Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases

Abstract
:1. Introduction
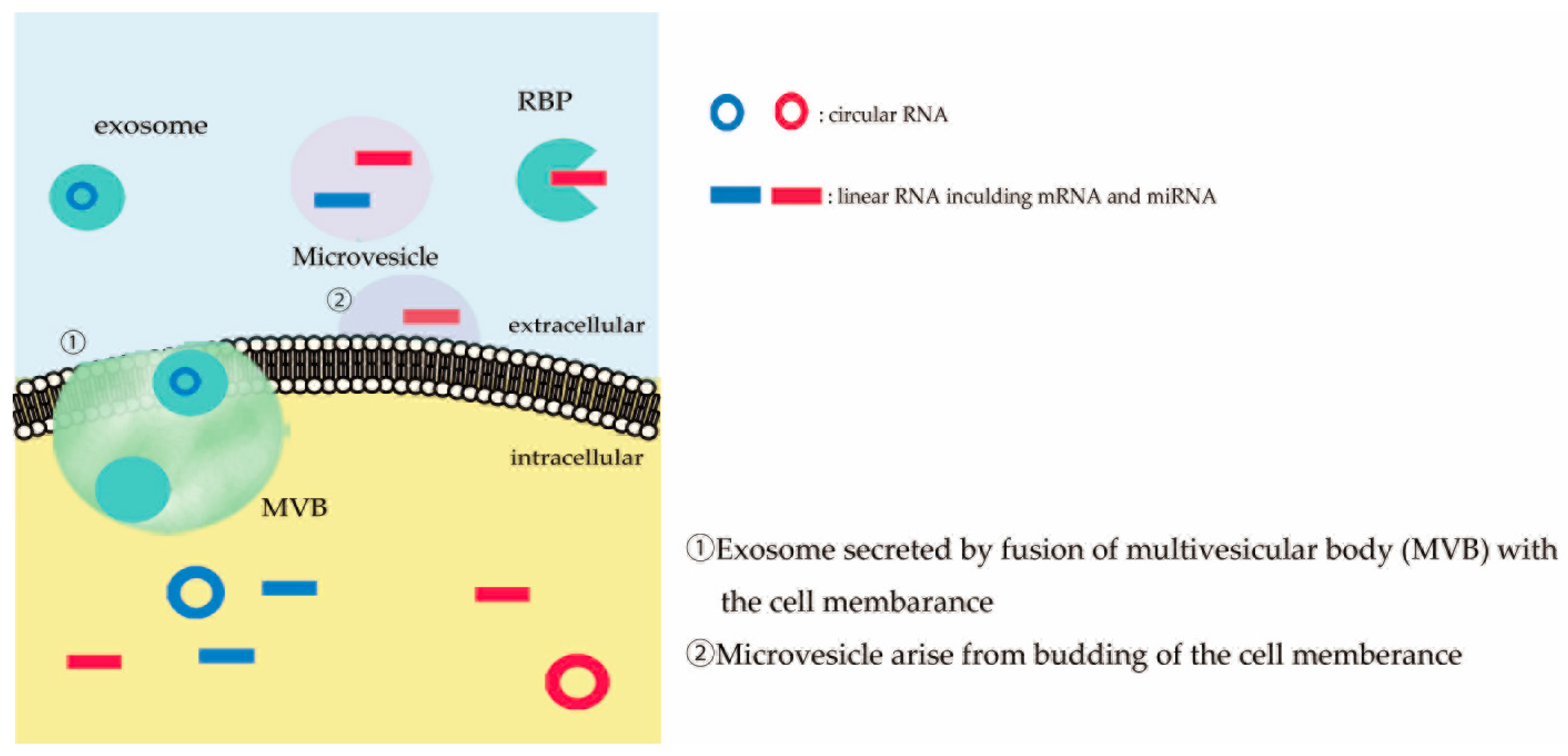
2. Extracellular RNAs
3. Liquid Biopsy in Neurodegenerative Diseases
3.1. Alzheimer Disease
3.2. Parkinson Disease
4. Liquid Biopsy in Sporadic ALS
4.1. Diagnostic Biomarkers
4.2. Other Types of Biomarkers
5. RNAs as Biomarker Candidates of Sporadic ALS
5.1. Diagnostic Biomarkers
5.2. Prognostic Biomarkers
5.3. Disease Progression Biomarkers
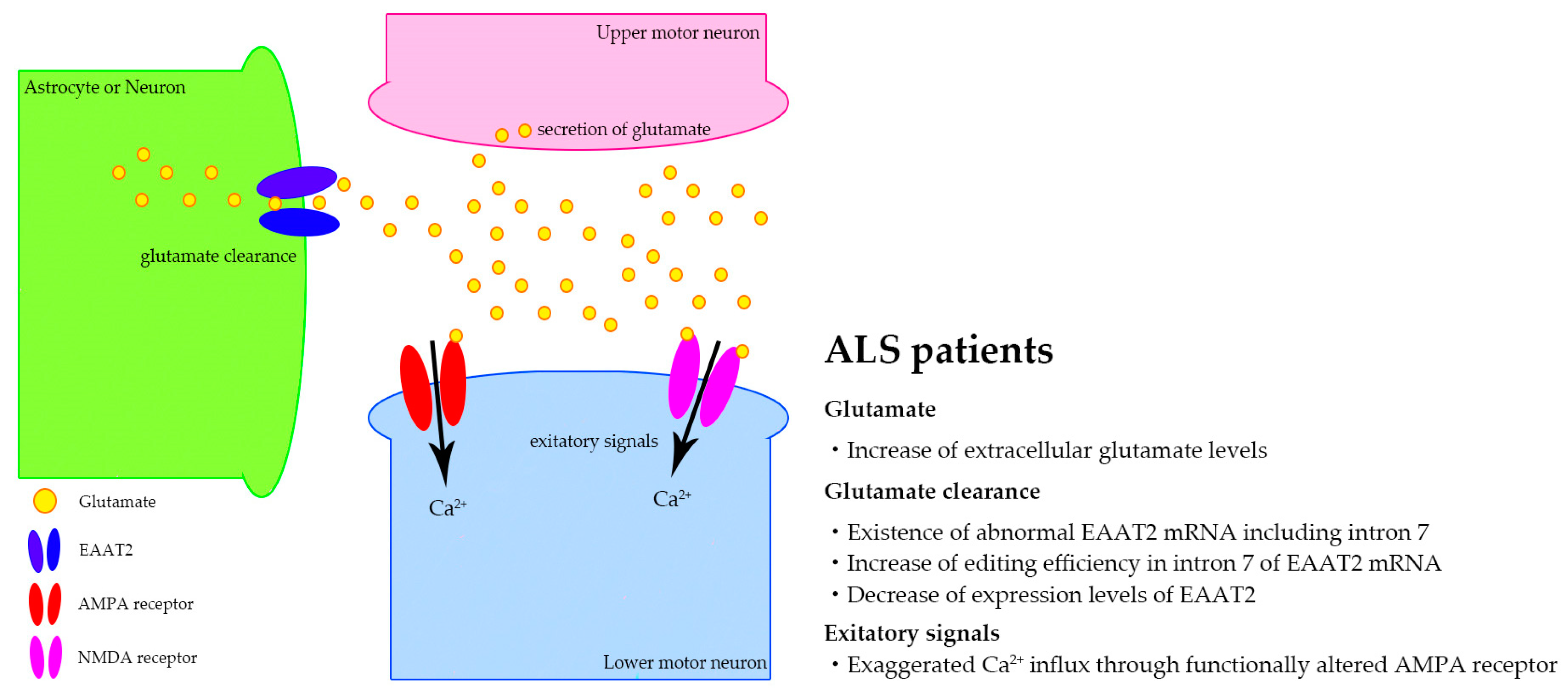
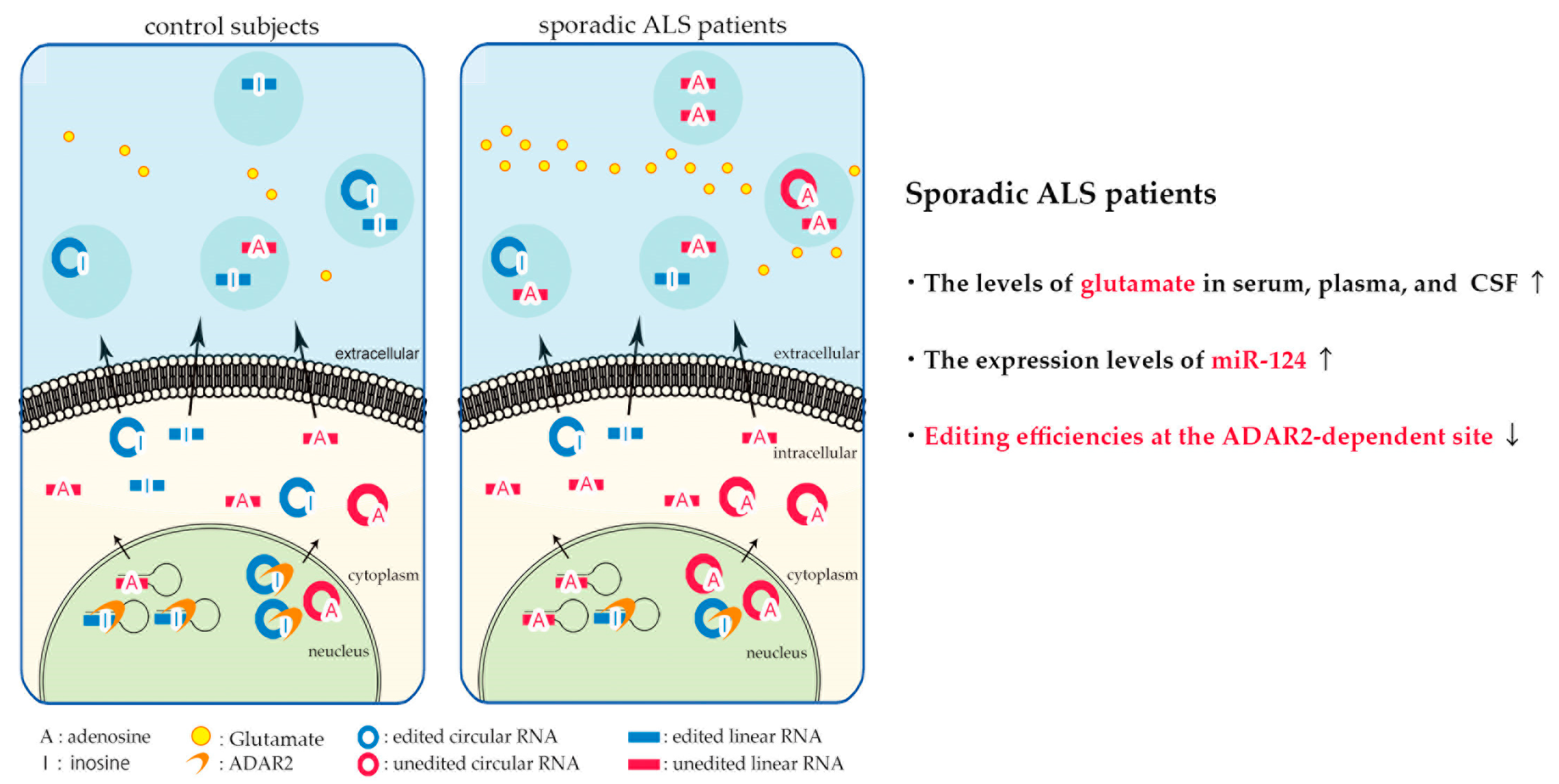
6. Biomarker Candidates Based on Excitotoxicity in Sporadic ALS
7. Conclusion and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| PD | Parkinson disease |
| ALS | amyotrophic lateral sclerosis |
| CNS | central nervous system |
| TDP-43 | transactive response DNA/RNA binding protein of 43kDa |
| CSF | cerebrospinal fluids |
| Aβ | β-amyloid |
| SOD-1 | superoxide dismutase 1 |
| FUS | fused in sarcoma |
| C9ORF72 | chromosome 9 open reading frame 72 |
| miRNA | micro RNA |
| circRNA | circular RNA |
| RBP | RNA binding protein |
| MVBs | multivesicular bodies |
| A-to-I | adenosine-to-inosine |
| RNA editing | A-to-I conversion of RNA |
| ADAR1 | adenosine deaminase acting on RNA 1 |
| PET | positron emission tomography |
| DAT-SPECT | dopamine transporter single-photon emission computed tomography |
| MIBG | meta-iodobenzylguanidine |
| Aβ42 | 42 amino acid form of Aβ |
| T-tau | total tau |
| P-tau | phosphorylated tau |
| NEFL | neurofilament light chain |
| NSE | neuron-specific enolase |
| VILIP-1 | visinin-like protein 1 |
| H-FABP | heart fatty acid binding protein |
| YKL-40 | chitinase-3-like protein 1 |
| MS | multiple sclerosis |
| PSP | progressive supranuclear palsy |
| MSA | multiple system atrophy |
| DJ-1 | parkinsonism associated deglycase |
| BDNF | brain derived neurotrophic factor |
| PBMCs | peripheral blood mononuclear cells |
| NEFH | neurofilament heavy chain |
| p75ECD | extracellular domain of the common neurotrophin receptor p75 |
| CMT2 | Charcot-Marie-Tooth disease type 2 |
| pNEFH | phosphorylated NEFH |
| CIDP | chronic inflammatory demyelinating polyneuropathy |
| ALSFS-R | ALS functional rating scale-revised |
| ATXN2 | ataxin 2 |
| VEGFA | vascular endothelial growth factor-A |
| NEK1 | NIMA-related kinase 1 |
| MATR3 | matrin 3 |
| qPCR | quantitative polymerase chain reaction |
| CCL2 | C-C motif Chemokine ligand 2 |
| KIF5C | kinesin family member 5C |
| DCTN1 | dynactin subunit 1 |
| RNAseq | RNA sequencing |
| NTRK2 | neurotrophic receptor tyrosine kinase 2 |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| AKT1 | AKT serine/threonine kinase 1 |
| GSK3β | glycogen synthase kinase 3β |
| NFκB | nuclear factor κB |
| FASLG | Fas ligand |
| CYFIP2 | cytoplasmic fragile X mental retardation interacting protein 2 |
| RBBP9 | retinoblastoma binding protein 9 |
| E2F1 | E2F transcription factor 1 |
| PBL | peripheral blood leukocyte |
| MAP1B | microtubule associated protein 1B |
| OC1 | onecut1 |
| FoxP1 | forkhead box P1 |
| MCPIP1 | monocyte chemotactic protein-1-induced protein-1 |
| REST | repressor element-1 silencing transcription factor |
| EAAT2 | excitatory amino acid transporter 2 |
| RhoA | Ras homolog family member A |
| PDZ-RhoGEF | Rho guanine nucleotide exchange factor |
| mTOR | mechanistic target of rapamycin |
| lncRNAs | long non-coding RNAs |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| NMDA | N-methyl-D-aspartate |
| Q/R | glutamine/arginine |
| ADAR2 | adenosine deaminase acting on RNA 2 |
| SBMA | spinal and bulbar muscle atrophy |
References
- Quinlan, S.; Kenny, A.; Medina, M.; Engel, T.; Jimenez-Mateos, E.M. Micrornas in neurodegenerative diseases. Int. Rev. Cell Mol. Biol. 2017, 334, 309–343. [Google Scholar] [PubMed]
- Maniati, M.S.; Maniati, M.; Yousefi, T.; Ahmadi-Ahangar, A.; Tehrani, S.S. New insights into the role of micrornas and long noncoding rnas in most common neurodegenerative diseases. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Quek, C.; Hill, A.F. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2017, 483, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Van den Berg, L.H.; Sorenson, E.; Gronseth, G.; Macklin, E.A.; Andrews, J.; Baloh, R.H.; Benatar, M.; Berry, J.D.; Chio, A.; Corcia, P.; et al. Revised airlie house consensus guidelines for design and implementation of als clinical trials. Neurology 2019. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Puglisi, C.; Santos, M.F.; Forte, S.; Memeo, L.; Lorico, A. Extracellular vesicles from thyroid carcinoma: The new frontier of liquid biopsy. Int. J. Mol. Sci. 2019, 20, 1114. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario, K.M.; Steindler, D.A. The role of extracellular vesicles in the progression of neurodegenerative disease and cancer. Trends Mol. Med. 2014, 20, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular vesicle-mediated cell(-)cell communication in the nervous system: Focus on neurological diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in als: A critical review. Amyotroph. Lateral. Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of als: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding als: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Recabarren-Leiva, D.; Alarcon, M. New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci. 2018, 193, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. Tdp-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated tdp-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- De Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of als. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Chiò, A. Risk factors in the early diagnosis of als: European epidemiological studies. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, S13–S18. [Google Scholar] [CrossRef]
