human patients in several clinical studies for solid and hematological tumors [41,42]. Thus, the employment of these drugs in aggressive TCs could exert a double activity, directly blocking the effect of BRD4 on TERT promoters and indirectly by restraining the expression and transcriptional activity of c-Myc on TERT regulation.
It has been reported that the nuclear factor kappa B (Nf-KB) pathway is able to enhance c-Myc and Sp-1 gene expression driving TERT activation in several hematological cell lines [43,44]. Furthermore, an NF-κB-responsive element is present in the TERT promoter 600 bp upstream of the transcription starting site [45]. However, in TCs, NF-κB signaling may have marginal effect on aggressiveness [46]. Indeed, a recent study used different NF-κB targeting strategies (genetics and pharmacologic) in different TC cell lines to demonstrate that NF-κB inhibitors are unlikely to be beneficial, even in combination with chemotherapy or radioiodine therapy [47].
TERT promoter also presents binding sites for signal transducer and activator of transcription 3 (STAT3), located about 2000 bp upstream of the transcription starting site. Published studies have suggested that STAT3, a TF activated in response to various cytokines and growth factors, in TC may function as onco-suppressor rather than as cancer promoting factor. Indeed, although STAT3 expression is detected in PTCs [48], the majority of the studies reported an increased activity in normal tissues compared to cancer lesions and an inverse correlation between STAT3 expression and tumor size [49,50,51].
Paired box 8 (PAX8) is an important TF in thyroid tumorigenesis. Additionally, it is known to be a marker of thyroid cell differentiation; PAX8 is among the more reliable markers of thyroid specification in advanced TCs, and indeed its expression has been detected in biopsies of ATC patients [52]. PAX8 responsive elements are found in TERT promoters, and it has been demonstrated that this TF positively regulates the expression of TERC raising the hypothesis that it plays a role in telomere regulation [53].
Recently, we described a relevant role for activator protein-1 (AP1) in driving thyroid tumorigenesis by controlling the expression of the pro-oncogenic factor Runt-related transcription factor 2 (RUNX2). AP-1 is a leucine zipper heterodimeric complex and the Jun-Fos dimer is the most common form of AP-1 protein in human cells. We reported the collaboration of c-JUN and BRD4 in the regulation of RUNX2 where they orchestrate the cooperation of three distal active enhancers (ENHs) with the RUNX2 promoter. Two hypotheses for their interplay have been formulated: either BRD4 binding to acetylated regions may function as a docking site for the recruitment of c-JUN on RUNX2 regulatory regions or c-JUN binding to ENHs favors BRD4 recruitment [54]. A similar mechanism of cooperation between these two factors can occur also on TERT promoter. Indeed, AP-1 can directly bind to the TERT promoter through binding sites at positions −1655 and −718 [55]. Additionally, BRD4 has been reported to interact with the mutated form of the TERT promoter by long-range chromatin interaction, the same ability BRD4 has to modify chromatin structure observed in the RUNX2 locus [34]. However, AP-1 recruitment at TERT promoter seems to have an inhibitory effect on TERT expression. Consistently, the mutations at both AP-1 binding sites eliminated about 70% of the suppressive effect caused by AP-1 [55]. Contrasting evidence links AP-1 expression to TC features in vivo. Some reports indicate that the expression of AP-1 was negative correlated with tumor size in PTCs [56]. Conversely, in a very recently published work, the level of AP-1 protein was found to be significantly up-regulated in PTCs compared with surrounding normal thyroid tissue by immunohistochemistry and was positively correlated with tumor size [57]. Thus, it remains to be established whether the activity of AP-1 in favoring TC progression partially relies on impairing telomere homeostasis and if yes with which mechanisms.
A comprehensive description of TERT promoter consensus sequences has been reviewed by Ramlee and colleagues, and may be used as a further hint to study possible players in telomerase regulation in aggressive TCs [45].
4. Telomere Length Involvement in Thyroid Cancer Development
In the spectrum of thyroid tumors, familial forms of non-medullary TCs (NMTCs) have also been reported. Familial NMTC patients show progressively increased disease aggressiveness in subsequent generations. This phenomenon is called “genetic anticipation”, and it was defined and associated with several inherited benign and malignant disorders. In the case of NMTCs, the later generations when compared to the first one showed an earlier age of insurgence, higher rate of lymph node metastases at surgery, and a worse outcome [58,59]. Familial NMTCs are generally more aggressive than the sporadic forms [60] and are associated with a 5–10-fold higher risk of developing the tumor for first-degree relatives compared to the general population. In recent years, several groups have collected and investigated pedigrees of familial NMTC patients in order to find the predisposing genetic loci, but this is probably a complex disease to the onset of which several genetic and environmental factors contribute [59]. Telomere length is a strong hereditable tract of these patients, and progressive telomere shortening through generations may represent the basis of anticipation. In line with this, anticipation is a typical condition of familial telomere diseases, a spectrum of genetic degenerative and age-dependent disease caused by premature senescence of stem cell compartments and determining increased rates of organ failure and cancer [61]. Indeed, short telomeres induce genomic instability and activate pathways of DNA damage response, whose functionality is crucial for cancer evolution [62]. The presence of short telomeres is reported in the blood of patients affected by different types of cancers, such as head and neck, bladder, lung, renal and breast cancer [63,64], enforcing the hypothesis that telomere dysfunction may represent a risk factor for cancer development. Confirming these data, familial PTCs also display short telomeres in peripheral leukocytes [63,65]. Interestingly, unaffected familial members present telomere lengths similar to those of relatives affected by PTC, suggesting that they likely have a genetic background prone to cancer development and probably need a lower number of additional factors to achieve tumor onset compared to sporadic cases [66]. Cantara and colleagues reported similar evidence in phytohemagglutinin-stimulated T-lymphocytes of both sporadic and familial PTC patients in which shorter telomeres with respect to healthy subjects were observed. Despite short telomeres, high telomerase activity and TERT expression were observed in PTCs as in other human cancer tissues [67,68]. Thus, telomerase activation is probably necessary as a telomere maintenance mechanism, likely not for inducing extensive telomere lengthening, but in order to conserve the minimal telomere length indispensable to save DNA-damaged cells from apoptosis, therefore contributing to their genomic instability and immortalization. Consistently, study on familial PTCs has demonstrated that these patients are characterized by abundant acentric fragments presenting telomeric sequences derived from subtelomeric chromosomal breaking, and thus, they show a frequency of chromosome fragility higher than healthy subjects and sporadic PTCs [66]. Overall, we can speculate that short telomeres are predisposed to genomic instability, and hence the occurrence of cancer-driving mutations, which include TERT promoter mutations [69,70]; subsequently, telomerase activation maintains telomere length and sustains cancer evolution and progression.
5. Telomere-Independent Cancer Supportive Mechanisms of TERT
Telomere-independent activities of TERT have been reported, and several studies have shown that this protein is engaged in processes such as regulation of DNA damage response, repression of apoptosis, regulation of chromatin state, and enhanced cell proliferation [71,72,73]. Furthermore, mounting evidence indicates a role of TERT in supporting metastatic spreading of cancer by the activation of the epithelial to mesenchymal transition (EMT) [74,75]. EMT is a process which allows epithelial cells to transiently transdifferentiate and acquire a mesenchymal-like phenotype. During this transition, epithelial cells lose their differentiated characteristics, including cell–cell adhesion, polarity, and lack of motility, and acquire mesenchymal features, including motility, invasiveness, and resistance to apoptosis [76,77]. Activation of the EMT program is triggered by many signaling factors and extracellular stimuli among which transforming growth factor beta (TGF-β) is considered a master player. Once engaged on its target receptors, TGF-β signals act within the cells through three major pathways: (1) inducing nuclear translocation of the SMAD family member 2/3 (SMAD2/3) proteins, (2) activating the MAPK, and (3) Phosphatidylinositol-4,5-Bisphosphate 3-Kinase—AKT serine/threonine kinase 1 (PI3K-AKT), signaling cascades. These pathways converge on the regulation of several TFs that together orchestrate gene expression silencing of epithelial-associated genes (like epithelial cadherin and cell–cell adhesion molecules) while promoting the expression of mesenchymal genes (including mesenchymal-cadherins, cell-matrix interacting molecules and cytoskeleton filaments) [78]. In addition to inducing migratory and invasive capacities, EMT also allows cancer cells to acquire higher resistance to apoptosis and immunosuppression. EMT is also crucial in TC, where it has been suggested to play a function both in the transient transdifferentiation of well-differentiated lesions during the metastatic spreading, as well as in the constitutive de-differentiation of highly aggressive ATCs. We recently reported that in PTCs, Cadherin 6 (CDH6), a class II cadherin, is induced by TGF-β and promotes the activation of EMT by restraining autophagy [79,80,81]. Strikingly, we also reported that CDH6 expression in human PTCs is restricted to highly metastatic lesions, in which we also found a significantly higher incidence of TERT promoter mutations and TERT locus amplification, raising the hypothesis of a functional interconnection between these molecular events [15,23]. Indeed, evidence exists that TERT takes part in the activation of EMT and this activity is independent by its function in maintaining telomere length [75].
Liu et al. reported that overexpression of TERT promotes the acquisition of mesenchymal features in gastric cancer. Conversely, its knockdown causes loss of mesenchymal markers and reduced invasiveness. In this model, TERT mediates the TGF-β signaling and in TERT KD cells TGF-β induction of target genes expression is impaired. Indeed, TERT interacts with beta-catenin (one of the EMT associated TFs and mediator of the Wnt-signaling) and together associate with mesenchymal marker promoters to drive their expression. This effect was largely independent of the telomere-lengthening activity of TERT, since overexpression of TERT mutants lacking these properties are still able to induce expression of EMT associated genes [75]. The transcriptional activity of TERT and its role in EMT are consistent in other models. Qin and colleagues reported that TERT promotes EMT in colon cancer through the Zinc finger E-box binding homeobox 1 (ZEB1) pathway. ZEB1 is a TGF-β target and a master regulator of EMT. The authors showed that TERT forms a complex with ZEB1 which binds to E-Cadherin promoter and represses its transcription leading to EMT [82]. The activity of TERT in this context is ZEB1 dependent, since overexpression of TERT fails to repress E-Cadherin in cells where ZEB1 expression is compromised. Intriguingly, EMT-related TFs seem to feedback on telomere homeostasis in a positive feedback loop. Snail Family Transcriptional Repressor 1 (Snail1) is an EMT TFs that belongs to the zinc finger TFs family. The relevance of Snail1 in supporting EMT is highlighted by a large amount of evidence. Recently, a role of Snail1 in controlling telomere integrity has been proposed. Mazzolini et al. reported that Snail1-depleted mesenchymal stem cells show a dramatic increase in telomere alterations and a significant reduction of telomere length. However, activity of telomerase is higher in Snail1-depleted cells, as is the expression of TERRA, a telomere-associated long non-coding RNA [74]. TERRA is a highly heterogeneous family of transcripts transcribed from subtelomeric regions. Loss of function as well as de-localization of TERRA leads to severe telomere dysfunction indicating that both expression and localization of this lncRNA are strictly necessary for telomere maintenance [83]. In the context of mesenchymal stem cells, Snail1 overexpression leads to a strong inhibition of TElomeric Repeat-containing RNA (TERRA) and TERT expression, coherently with the increased telomerase activity observed in Snail1-depleted cells [74]. In particular, the observation that Snail1 directly controls TERRA identified this TF as one of the factors controlling transcription at telomeric regions. TERRA expression is repressed by TGF-β concomitantly with Snail1 upregulation. Furthermore, RNA-sequencing profiling in a TERRA overexpressing model of mammary epithelial cells, showed that TERRA impaired TGF-β signaling by altering the response of a large number of EMT-related genes to the TGF-β stimulation. Together, these data indicate that expression of TERRA prevent activation of EMT and that the Snail1-mediated inhibition of this lncRNA is required to drive EMT [74].