- Cellura, E.; Spataro, R.; Taiello, A.C.; La Bella, V. Factors affecting the diagnostic delay in amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 2012, 114, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Swash, M.; Ingram, D. Preclinical and subclinical events in motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; Eurals, E. Incidence of amyotrophic lateral sclerosis in europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. Rna in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.M.; Buck, A.H. Extracellular small rnas: What, where, why? Biochem. Soc. Trans. 2012, 40, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microrna profiles in cerebrospinal fluid exosome in parkinson disease and alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Marzocchi, C.; Battistini, S. Micrornas as biomarkers in amyotrophic lateral sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Butti, Z.; Patten, S.A. Rna dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Köppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microrna-126 by apoptotic bodies induces cxcl12-dependent vascular protection. Sci. Signal 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (ev): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. Micrornas are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating micrornas independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of micrornas and microrna-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Dluzen, D.F.; Noren Hooten, N.; Evans, M.K. Extracellular rna in aging. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular rna released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microrna biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Idda, M.L.; Munk, R.; Abdelmohsen, K.; Gorospe, M. Noncoding rnas in alzheimer’s disease. Wiley Interdiscip. Rev. RNA 2018, 9, e1463. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-i editing of coding and non-coding rnas by adars. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; De Strooper, B. Non-coding rnas with essential roles in neurodegenerative disorders. Lancet Neurol. 2012, 11, 189–200. [Google Scholar] [CrossRef]
- Wilusz, J.E. A 360 degrees view of circular rnas: From biogenesis to functions. Wiley Interdiscip. Rev. RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular rna is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.L.; Yang, L.; Chen, L.L. The biogenesis of nascent circular rnas. Cell Rep. 2016, 15, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular rna biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfo, R.; Peruzzi, G.; et al. Fus affects circular rna expression in murine embryonic stem cell-derived motor neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular rnas are abundant, conserved, and associated with alu repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular rnas are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glazar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular rnas in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef]
- Gruner, H.; Cortes-Lopez, M.; Cooper, D.A.; Bauer, M.; Miura, P. Circrna accumulation in the aging mouse brain. Sci. Rep. 2016, 6, 38907. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. Csf and blood biomarkers for the diagnosis of alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef]
- De Souza, L.C.; Lamari, F.; Belliard, S.; Jardel, C.; Houillier, C.; De Paz, R.; Dubois, B.; Sarazin, M. Cerebrospinal fluid biomarkers in the differential diagnosis of alzheimer’s disease from other cortical dementias. J. Neurol. Neurosurg. Psychiatry 2011, 82, 240–246. [Google Scholar] [CrossRef]
- Tzen, K.Y.; Yang, S.Y.; Chen, T.F.; Cheng, T.W.; Horng, H.E.; Wen, H.P.; Huang, Y.Y.; Shiue, C.Y.; Chiu, M.J. Plasma abeta but not tau is related to brain pib retention in early alzheimer’s disease. ACS Chem. Neurosci. 2014, 5, 830–836. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-beta biomarkers for alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Zetterberg, H.; Wilson, D.; Andreasson, U.; Minthon, L.; Blennow, K.; Randall, J.; Hansson, O. Plasma tau levels in alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 9. [Google Scholar] [CrossRef]
- Mielke, M.M.; Hagen, C.E.; Xu, J.; Chai, X.; Vemuri, P.; Lowe, V.J.; Airey, D.C.; Knopman, D.S.; Roberts, R.O.; Machulda, M.M.; et al. Plasma phospho-tau181 increases with alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 2018, 14, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Graber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, L.E.; Karlsson, J.E.; Karlsson, J.O.; Persson, L.; Wikkelsø, C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in csf. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Weston, P.S.J.; Poole, T.; Ryan, N.S.; Nair, A.; Liang, Y.; Macpherson, K.; Druyeh, R.; Malone, I.B.; Ahsan, R.L.; Pemberton, H.; et al. Serum neurofilament light in familial alzheimer disease: A marker of early neurodegeneration. Neurology 2017, 89, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative. Association of plasma neurofilament light with neurodegeneration in patients with alzheimer disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Blood-based biomarkers for alzheimer’s disease-an update. J. Neurosci. Methods 2019, 319, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Janowitz, D.; Van der Auwera, S.; Wittfeld, K.; Nauck, M.; Friedrich, N.; Habes, M.; Davatzikos, C.; Terock, J.; Bahls, M.; et al. Association between serum neuron-specific enolase, age, overweight, and structural mri patterns in 901 subjects. Transl. Psychiatry 2017, 7, 1272. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Ekman, R. Neuron specific enolase in cerebrospinal fluid: A biochemical marker for neuronal degeneration in dementia disorders? J. Neural Transm. Park. Dis. Dement. Sect. 1994, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, B.; Siepi, D.; Sabalich, I.; Tranfaglia, C.; Parnetti, L. Cerebrospinal fluid neuron-specific enolase: A further marker of alzheimer’s disease? Funct. Neurol. 2008, 23, 93–96. [Google Scholar]
- Schmidt, F.M.; Mergl, R.; Stach, B.; Jahn, I.; Gertz, H.J.; Schonknecht, P. Elevated levels of cerebrospinal fluid neuron-specific enolase (nse) in alzheimer’s disease. Neurosci. Lett. 2014, 570, 81–85. [Google Scholar] [CrossRef]
- Parnetti, L.; Palumbo, B.; Cardinali, L.; Loreti, F.; Chionne, F.; Cecchetti, R.; Senin, U. Cerebrospinal fluid neuron-specific enolase in alzheimer’s disease and vascular dementia. Neurosci. Lett. 1995, 183, 43–45. [Google Scholar] [PubMed]
- Rosen, C.; Mattsson, N.; Johansson, P.M.; Andreasson, U.; Wallin, A.; Hansson, O.; Johansson, J.O.; Lamont, J.; Svensson, J.; Blennow, K.; et al. Discriminatory analysis of biochip-derived protein patterns in csf and plasma in neurodegenerative diseases. Front. Aging Neurosci. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Chaves, M.L.; Camozzato, A.L.; Ferreira, E.D.; Piazenski, I.; Kochhann, R.; Dall’Igna, O.; Mazzini, G.S.; Souza, D.O.; Portela, L.V. Serum levels of s100b and nse proteins in alzheimer’s disease patients. J. Neuroinflammation 2010, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Blennow, K.; Andreasen, N.; Laterza, O.; Modur, V.; Olander, J.; Gao, F.; Ohlendorf, M.; Ladenson, J.H. The brain injury biomarker vlp-1 is increased in the cerebrospinal fluid of alzheimer disease patients. Clin. Chem. 2008, 54, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; D’Angelo, G.; Macy, E.; Xiong, C.; Carter, D.; Cairns, N.J.; Fagan, A.M.; Head, D.; Mintun, M.A.; Ladenson, J.H.; et al. Visinin-like protein-1: Diagnostic and prognostic biomarker in alzheimer disease. Ann. Neurol. 2011, 70, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Hou, L.; Shi, H.; Zhong, X.; Zhang, Y.; Zheng, D.; Tan, Y.; Hu, G.; Mu, N.; Chan, J.; et al. Csf levels of the neuronal injury biomarker visinin-like protein-1 in alzheimer’s disease and dementia with lewy bodies. J. Neurochem. 2013, 127, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.H.; Alexopoulos, P.; Perneczky, R. Heart-type fatty acid binding protein and vascular endothelial growth factor: Cerebrospinal fluid biomarker candidates for alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Hertze, J.; Lautner, R.; Zetterberg, H.; Nagga, K.; Hoglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; et al. Microglial markers are elevated in the prodromal phase of alzheimer’s disease and vascular dementia. J. Alzheimers Dis. 2013, 33, 45–53. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. Ykl-40: A novel prognostic fluid biomarker for preclinical alzheimer’s disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef]
- Alcolea, D.; Carmona-Iragui, M.; Suarez-Calvet, M.; Sanchez-Saudinos, M.B.; Sala, I.; Anton-Aguirre, S.; Blesa, R.; Clarimon, J.; Fortea, J.; Lleo, A. Relationship between beta-secretase, inflammation and core cerebrospinal fluid biomarkers for alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 157–167. [Google Scholar] [CrossRef]
- Mattsson, N.; Tabatabaei, S.; Johansson, P.; Hansson, O.; Andreasson, U.; Mansson, J.E.; Johansson, J.O.; Olsson, B.; Wallin, A.; Svensson, J.; et al. Cerebrospinal fluid microglial markers in alzheimer’s disease: Elevated chitotriosidase activity but lack of diagnostic utility. Neuromolecular Med. 2011, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Fernandez, M.; Martin, R.; Rivera-Vallve, S.; Borras, E.; Chiva, C.; Julia, E.; Rovira, A.; Canto, E.; Alvarez-Cermeno, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133, 1082–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Reddy, P.H. Are circulating micrornas peripheral biomarkers for alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Zamzami, M.A.; Choudhry, H.; Shaik, M.M.; Tamargo, I.A.; Kamal, M.A. Mirnas as circulating biomarkers for alzheimer’s disease and parkinson’s disease. Med. Chem. 2016, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Fransquet, P.D.; Ryan, J. Micro rna as a potential blood-based epigenetic biomarker for alzheimer’s disease. Clin. Biochem. 2018, 58, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic review of mirna as biomarkers in alzheimer’s disease. Mol. Neurobiol. 2019, 1–12. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Muddashetty, R.S. Emerging role of micrornas in dementia. J. Mol. Biol. 2019, 431, 1743–1762. [Google Scholar] [CrossRef]
- Noyce, A.J.; Lees, A.J.; Schrag, A.E. The prediagnostic phase of parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 871–878. [Google Scholar] [CrossRef]
- Maass, F.; Schulz, I.; Lingor, P.; Mollenhauer, B.; Bahr, M. Cerebrospinal fluid biomarker for parkinson’s disease: An overview. Mol. Cell. Neurosci. 2018, 97, 60–66. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Bidinosti, M.; Shimshek, D.R.; Mollenhauer, B.; Marcellin, D.; Schweizer, T.; Lotz, G.P.; Schlossmacher, M.G.; Weiss, A. Novel one-step immunoassays to quantify alpha-synuclein: Applications for biomarker development and high-throughput screening. J. Biol. Chem. 2012, 287, 33691–33705. [Google Scholar] [CrossRef] [PubMed]
- Foulds, P.G.; Diggle, P.; Mitchell, J.D.; Parker, A.; Hasegawa, M.; Masuda-Suzukake, M.; Mann, D.M.; Allsop, D. A longitudinal study on alpha-synuclein in blood plasma as a biomarker for parkinson’s disease. Sci. Rep. 2013, 3, 2540. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Leverenz, J.B.; Baird, G.; et al. Dj-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of parkinson’s disease. Brain 2010, 133, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. A-synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, T.; Qureshi, M.M.; Ardah, M.T.; Varghese, S.; Shehab, S.A.; Kasai, T.; Ishigami, N.; Tamaoka, A.; Nakagawa, M.; El-Agnaf, O.M. Detection of elevated levels of α-synuclein oligomers in csf from patients with parkinson disease. Neurology 2010, 75, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a biochemical diagnosis of parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017, 74, 163–172. [Google Scholar] [CrossRef]