6. Are Telomeres Possible Targets for TC Therapy?
For many years, therapeutic options for TC patients were limited to surgery and radioiodine ablation (RAI). However, 15–20% of well-differentiated cases that generate distant metastasis, as well as most of ATCs, remain resistant to standard treatments or present contraindication for surgery [84,85].
However, in recent years, several new drugs have been approved for the use in TCs. In this section, we intend to discuss some of these new options and their possible interplay with telomere biology. Mutations in BRAF and other members of the MAPK pathway are frequently mutated in TCs. BRAF V600E specific (Vemurafenib) and BRAF-MEK dual inhibitors (Dabrafenib) were recently proposed as potential therapies for advanced forms of TC, including ATCs [86,87]. Furthermore, multiple kinase inhibitors (MKIs), like Sorafenib and Lenvatinib, were FDA approved for the treatment of RAI-refractory and/or recurrent TCs. Both of these drugs improved overall survival compared to placebo in phase III randomized clinical trials; however, their clinical benefits were lessened by induced systemic toxicity [88,89,90,91,92]. As previously discussed, MAPK signaling converges on the activation of many cancer-related TFs, including AP-1 and c-Myc [93,94]. Both these TFs were proposed as potential regulators of TERT promoter, and therefore the use of these drugs may impact TERT expression and consequently the biology of telomeres [38,55].
Immunotherapy, aiming to exploit immune response and restore its ability to eliminate tumor cells, represents one of the more recent frontiers for tumor treatment. Among others, Pembrolizumab, targeting PD-1 by avoiding the interaction with its ligand, was tested in a clinical trial involving different solid malignancies including thyroid tumors [95,96]. This approach seems promising since recently published data showed that the 70–90% of highly aggressive TCs are positive for PD-L1 expression with respect to normal thyroid and differentiated tumors resulting negative [97]. However, the clinical benefits of these drugs need to be maximized, and therefore, several clinical trials are testing the effects of combined therapies for advanced tumors. Among others, some trials are currently testing the interaction between immunotherapy and epigenetic drugs, including one investigating the relationship between BETi and PD1/PD-L1 signaling blockade [98]. In line with this, a recent study conducted in a mouse model of c-Myc driven lymphoma showed that simultaneous inhibition of the PD-1/PD-L1 axis and treatment with the BETi JQ1 caused synergistic responses, demonstrating an important functional interaction between BETi and the host immune system [99]. For a more complete overview of the studies conducted up to date, Naoum and colleagues provided a detailed list of all clinical trials currently being developed for the treatment of highly aggressive TCs [85]. The observations collected in this review pave the way for new interesting speculations for the employment of alternative combined treatments. Solid evidence indicates that cancers with a high degree of genomic damage e/o mutational load are more sensitive to immunotherapy [100,101,102]. The reasons for this phenomenon are still to be determined, but the implications of these observations are already under investigation in clinics. Noticeably, aberrant telomere regulation may affect genomic stability by increasing DNA damage and consequently may condition immunotherapy response in cancer. Furthermore, Telomerase-based immunotherapy has also been developed and relies on the possibility of using TERT-derived peptides processed and presented on the surface of cancer cells as cancer neo-antigens, stimulating immune-mediated cell-kill. Two approaches have been proposed: one based on immune activation in vivo through the direct injection of optimized TERT derived peptides, and the other ex vivo based on the activation and expansion of patient-derived dendritic cells and then transfected with peptide and re-administered to patients through intradermal injection. For both of these strategies, clinical trials are ongoing in different tumor settings, and preliminary results have shown good tolerability and encouraging responses [103,104,105,106,107,108]. Recent evidence demonstrates that combination of checkpoint inhibitors with traditional cancer vaccines enhances anti-tumor activity in some studies and improves response to immunotherapy, laying the basis for a promising use of Telomerase vaccines in combination with immunotherapy [109]. Further studies are needed to strengthen the link between TERT regulation and immunotherapy response. However, in this context, telomere biology should be exploited both as potential biomarker for response as well as potential co-target to improve immunotherapy response in combination therapy schedules.
Targeting telomerase and mechanisms regulating telomere homeostasis may be considered a promising anti-cancer strategy. However, drugs directly targeting the telomerase complex have shown effectiveness in preclinical studies, but in clinical trials show lower performance and high side effects of toxicity [110,111,112,113]. In this context, the possibility of counteracting telomere impairment with alternative strategies appears to be of great interest. Here, we propose the employment of BETi drugs to indirectly target telomerase [114]. Inhibiting BRD4 transcriptional activity through the use of BETi may directly reduce TERT expression, but may also indirectly condition its transcription by repressing other BRD4-dependent TFs like c-Myc. Furthermore, the involvement of BRD4, as well as HDAC (which also controls histone acetylation at the telomere level), in telomere regulation suggests the use of epigenetic drugs (such as BETi and HDAC inhibitors) to improve the effectiveness of telomerase-inhibiting compounds. Both of these drugs, when employed alone, failed to show substantial effects in clinical trials [115,116]. However, their use in combination with other compounds showed promising results [117]. We may intriguingly speculate as to the use of BETi and HDACi in combination with telomerase inhibitors. Their effects on TERT expression and telomeres regulation may synergize with the one of telomerase inhibitors and improving their effectiveness. This could potentially allow the use of telomerase inhibitors at lower doses, thus limiting the toxicity of these drugs.