- Hall, S.; Ohrfelt, A.; Constantinescu, R.; Andreasson, U.; Surova, Y.; Bostrom, F.; Nilsson, C.; Hakan, W.; Decraemer, H.; Nagga, K.; et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch. Neurol. 2012, 69, 1445–1452. [Google Scholar] [CrossRef]
- Eusebi, P.; Giannandrea, D.; Biscetti, L.; Abraha, I.; Chiasserini, D.; Orso, M.; Calabresi, P.; Parnetti, L. Diagnostic utility of cerebrospinal fluid alpha-synuclein in parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2017, 32, 1389–1400. [Google Scholar] [CrossRef]
- Magdalinou, N.K.; Paterson, R.W.; Schott, J.M.; Fox, N.C.; Mummery, C.; Blennow, K.; Bhatia, K.; Morris, H.R.; Giunti, P.; Warner, T.T.; et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1240–1247. [Google Scholar] [CrossRef]
- Hansson, O.; Janelidze, S.; Hall, S.; Magdalinou, N.; Lees, A.J.; Andreasson, U.; Norgren, N.; Linder, J.; Forsgren, L.; Constantinescu, R.; et al. Blood-based nfl: A biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017, 88, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Lotankar, S.; Prabhavalkar, K.S.; Bhatt, L.K. Biomarkers for parkinson’s disease: Recent advancement. Neurosci. Bull. 2017, 33, 585–597. [Google Scholar] [CrossRef]
- Andersen, A.D.; Binzer, M.; Stenager, E.; Gramsbergen, J.B. Cerebrospinal fluid biomarkers for parkinson’s disease - a systematic review. Acta Neurol. Scand. 2017, 135, 34–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sen, D. Micrornas in parkinson’s disease. Exp. Brain Res. 2017, 235, 2359–2374. [Google Scholar] [CrossRef] [PubMed]
- Roser, A.E.; Caldi Gomes, L.; Schunemann, J.; Maass, F.; Lingor, P. Circulating mirnas as diagnostic biomarkers for parkinson’s disease. Front. Neurosci. 2018, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M.; Ungar, L.H.; Lange, D.J.; Yemul, S.; Deng, H.; Yuan, X.; Brown, R.H.; Cudkowicz, M.E.; Newhall, K.; Peskind, E.; et al. Identification of potential csf biomarkers in als. Neurology 2006, 66, 1218–1222. [Google Scholar] [CrossRef]
- Von Neuhoff, N.; Oumeraci, T.; Wolf, T.; Kollewe, K.; Bewerunge, P.; Neumann, B.; Brors, B.; Bufler, J.; Wurster, U.; Schlegelberger, B.; et al. Monitoring csf proteome alterations in amyotrophic lateral sclerosis: Obstacles and perspectives in translating a novel marker panel to the clinic. PLoS ONE 2012, 7, e44401. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.H.; Wu, J.J.; Ren, H.M.; Wang, J.; Ding, Z.T.; Jiang, Y.P. Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp. Ther. Med. 2016, 11, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Leoni, E.; Bremang, M.; Mitra, V.; Zubiri, I.; Jung, S.; Lu, C.H.; Adiutori, R.; Lombardi, V.; Russell, C.; Koncarevic, S.; et al. Combined tissue-fluid proteomics to unravel phenotypic variability in amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 4478. [Google Scholar] [CrossRef]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.; et al. Tdp-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.; Allsop, D.; Nakagawa, M. Increased tdp-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009, 117, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Noto, Y.; Shibuya, K.; Sato, Y.; Kanai, K.; Misawa, S.; Sawai, S.; Mori, M.; Uchiyama, T.; Isose, S.; Nasu, S.; et al. Elevated csf tdp-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph. Lateral Scler. 2011, 12, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Arai, T.; Yamashita, M.; Tsuji, H.; Nonaka, T.; Masuda-Suzukake, M.; Tamaoka, A.; Hasegawa, M.; Akiyama, H. Differential diagnosis of amyotrophic lateral sclerosis from guillain-barre syndrome by quantitative determination of tdp-43 in cerebrospinal fluid. Int. J. Neurosci. 2014, 124, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.R.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free tdp-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014, 15, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Poesen, K.; Van Damme, P. Diagnostic and prognostic performance of neurofilaments in als. Front. Neurol. 2019, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Petzold, A.; Süssmuth, S.D.; Ludolph, A.C.; Tumani, H. Axonal damage markers in cerebrospinal fluid are increased in als. Neurology 2006, 66, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K.; Yang, C.; Crook, J.; Overstreet, K.; Heckman, M.; Wang, Y.; Borchelt, D.; Shaw, G. Immunoreactivity of the phosphorylated axonal neurofilament h subunit (pnf-h) in blood of als model rodents and als patients: Evaluation of blood pnf-h as a potential als biomarker. J. Neurochem. 2009, 111, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Feneberg, E.; Weishaupt, J.; Brettschneider, J.; Tumani, H.; Andersen, P.M.; von Arnim, C.A.; Bohm, S.; Kassubek, J.; Kubisch, C.; et al. Neurofilaments in the diagnosis of motoneuron diseases: A prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for als correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Oeckl, P.; Steinacker, P.; Verde, F.; Barro, C.; Van Damme, P.; Gray, E.; Grosskreutz, J.; Jardel, C.; Kuhle, J.; et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology 2018, 90, e22–e30. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Ren, H.; Jeromin, A.; Liu, M.; Shen, D.; Tai, H.; Ding, Q.; Li, X.; Cui, L. Diagnostic performance of neurofilaments in chinese patients with amyotrophic lateral sclerosis: A prospective study. Front. Neurol. 2018, 9, 726. [Google Scholar] [CrossRef] [PubMed]
- Gille, B.; De Schaepdryver, M.; Goossens, J.; Dedeene, L.; De Vocht, J.; Oldoni, E.; Goris, A.; Van Den Bosch, L.; Depreitere, B.; Claeys, K.G.; et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2019, 45, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Steinacker, P.; Weishaupt, J.H.; Kassubek, J.; Oeckl, P.; Halbgebauer, S.; Tumani, H.; von Arnim, C.A.F.; Dorst, J.; Feneberg, E.; et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Matusica, D.; Alfonsi, F.; Turner, B.J.; Butler, T.J.; Shepheard, S.R.; Rogers, M.L.; Skeldal, S.; Underwood, C.K.; Mangelsdorf, M.; Coulson, E.J. Inhibition of motor neuron death in vitro and in vivo by a p75 neurotrophin receptor intracellular domain fragment. J. Cell Sci. 2016, 129, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Shepheard, S.R.; Chataway, T.; Schultz, D.W.; Rush, R.A.; Rogers, M.L. The extracellular domain of neurotrophin receptor p75 as a candidate biomarker for amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e87398. [Google Scholar] [CrossRef] [PubMed]
- Shepheard, S.R.; Wuu, J.; Cardoso, M.; Wiklendt, L.; Dinning, P.G.; Chataway, T.; Schultz, D.; Benatar, M.; Rogers, M.L. Urinary p75ecd: A prognostic, disease progression, and pharmacodynamic biomarker in als. Neurology 2017, 88, 1137–1143. [Google Scholar] [CrossRef]
- Jia, R.; Shepheard, S.; Jin, J.; Hu, F.; Zhao, X.; Xue, L.; Xiang, L.; Qi, H.; Qu, Q.; Guo, F.; et al. Urinary extracellular domain of neurotrophin receptor p75 as a biomarker for amyotrophic lateral sclerosis in a chinese cohort. Sci. Rep. 2017, 7, 5127. [Google Scholar] [CrossRef]
- Ibanez, C.F.; Simi, A. P75 neurotrophin receptor signaling in nervous system injury and degeneration: Paradox and opportunity. Trends Neurosci. 2012, 35, 431–440. [Google Scholar] [CrossRef]
- Kumar, V.; Hasan, G.M.; Hassan, M.I. Unraveling the role of rna mediated toxicity of c9orf72 repeats in c9-ftd/als. Front. Neurosci. 2017, 11, 711. [Google Scholar] [CrossRef]
- Gibson, S.B.; Downie, J.M.; Tsetsou, S.; Feusier, J.E.; Figueroa, K.P.; Bromberg, M.B.; Jorde, L.B.; Pulst, S.M. The evolving genetic risk for sporadic als. Neurology 2017, 89, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.J.; van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; van Es, M.A.; Blauw, H.M.; van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLoS ONE 2018, 13, e0198874. [Google Scholar] [CrossRef] [PubMed]
- Saris, C.G.; Horvath, S.; van Vught, P.W.; van Es, M.A.; Blauw, H.M.; Fuller, T.F.; Langfelder, P.; DeYoung, J.; Wokke, J.H.; Veldink, J.H.; et al. Weighted gene co-expression network analysis of the peripheral blood from amyotrophic lateral sclerosis patients. BMC Genom. 2009, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding rnas characterization in peripheral blood mononuclear cells and spinal cord from amyotrophic lateral sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Prabhakar, S.; Abburi, C.; Sharma, N.K.; Anand, A. Vascular endothelial growth factor-a and chemokine ligand (ccl2) genes are upregulated in peripheral blood mononuclear cells in indian amyotrophic lateral sclerosis patients. J. Neuroinflammation 2011, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Kuzma-Kozakiewicz, M.; Kazmierczak, B.; Chudy, A.; Gajewska, B.; Baranczyk-Kuzma, A. Alteration of motor protein expression involved in bidirectional transport in peripheral blood mononuclear cells of patients with amyotrophic lateral sclerosis. Neurodegener. Dis. 2016, 16, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Nachmany, H.; Wald, S.; Abekasis, M.; Bulvik, S.; Weil, M. Two potential biomarkers identified in mesenchymal stem cells and leukocytes of patients with sporadic amyotrophic lateral sclerosis. Dis. Markers 2012, 32, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sadanand, A.; Janardhanan, A.; Vanisree, A.J.; Pavai, T. Neurotrophin expression in lymphocytes: A powerful indicator of degeneration in parkinson’s disease, amyotrophic lateral sclerosis and ataxia. J. Mol. Neurosci. 2018, 64, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Storkebaum, E.; Lambrechts, D.; Carmeliet, P. Vegf: Once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays 2004, 26, 943–954. [Google Scholar] [CrossRef]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J.L.; Leza, J.C.; Polak, P.; Kalinin, S.; Feinstein, D.L. Astrocyte-derived mcp-1 mediates neuroprotective effects of noradrenaline. J. Neurosci. 2009, 29, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. Kif5c, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef] [PubMed]
- Kuzma-Kozakiewicz, M.; Chudy, A.; Kazmierczak, B.; Dziewulska, D.; Usarek, E.; Baranczyk-Kuzma, A. Dynactin deficiency in the cns of humans with sporadic als and mice with genetically determined motor neuron degeneration. Neurochem. Res. 2013, 38, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Knaepen, K.; Goekint, M.; Heyman, E.M.; Meeusen, R. Neuroplasticity—exercise-induced response of peripheral brain-derived neurotrophic factor: A systematic review of experimental studies in human subjects. Sports Med. 2010, 40, 765–801. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.S., 2nd; Cho, Y.J.; Stein, S.; Liang, P. Cyfip2, a direct p53 target, is leptomycin-b sensitive. Cell Cycle 2007, 6, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Woitach, J.T.; Zhang, M.; Niu, C.H.; Thorgeirsson, S.S. A retinoblastoma-binding protein that affects cell-cycle control and confers transforming ability. Nat. Genet. 1998, 19, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Cho, J. Role of chemokine ccl2 and its receptor ccr2 in neurodegenerative diseases. Arch. Pharm. Res. 2013, 36, 1039–1050. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, W.; Zhou, B.; Zhang, J.; for Alzheimer’s Disease Neuroimaging Initiative. Association of vascular endothelial growth factor levels in csf and cerebral glucose metabolism across the alzheimer’s disease spectrum. Neurosci. Lett. 2018, 687, 276–279. [Google Scholar] [CrossRef]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem brain, cerebrospinal fluid, and blood neurotrophic factor levels in alzheimer’s disease: A systematic review and meta-analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef]