7. Conclusions
TC represents a paradigmatic example of the importance of telomere regulation in malignancies. Currently, mutations in TERT promoter are among the more common genetic abnormalities observed in cancers and the most frequent alterations observed in aggressive thyroid tumors. Mutations in TERT promoter are generally associated with increased TERT expression and Telomerase activity and although they are not always connected to increased telomere length [118], they are close related to bad prognosis, metastatic spreading and low patients overall survival [15,17,21,22,23,43,44,59,76,106]. We also explored the wide spectrum of signaling pathways converging on TERT promoter and the possible still poorly understood connections with the biological mechanisms already described in thyroid cancer biological settings. Overall, this evidence highlights the feasible employment of BETi for the treatment of thyroid tumors. Indeed, these drugs seem to hamper tumor spreading by different sides and having beneficial effects both in tumor harboring TERT promoter mutations and in the wild type ones. Here, we also discussed the available evidence showing the ability of BETi to disrupt long-range chromatin interaction on mutated TERT promoter such as to inhibit c-Myc and AP-1 transcriptional activities independently from tumor genetic background (Figure 1) [34,41,42,54].
The role of Shelterin proteins in the regulation of telomeres has not been studied in deep in TC setting. A single paper reported no mutations or polymorphisms in Shelterin complex genes in familial PTCs [119]. However further studies are necessary to define the structural organization of telomere proteins in TC lesions and whether TERT is the major players of telomere regulation or other mechanisms intervene. Conversely, ALT mechanisms as determined by mutations in ATRX and DAXX, are not reported in non-medullary cells derived thyroid cancers in many different studies [6,11,118], thus an involvement of these mechanisms in telomere deregulation may be likely excluded in this pathogenic environment.
In conclusion, these data enforce the role of telomerase dependent mechanisms in sustaining TC progression. Thus, telomerase regulation represents a major challenge and an interesting hint for studies investigating the pathogenesis of TC and/or the employment of new combined treatments.
Author Contributions
B.D. and A.C. wrote and revised the manuscript. All the authors have approved the final manuscript.
Acknowledgments
We wish to thank all members of the Lab for useful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Ann. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. The structural biology of the shelterin complex. Biol. Chem. 2019, 400, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K. How long does telomerase extend telomeres? Regulation of telomerase release and telomere length homeostasis. Curr. Genet. 2018, 64, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin-Mediated Telomere Protection. Ann. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Cacchione, S.; Biroccio, A.; Rizzo, A. Emerging roles of telomeric chromatin alterations in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 21. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Kim, J.Y.; Brosnan-Cashman, J.A.; An, S.; Kim, S.J.; Song, K.B.; Kim, M.S.; Kim, M.J.; Hwang, D.W.; Meeker, A.K.; Yu, E.; et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin. Cancer Res. 2017, 23, 1598–1606. [Google Scholar] [CrossRef]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.M.; Dagg, R.A.; Lau, L.M.S.; Lee, J.H.; Napier, C.E.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Cerone, M.A.; Londono-Vallejo, J.A.; Bacchetti, S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum. Mol. Genet. 2001, 10, 1945–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisei, R.; Molinaro, E.; Agate, L.; Bottici, V.; Masserini, L.; Ceccarelli, C.; Lippi, F.; Grasso, L.; Basolo, F.; Bevilacqua, G.; et al. Are the clinical and pathological features of differentiated thyroid carcinoma really changed over the last 35 years? Study on 4187 patients from a single Italian institution to answer this question. J. Clin. Endocrinol. Metabol. 2010, 95, 1516–1527. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, G.; Ragazzi, M.; de Biase, D.; Visani, M.; Zanetti, E.; Torricelli, F.; Sancisi, V.; Gugnoni, M.; Manzotti, G.; Braglia, L.; et al. Genome-wide profiling identifies the THYT1 signature as a distinctive feature of widely metastatic Papillary Thyroid Carcinomas. Oncotarget 2018, 9, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Matrone, A.; Valerio, L.; Pieruzzi, L.; Giani, C.; Cappagli, V.; Lorusso, L.; Agate, L.; Puleo, L.; Viola, D.; Bottici, V.; et al. Protein kinase inhibitors for the treatment of advanced and progressive radiorefractory thyroid tumors: From the clinical trials to the real life. Best Pract. Res. Clin. Endocrinol. Metabol. 2017, 31, 319–334. [Google Scholar] [CrossRef]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metabol. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metabol. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef]
- Shi, X.; Liu, R.; Qu, S.; Zhu, G.; Bishop, J.; Liu, X.; Sun, H.; Shan, Z.; Wang, E.; Luo, Y.; et al. Association of TERT promoter mutation 1,295,228 C>T with BRAF V600E mutation, older patient age, and distant metastasis in anaplastic thyroid cancer. J. Clin. Endocrinol. Metabol. 2015, 100, E632–E637. [Google Scholar] [CrossRef]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzza, M.; Colombo, C.; Rossi, S.; Tosi, D.; Cirello, V.; Perrino, M.; De Leo, S.; Magnani, E.; Pignatti, E.; Vigo, B.; et al. Telomerase in differentiated thyroid cancer: Promoter mutations, expression and localization. Mol. Cell. Endocrinol. 2015, 399, 288–295. [Google Scholar] [CrossRef]
- Gandolfi, G.; Ragazzi, M.; Frasoldati, A.; Piana, S.; Ciarrocchi, A.; Sancisi, V. TERT promoter mutations are associated with distant metastases in papillary thyroid carcinoma. Eur. J. Endocrinol. 2015, 172, 403–413. [Google Scholar] [CrossRef]
- Chien, M.N.; Yang, P.S.; Hsu, Y.C.; Liu, T.P.; Lee, J.J.; Cheng, S.P. Transcriptome analysis of papillary thyroid cancer harboring telomerase reverse transcriptase promoter mutation. Head Neck 2018, 40, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- de Biase, D.; Gandolfi, G.; Ragazzi, M.; Eszlinger, M.; Sancisi, V.; Gugnoni, M.; Visani, M.; Pession, A.; Casadei, G.; Durante, C.; et al. TERT Promoter Mutations in Papillary Thyroid Microcarcinomas. Thyroid 2015, 25, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- de Biase, D.; Torricelli, F.; Ragazzi, M.; Donati, B.; Khun, E.; Visani, M.; Acquaviva, G.; Pession, A.; Tallini, G.; Piana, S.; et al. Not the same thing: Metastatic PTCs have a different background than ATCs. Endocr. Connect. 2018, 7, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Liu, R.; Liu, X.; Murugan, A.K.; Zhu, G.; Zeiger, M.A.; Pai, S.; Bishop, J. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J. Clin. Oncol. 2014, 32, 2718–2726. [Google Scholar] [CrossRef]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Ferris, M.W.; Hull, M.A.; Chae, H.; Kim, S.; Graves, B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011, 25, 2147–2157. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Mu, N.; Wang, N.; Strååt, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2019, 38, 965–979. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Yoo, S.K.; Kim, H.H.; Jung, G.; Oh, A.R.; Cha, J.Y.; Kim, S.J.; Cho, S.W.; Lee, K.E.; Seo, J.S.; et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endor. Relat. Cancer 2019, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Sun, H. Bromodomaincontaining protein 4 is critical for the antiproliferative and proapoptotic effects of gambogic acid in anaplastic thyroid cancer. Int. J. Mol. Med. 2018, 42, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Enomoto, K.; Zhao, L.; Zhu, Y.J.; Willingham, M.C.; Meltzer, P.; Qi, J.; Cheng, S.Y. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clin. Cancer Res. 2017, 23, 430–440. [Google Scholar] [CrossRef]
- Wang, S.; Pike, A.M.; Lee, S.S.; Strong, M.A.; Connelly, C.J.; Greider, C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017, 45, 8403–8410. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Sakr, H.I.; Chute, D.J.; Nasr, C.; Sturgis, C.D. cMYC expression in thyroid follicular cell-derived carcinomas: A role in thyroid tumorigenesis. Diagn. Pathol. 2017, 12, 71. [Google Scholar] [CrossRef]
- Hu, Y.J.; Luo, X.Y.; Yang, Y.; Chen, C.Y.; Zhang, Z.Y.; Guo, X. Characterization and significance of MUC1 and c-myc expression in elderly patients with papillary thyroid carcinoma. Genet. Res. 2015, 14, 15325–15330. [Google Scholar] [CrossRef]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Willingham, M.C.; Qi, J.; Cheng, S.Y. Metformin and JQ1 synergistically inhibit obesity-activated thyroid cancer. Endocr. Relat. Cancer 2018, 25, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Sinha-Datta, U.; Horikawa, I.; Michishita, E.; Datta, A.; Sigler-Nicot, J.C.; Brown, M.; Kazanji, M.; Barrett, J.C.; Nicot, C. Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-I-transformed cells. Blood 2004, 104, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.Y.; Chen, Y.R.; Wang, T.C. A major role of PKC theta and NFkappaB in the regulation of hTERT in human T lymphocytes. FEBS Lett. 2006, 580, 6819–6824. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef]