- Hawley, Z.C.E.; Campos-Melo, D.; Droppelmann, C.A.; Strong, M.J. Motomirs: Mirnas in motor neuron function and disease. Front. Mol. Neurosci. 2017, 10, 127. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched micrornas as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small rna sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed mirnas related to neural and glial activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Vrabec, K.; Bostjancic, E.; Koritnik, B.; Leonardis, L.; Dolenc Groselj, L.; Zidar, J.; Rogelj, B.; Glavac, D.; Ravnik-Glavac, M. Differential expression of several mirnas and the host genes aatk and dnm2 in leukocytes of sporadic als patients. Front. Mol. Neurosci. 2018, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microrna gene signature ameliorates murine als. J. Clin. Invest. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating micrornas as biomarkers of muscle differentiation and atrophy in als. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of micrornas and target genes networks in peripheral blood of patients with sporadic amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, Q.; Chen, X.; Li, C.; Cao, B.; Ou, R.; Hadano, S.; Shang, H.F. Aberration of mirnas expression in leukocytes from sporadic amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2016, 9, 69. [Google Scholar] [CrossRef]
- Toivonen, J.M.; Manzano, R.; Olivan, S.; Zaragoza, P.; Garcia-Redondo, A.; Osta, R. Microrna-206: A potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum mirnas mir-206, 143-3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (als). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef]
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M. A systematic review of suggested molecular strata, biomarkers and their tissue sources in als. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.; Bonev, B.; Garcez, P.; Stanley, P.; Guillemot, F.; Papalopulu, N. Microrna-9 regulates axon extension and branching by targeting map1b in mouse cortical neurons. Nat. Neurosci. 2012, 25, 697. [Google Scholar] [CrossRef] [PubMed]
- Otaegi, G.; Pollock, A.; Hong, J.; Sun, T. Microrna mir-9 modifies motor neuron columns by a tuning regulation of foxp1 levels in developing spinal cords. J. Neurosci. 2011, 31, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Luxenhofer, G.; Helmbrecht, M.S.; Langhoff, J.; Giusti, S.A.; Refojo, D.; Huber, A.B. Microrna-9 promotes the switch from early-born to late-born motor neuron populations by regulating onecut transcription factor expression. Dev. Biol. 2014, 386, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.M.; Wirguin, I.; et al. Mirna malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; An, B.Y.; Xi, X.B.; Li, Z.W.; Li, F.Y. Microrna-9 controls apoptosis of neurons by targeting monocyte chemotactic protein-induced protein 1 expression in rat acute spinal cord injury model. Brain Res. Bull. 2016, 121, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, J.; Lee, S.; Lee, B.; Lee, J.W.; Lee, S.K. The microrna mir-124 antagonizes the anti-neural rest/scp1 pathway during embryonic cns development. Genes Dev. 2007, 21, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal mirna-dependent translational regulation of astroglial glutamate transporter glt1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed]
- Ishtiaq, M.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Analysis of novel nefl mrna targeting micrornas in amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e85653. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhou, Z.; Fink, D.J.; Mata, M. Hspb1 silences translation of pdz-rhogef by enhancing mir-20a and mir-128 expression to promote neurite extension. Mol. Cell. Neurosci. 2013, 57, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Goncalves Ido, C.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. Smn regulates axonal local translation via mir-183/mtor pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. Mir-206, a key modulator of skeletal muscle development and disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. Mir-338-3p is over-expressed in blood, cfs, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A mirna signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A microrna feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.C.; Zheng, J.Y.; Tang, L.J.; Huang, B.S.; Li, K.; Tao, Y.; Yu, W.; Zhu, R.L.; Li, S.; Li, L.X. Mir-133b promotes neurite outgrowth by targeting rhoa expression. Cell. Physiol. Biochem. 2015, 35, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Raheja, R.; Regev, K.; Healy, B.C.; Mazzola, M.A.; Beynon, V.; Von Glehn, F.; Paul, A.; Diaz-Cruz, C.; Gholipour, T.; Glanz, B.I.; et al. Correlating serum micrornas and clinical parameters in amyotrophic lateral sclerosis. Muscle Nerve 2018, 58, 261–269. [Google Scholar] [CrossRef]
- Calvo, A.C.; Cibreiro, G.A.; Merino, P.T.; Roy, J.F.; Galiana, A.; Rufian, A.J.; Cano, J.M.; Martin, M.A.; Moreno, L.; Larrode, P.; et al. Collagen xix alpha 1 improves prognosis in amyotrophic lateral sclerosis. Aging Dis. 2019, 10, 278–292. [Google Scholar] [CrossRef]
- De Andrade, H.M.; de Albuquerque, M.; Avansini, S.H.; de, S.R.C.; Dogini, D.B.; Nucci, A.; Carvalho, B.; Lopes-Cendes, I.; Franca, M.C., Jr. Micrornas-424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J. Neurol. Sci. 2016, 368, 19–24. [Google Scholar] [CrossRef]
- Di Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for als: Mir206, mir208b and mir499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef]