- Bauerle, K.T.; Schweppe, R.E.; Haugen, B.R. Inhibition of nuclear factor-kappa B differentially affects thyroid cancer cell growth, apoptosis, and invasion. Mol. Cancer 2010, 9, 117. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Berlinberg, A.; Zhou, Q.; Wuensch, K.; Shibata, H.; Wood, W.M.; Haugen, B.R. Targeting the NF-kappaB Pathway as a Combination Therapy for Advanced Thyroid Cancer. PLoS ONE 2015, 10, e0134901. [Google Scholar] [CrossRef]
- Trovato, M.; Grosso, M.; Vitarelli, E.; Ruggeri, R.M.; Alesci, S.; Trimarchi, F.; Barresi, G.; Benvenga, S. Distinctive expression of STAT3 in papillary thyroid carcinomas and a subset of follicular adenomas. Histol. Histopathol. 2003, 18, 393–399. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Kim, W.G.; Choi, H.J.; Kim, W.B.; Kim, E.Y.; Yim, J.H.; Kim, T.Y.; Gong, G.; Kim, S.Y.; Chung, N.; Shong, Y.K. Basal STAT3 activities are negatively correlated with tumor size in papillary thyroid carcinomas. J. Endocrinol. Investig. 2012, 35, 413–418. [Google Scholar] [CrossRef]
- Sosonkina, N.; Starenki, D.; Park, J.I. The Role of STAT3 in Thyroid Cancer. Cancers 2014, 6, 526–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, E.; Ragazzi, M.; Ciarrocchi, A.; Torricelli, F.; de Biase, D. Angiosarcoma and anaplastic carcinoma of the thyroid are two distinct entities: A morphologic, immunohistochemical, and genetic study. Mod. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Campbell, H.G.; Wiles, A.K.; Eccles, M.R.; Reddel, R.R.; Braithwaite, A.W.; Royds, J.A. PAX8 regulates telomerase reverse transcriptase and telomerase RNA component in glioma. Cancer Res. 2008, 68, 5724–5732. [Google Scholar] [CrossRef] [PubMed]
- Sancisi, V.; Manzotti, G.; Gugnoni, M.; Rossi, T.; Gandolfi, G.; Gobbi, G.; Torricelli, F.; Catellani, F.; Faria do Valle, I.; Remondini, D.; et al. RUNX2 expression in thyroid and breast cancer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017, 45, 11249–11267. [Google Scholar] [CrossRef] [PubMed]
- Takakura, M.; Kyo, S.; Inoue, M.; Wright, W.E.; Shay, J.W. Function of AP-1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse cells. Mol. Cell. Biol. 2005, 25, 8037–8043. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, W.; Chen, X.; Gong, X. Roles of phosphatidylinositol 3-kinase regulatory subunit alpha, activator protein-1, and programmed cell death 4 in diagnosis of papillary thyroid carcinoma. Tumor Biol. 2016, 37, 6519–6526. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Huang, Y.; Gao, Q.; Feng, Z.; Li, Q.; Liu, Z. Expression of activator protein-1 in papillary thyroid carcinoma and its clinical significance. World J. Surg. Oncol. 2019, 17, 25. [Google Scholar] [CrossRef]
- Capezzone, M.; Marchisotta, S.; Cantara, S.; Busonero, G.; Brilli, L.; Pazaitou-Panayiotou, K.; Carli, A.F.; Caruso, G.; Toti, P.; Capitani, S.; et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr. Relat. Cancer 2008, 15, 1075–1081. [Google Scholar] [CrossRef]
- Bonora, E.; Tallini, G.; Romeo, G. Genetic Predisposition to Familial Nonmedullary Thyroid Cancer: An Update of Molecular Findings and State-of-the-Art Studies. J. Oncol. 2010, 2010, 385206. [Google Scholar] [CrossRef]
- Tavarelli, M.; Russo, M.; Terranova, R.; Scollo, C.; Spadaro, A.; Sapuppo, G.; Malandrino, P.; Masucci, R.; Squatrito, S.; Pellegriti, G. Familial Non-Medullary Thyroid Cancer Represents an Independent Risk Factor for Increased Cancer Aggressiveness: A Retrospective Analysis of 74 Families. Front. Endocrinol. 2015, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomere loss as a mechanism for chromosome instability in human cancer. Cancer Res. 2010, 70, 4255–4259. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metabol. 2008, 93, 3950–3957. [Google Scholar] [CrossRef]
- Shen, J.; Terry, M.B.; Gurvich, I.; Liao, Y.; Senie, R.T.; Santella, R.M. Short telomere length and breast cancer risk: A study in sister sets. Cancer Res. 2007, 67, 5538–5544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte telomere length and risk of papillary thyroid carcinoma. J. Clin. Endocrinol. Metabol. 2019, 104, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Pisu, M.; Frau, D.V.; Caria, P.; Dettori, T.; Capezzone, M.; Capuano, S.; Vanni, R.; Pacini, F. Telomere abnormalities and chromosome fragility in patients affected by familial papillary thyroid cancer. J. Clin. Endocrinol. Metabol. 2012, 97, E1327–E1331. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Rangel, H.A.; Sanchez-Suarez, P.; Castellanos-Juarez, E.; Penaroja-Flores, R.; Arenas-Aranda, D.J.; Gariglio, P.; Benitez-Bribiesca, L. Shorter telomeres and high telomerase activity correlate with a highly aggressive phenotype in breast cancer cell lines. Tumor Biol. 2016, 37, 11917–11926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meeker, A.K.; Kowalski, J.; Tsai, H.L.; Somervell, H.; Heaphy, C.; Sangenario, L.E.; Prasad, N.; Westra, W.H.; Zeiger, M.A.; et al. Telomere length is related to alternative splice patterns of telomerase in thyroid tumors. Am. J. Pathol. 2011, 179, 1415–1424. [Google Scholar] [CrossRef]