- Bede, P.; Bokde, A.L.; Byrne, S.; Elamin, M.; Fagan, A.J.; Hardiman, O. Spinal cord markers in als: Diagnostic and biomarker considerations. Amyotroph. Lateral Scler. 2012, 13, 407–415. [Google Scholar] [CrossRef]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A network of noncoding regulatory rnas acts in the mammalian brain. Cell 2018, 174, 350–362 e317. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Dewil, M.; Robberecht, W.; Van Den Bosch, L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 147–159. [Google Scholar] [PubMed]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in als: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Constantakakis, E.; Smith, J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann. Neurol. 1988, 24, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. Csf and plasma amino acid levels in motor neuron disease: Elevation of csf glutamate in a subset of patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new hplc method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Ferrarese, C.; Sala, G.; Riva, R.; Begni, B.; Zoia, C.; Tremolizzo, L.; Galimberti, G.; Millul, A.; Bastone, A.; Mennini, T.; et al. Decreased platelet glutamate uptake in patients with amyotrophic lateral sclerosis. Neurology 2001, 56, 270–272. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic analysis of serum by (1) h nmr spectroscopy in amyotrophic lateral sclerosis. Clin. Chim. Acta 2010, 411, 563–567. [Google Scholar] [CrossRef]
- Andreadou, E.; Kapaki, E.; Kokotis, P.; Paraskevas, G.P.; Katsaros, N.; Libitaki, G.; Zis, V.; Sfagos, C.; Vassilopoulos, D. Plasma glutamate and glycine levels in patients with amyotrophic lateral sclerosis: The effect of riluzole treatment. Clin. Neurol. Neurosurg. 2008, 110, 222–226. [Google Scholar] [CrossRef]
- Tarasiuk, J.; Kułakowska, A.; Drozdowski, W.; Kornhuber, J.; Lewczuk, P. Csf markers in amyotrophic lateral sclerosis. J. Neural Transm. (Vienna) 2012, 119, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.E.; Li, N.; Guo, D.Z.; Pan, S.Y.; Li, H.; Yang, C. High plasma glutamate levels are associated with poor functional outcome in acute ischemic stroke. Cell. Mol. Neurobiol. 2015, 35, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majlath, Z.; Vecsei, L. Excitotoxins, mitochondrial and redox disturbances in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 353. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter glt-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant rna processing in a neurodegenerative disease: The cause for absent eaat2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef]
- Flomen, R.; Makoff, A. Increased rna editing in eaat2 pre-mrna from amyotrophic lateral sclerosis patients: Involvement of a cryptic polyadenylation site. Neurosci. Lett. 2011, 497, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Jin, L.; Dykes-Hoberg, M.; Kuncl, R.W. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 6591–6595. [Google Scholar] [CrossRef]
- Velasco, I.; Tapia, R.; Massieu, L. Inhibition of glutamate uptake induces progressive accumulation of extracellular glutamate and neuronal damage in rat cortical cultures. J. Neurosci. Res. 1996, 44, 551–561. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter glt-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Corona, J.C.; Tapia, R. Ampa receptor activation, but not the accumulation of endogenous extracellular glutamate, induces paralysis and motor neuron death in rat spinal cord in vivo. J. Neurochem. 2004, 89, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Tovar, Y.R.L.B.; Santa-Cruz, L.D.; Zepeda, A.; Tapia, R. Chronic elevation of extracellular glutamate due to transport blockade is innocuous for spinal motoneurons in vivo. Neurochem. Int. 2009, 54, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Beart, P.M.; O’Shea, R.D. Transporters for l-glutamate: An update on their molecular pharmacology and pathological involvement. Br. J. Pharmacol. 2009, 150, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Fromm, A.; Münch, C.; Schwalenstöcker, B.; Fray, A.E.; Ince, P.G.; Stamm, S.; Grön, G.; Ludolph, A.C.; Shaw, P.J. The rna of the glutamate transporter eaat2 is variably spliced in amyotrophic lateral sclerosis and normal individuals. J. Neurol. Sci. 1999, 170, 45–50. [Google Scholar] [CrossRef]
- Ferrarese, C.; Begni, B.; Canevari, C.; Zoia, C.; Piolti, R.; Frigo, M.; Appollonio, I.; Frattola, L. Glutamate uptake is decreased in platelets from alzheimer’s disease patients. Ann. Neurol. 2000, 47, 641–643. [Google Scholar] [CrossRef]
- Ferrarese, C.; Tremolizzo, L.; Rigoldi, M.; Sala, G.; Begni, B.; Brighina, L.; Ricci, G.; Albizzati, M.G.; Piolti, R.; Crosti, F.; et al. Decreased platelet glutamate uptake and genetic risk factors in patients with parkinson’s disease. Neurol. Sci. 2001, 22, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Bos, I.W.; Hoogland, G.; Meine Jansen, C.F.; Willigen, G.; Spierenburg, H.A.; van den Berg, L.H.; de Graan, P.N. Increased glutamine synthetase but normal eaat2 expression in platelets of als patients. Neurochem. Int. 2006, 48, 306–311. [Google Scholar] [CrossRef]
- Heath, P.R.; Shaw, P.J. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 438–458. [Google Scholar] [CrossRef]
- Carriedo, S.G.; Yin, H.Z.; Weiss, J.H. Motor neurons are selectively vulnerable to ampa/kainate receptor-mediated injury in vitro. J. Neurosci. 1996, 16, 4069–4079. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Burnashev, N.; Monyer, H.; Seeburg, P.H.; Sakmann, B. Divalent ion permeability of ampa receptor channels is dominated by the edited form of a single subunit. Neuron 1992, 8, 189–198. [Google Scholar] [CrossRef]