- Caria, P.; Pillai, R.; Dettori, T.; Frau, D.V.; Zavattari, P.; Riva, G.; Romano, G.; Pani, F.; Bentivegna, A.; Giovannoni, R.; et al. Thyrospheres from B-CPAP Cell Line with BRAF and TERT Promoter Mutations have Different Functional and Molecular Features than Parental Cells. J. Cancer 2017, 8, 1629–1639. [Google Scholar] [CrossRef]
- Caria, P.; Cantara, S.; Frau, D.V.; Pacini, F.; Vanni, R.; Dettori, T. Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma. Int. J. Mol. Sci. 2016, 17, 1759. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzolini, R.; Gonzalez, N.; Garcia-Garijo, A.; Millanes-Romero, A.; Peiro, S.; Smith, S.; Garcia de Herreros, A.; Canudas, S. Snail1 transcription factor controls telomere transcription and integrity. Nucleic Acids Res. 2018, 46, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Bjorkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Ciarrocchi, A. Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Ciarrocchi, A.; Piana, S.; Valcavi, R.; Gardini, G.; Casali, B. Inhibitor of DNA binding-1 induces mesenchymal features and promotes invasiveness in thyroid tumour cells. Eur. J. Cancer 2011, 47, 934–945. [Google Scholar] [CrossRef]
- Sancisi, V.; Gandolfi, G.; Ragazzi, M.; Nicoli, D.; Tamagnini, I.; Piana, S.; Ciarrocchi, A. Cadherin 6 is a new RUNX2 target in TGF-beta signalling pathway. PLoS ONE 2013, 8, e75489. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Tang, B.; Hu, C.J.; Xiao, Y.F.; Xie, R.; Yong, X.; Wu, Y.Y.; Dong, H.; Yang, S.M. An hTERT/ZEB1 complex directly regulates E-cadherin to promote epithelial-to-mesenchymal transition (EMT) in colorectal cancer. Oncotarget 2016, 7, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F.; Castagna, M.G.; Brilli, L.; Pentheroudakis, G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii110–vii119. [Google Scholar] [CrossRef] [PubMed]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.; Naing, A.; Piha-Paul, S.; Waguespack, S.G.; Cabanillas, M.E.; Sherman, S.I.; Ma, B.; Curtis, M.; et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Fu, S.; Kim, K.B.; Davies, M.A.; Nguyen, L.M.; George, G.C.; et al. Phase I Dose-Escalation Study of the Multikinase Inhibitor Lenvatinib in Patients with Advanced Solid Tumors and in an Expanded Cohort of Patients with Melanoma. Clin. Cancer Res. 2015, 21, 4801–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brose, M.S.; Worden, F.P.; Newbold, K.L.; Guo, M.; Hurria, A. Effect of Age on the Efficacy and Safety of Lenvatinib in Radioiodine-Refractory Differentiated Thyroid Cancer in the Phase III SELECT Trial. J. Clin. Oncol. 2017, 35, 2692–2699. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.M.; Lee, E.K.; Hwangbo, Y.; Lee, Y.J.; Cho, S.W.; Park, D.J.; Lee, Y.; Park, Y.J. Tumor doubling time predicts response to sorafenib in radioactive iodine-refractory differentiated thyroid cancer. Endocr. J. 2019. [Google Scholar] [CrossRef]
- Yu, S.T.; Ge, J.N.; Luo, J.Y.; Wei, Z.G.; Sun, B.H.; Lei, S.T. Treatment-related adverse effects with TKIs in patients with advanced or radioiodine refractory differentiated thyroid carcinoma: A systematic review and meta-analysis. Cancer Manag. Res. 2019, 11, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Bertelli, E.; Occhini, R.; Regoli, M.; Brilli, L.; Pacini, F.; Castagna, M.G.; Toti, P. Blockade of the programmed death ligand 1 (PD-L1) as potential therapy for anaplastic thyroid cancer. Endocrine 2019, 64, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Papadimitraki, E.; Aggouraki, D.; Konsolakis, G.; Mela, M.E.; Kotsakis, A.; Christou, S.; Patramani, S.; Alefantinou, M.; Kaskara, A.; et al. Sequential administration of the native TERT572 cryptic peptide enhances the immune response initiated by its optimized variant TERT(572Y) in cancer patients. J. Immunother. 2011, 34, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Konsolakis, G.; Aggouraki, D.; Kotsakis, A.; Papadimitraki, E.; Christou, S.; Menez-Jamet, J.; Kosmatopoulos, K.; Georgoulias, V.; Mavroudis, D. Immunological responses in cancer patients after vaccination with the therapeutic telomerase-specific vaccine Vx-001. Cancer Immunol. Immunother. 2012, 61, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mavroudis, D.; Bolonakis, I.; Cornet, S.; Myllaki, G.; Kanellou, P.; Kotsakis, A.; Galanis, A.; Nikoloudi, I.; Spyropoulou, M.; Menez, J.; et al. A phase I study of the optimized cryptic peptide TERT(572y) in patients with advanced malignancies. Oncology 2006, 70, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, D.; Traverso, P.; Parodi, A.; Tomasello, L.; Negrini, S.; Kalli, F.; Battaglia, F.; Ferrera, F.; Sciallero, S.; Murdaca, G.; et al. A multi-peptide, dual-adjuvant telomerase vaccine (GX301) is highly immunogenic in patients with prostate and renal cancer. Cancer Immunol. Immunother. 2013, 62, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dannull, J.; Yang, B.K.; Dahm, P.; Coleman, D.; Yancey, D.; Sichi, S.; Niedzwiecki, D.; Boczkowski, D.; Gilboa, E.; et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J. Immunol. 2005, 174, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Collins, R.H., Jr.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D., 3rd; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.A.; Maples, P.B.; Chen, S.; et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Goldkorn, A. Telomere and Telomerase Therapeutics in Cancer. Genes 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Mender, I.; Senturk, S.; Ozgunes, N.; Akcali, K.C.; Kletsas, D.; Gryaznov, S.; Can, A.; Shay, J.W.; Dikmen, Z.G. Imetelstat (a telomerase antagonist) exerts offtarget effects on the cytoskeleton. Int. J. Oncol. 2013, 42, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Nilubol, N.; Merkel, R.; Yang, L.; Patel, D.; Reynolds, J.C.; Sadowski, S.M.; Neychev, V.; Kebebew, E. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin. Endocrinol. 2017, 86, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Pugliese, M.; Gallo, M.; Brignardello, E.; Milla, P.; Orlandi, F.; Limone, P.P.; Arvat, E.; Boccuzzi, G.; Piovesan, A. Valproic Acid, a Histone Deacetylase Inhibitor, in Combination with Paclitaxel for Anaplastic Thyroid Cancer: Results of a Multicenter Randomized Controlled Phase II/III Trial. Int. J. Endocrinol. 2016, 2016, 2930414. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Cantara, S.; Capuano, S.; Capezzone, M.; Benigni, M.; Pisu, M.; Marchisotta, S.; Pacini, F. Lack of mutations of the telomerase RNA component in familial papillary thyroid cancer with short telomeres. Thyroid 2012, 22, 363–368. [Google Scholar] [CrossRef]
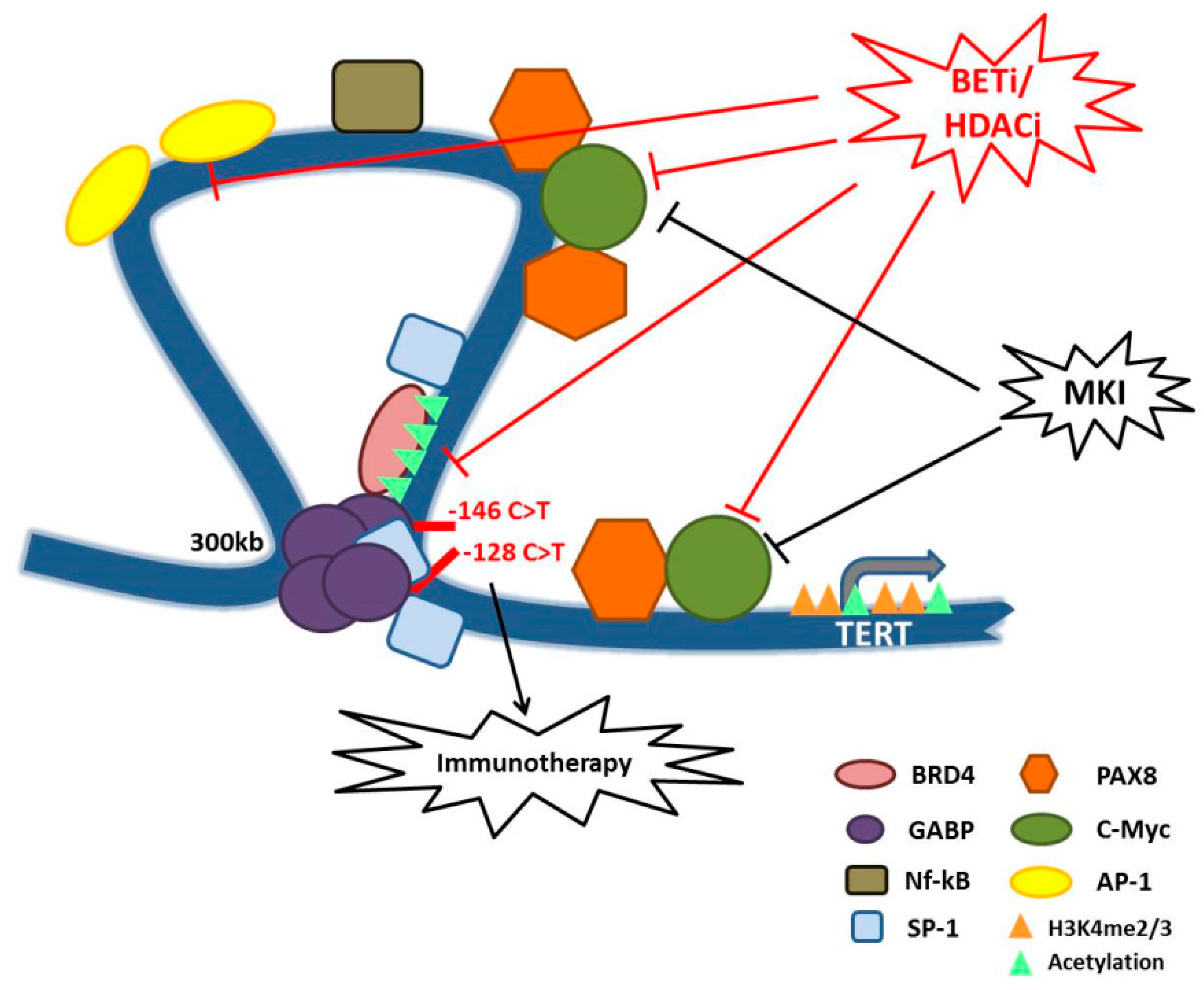
Figure 1.
Schematic model representing TERT promoter regulation in TCs and some hypotheses of therapeutic interventions. The figure shows the long-range chromatin interaction mediated by GABP factors and BRD4 on the mutated TERT promoter, and all the transcription factors positively influencing TERT expression and for whom we described a possible connection with TERT promoter in TCs. We also summarized the new proposed therapies for aggressive TCs, that may also rely on telomerase inhibition. In particular, MKI that acts on MAPK signaling and epigenetic drugs such as BETi and HDACi that may counteract TERT expression by impairing the function of TFs such as c-Myc and BRD4. Moreover, telomeres deregulation and the subsequent genomic instability, here represented by TERT promoter mutations, may influence the response to immunotherapeutic treatments. Overall, this model shows that there are many possible therapeutic combinations to counteract the aberrant activity of telomerase in TCs and consequently to inhibit one of the major trigger of TC aggressiveness.
Figure 1.
Schematic model representing TERT promoter regulation in TCs and some hypotheses of therapeutic interventions. The figure shows the long-range chromatin interaction mediated by GABP factors and BRD4 on the mutated TERT promoter, and all the transcription factors positively influencing TERT expression and for whom we described a possible connection with TERT promoter in TCs. We also summarized the new proposed therapies for aggressive TCs, that may also rely on telomerase inhibition. In particular, MKI that acts on MAPK signaling and epigenetic drugs such as BETi and HDACi that may counteract TERT expression by impairing the function of TFs such as c-Myc and BRD4. Moreover, telomeres deregulation and the subsequent genomic instability, here represented by TERT promoter mutations, may influence the response to immunotherapeutic treatments. Overall, this model shows that there are many possible therapeutic combinations to counteract the aberrant activity of telomerase in TCs and consequently to inhibit one of the major trigger of TC aggressiveness.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Donati, B.; Ciarrocchi, A. Telomerase and Telomeres Biology in Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 2887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122887
AMA Style
Donati B, Ciarrocchi A. Telomerase and Telomeres Biology in Thyroid Cancer. International Journal of Molecular Sciences. 2019; 20(12):2887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122887
Chicago/Turabian StyleDonati, Benedetta, and Alessia Ciarrocchi. 2019. "Telomerase and Telomeres Biology in Thyroid Cancer" International Journal of Molecular Sciences 20, no. 12: 2887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122887
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.