- Melcher, T.; Maas, S.; Herb, A.; Sprengel, R.; Seeburg, P.H.; Higuchi, M. A mammalian rna editing enzyme. Nature 1996, 379, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of rna editing by adar deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, D.; Kask, K.; Brusa, R.; Kornau, H.C.; Kolhekar, R.; Rozov, A.; Burnashev, N.; Jensen, V.; Hvalby, O.; Sprengel, R.; et al. Neurological dysfunctions in mice expressing different levels of the q/r site-unedited ampar subunit glur-b. Nat. Neurosci. 1999, 2, 57–64. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an ampa receptor gene rescues lethality in mice deficient in the rna-editing enzyme adar2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Seeburg, P.H. A-to-i editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ito, K.; Sun, H.; Aizawa, H.; Kanazawa, I.; Kwak, S. Glutamate receptors: Rna editing and death of motor neurons. Nature 2004, 427, 801. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound downregulation of the rna editing enzyme adar2 in als spinal motor neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef]
- Kawahara, Y.; Sun, H.; Ito, K.; Hideyama, T.; Aoki, M.; Sobue, G.; Tsuji, S.; Kwak, S. Underediting of glur2 mrna, a neuronal death inducing molecular change in sporadic als, does not occur in motor neurons in als1 or sbma. Neurosci. Res. 2006, 54, 11–14. [Google Scholar] [CrossRef]
- Aizawa, H.; Sawada, J.; Hideyama, T.; Yamashita, T.; Katayama, T.; Hasebe, N.; Kimura, T.; Yahara, O.; Kwak, S. Tdp-43 pathology in sporadic als occurs in motor neurons lacking the rna editing enzyme adar2. Acta Neuropathol. 2010, 120, 75–84. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of tdp-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Yamashita, T.; Teramoto, S.; Hirose, N.; Tamaoka, A.; Kwak, S. Adar2-dependent a-to-i rna editing in the extracellular linear and circular rnas. Neurosci. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Hideyama, T.; Yamashita, T.; Kimura, T.; Suzuki, N.; Aoki, M.; Kwak, S. Deficient rna-editing enzyme adar2 in an amyotrophic lateral sclerosis patient with a fus(p525l) mutation. J. Clin. Neurosci. 2016, 32, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9orf72 repeat expansion causes vulnerability of motor neurons to ca(2+)-permeable ampa receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Alsop, E.; Lorenzini, I.; Starr, A.; Rabichow, B.E.; Mendez, E.; Levy, J.L.; Burciu, C.; Reiman, R.; Chew, J.; et al. Adar2 mislocalization and widespread rna editing aberrations in c9orf72-mediated als/ftd. Acta Neuropathol. 2019, 138, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Chai, H.L.; Teramoto, S.; Tsuji, S.; Shimazaki, K.; Muramatsu, S.; Kwak, S. Rescue of amyotrophic lateral sclerosis phenotype in a mouse model by intravenous aav9-adar2 delivery to motor neurons. EMBO Mol. Med. 2013, 5, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, M.; Yamashita, T.; Hirose, N.; Teramoto, S.; Kwak, S. The ampa receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (als) pathology in sporadic als model mice. Sci. Rep. 2016, 6, 28649. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Proteins | Changes in Levels | Kinds of Body Fluids | Biomarkers | Patients (Origin and Number) | Disease Specificity | Reference | |
|---|---|---|---|---|---|---|---|
| TDP-43 | Increase | CSF | Diagnostic | Germany | 15 sporadic ALS | Low (not disease-specific) | [110] |
| 12 FTLD | |||||||
| 9 ALS + FTLD | |||||||
| 13 disease controls | |||||||
| Japanese | 30 sporadic ALS | [111] | |||||
| 29 disease controls | |||||||
| Japanese | 27 sporadic ALS | [112] | |||||
| 50 disease controls | |||||||
| Japanese | 13 sporadic ALS | [113] | |||||
| 7 GBS | |||||||
| Germany | 9 sporadic ALS | [114] | |||||
| 4 FTLD | |||||||
| 8 control subjects | |||||||
| Prognostic | Japanese | 29 disease controls | High (correlation with survival time and disease duration) | [111] | |||
| 27 sporadic ALS | |||||||
| Japanese | 27 sporadic ALS | [112] | |||||
| 50 disease controls | |||||||
| Japanese | 13 sporadic ALS | [113] | |||||
| 7 GBS | |||||||
| Neurofilament (NEFL or pNEFH) | Increase | CSF | Diagnostic | Germany | 67 sporadic ALS | Low (not disease-specific) | [117] |
| 2 familial ALS | |||||||
| 33 control subjects | |||||||
| American | 20 sporadic ALS | [118] | |||||
| 20 control subjects | |||||||
| Germany | 222 sporadic ALS | [119] | |||||
| 20 familial ALS | |||||||
| 199 disease controls | |||||||
| Germany | 194 sporadic ALS | [120] | |||||
| 26 familial ALS | |||||||
| 316 disease controls | |||||||
| European | 176 sporadic ALS | [121] | |||||
| 63 disease controls | |||||||
| Chinese | 53 sporadic ALS | [122] | |||||
| 32 disease controls | |||||||
| American | 134 sporadic ALS | [123] | |||||
| 15 familial ALS | |||||||
| 101 disease controls | |||||||
| Germany | 124 sporadic ALS | [124] | |||||
| 109 disease controls | |||||||
| 50 control subjects | |||||||
| Prognostic | Germany | 67 sporadic ALS | High (correlation with survival time and disease duration) | [117] | |||
| 2 familial ALS | |||||||
| 33 control subjects | |||||||
| Germany | 222 sporadic ALS | [119] | |||||
| 20 familial ALS | |||||||
| 199 disease controls | |||||||
| Germany | 194 sporadic ALS | [120] | |||||
| 26 familial ALS | |||||||
| 316 disease controls | |||||||
| European | 176 sporadic ALS | [121] | |||||
| 63 disease controls | |||||||
| Chinese | 53 sporadic ALS | [122] | |||||
| 32 disease controls | |||||||
| Disease progression | Germany | 194 sporadic ALS | High (correlation with ALSFS-R) | [120,124] | |||
| 26 familial ALS | |||||||
| 316 disease controls | |||||||
| Germany | 124 sporadic ALS | [124] | |||||
| 109 disease controls | |||||||
| 50 control subjects | |||||||
| Serum | Diagnostic | Germany | 67 sporadic ALS | Low (not disease-specific) | [117,118,119,120,121,122,123,124] | ||
| 2 familial ALS | |||||||
| 33 control subjects | |||||||
| American | 20 sporadic ALS | [118] | |||||
| 20 control subjects | |||||||
| Germany | 222 sporadic ALS | [119] | |||||
| 20 familial ALS | |||||||
| 199 disease controls | |||||||
| Germany | 194 sporadic ALS | [120] | |||||
| 26 familial ALS | |||||||
| 316 disease controls | |||||||
| European | 176 sporadic ALS | [121] | |||||
| 63 disease controls | |||||||
| Chinese | 53 sporadic ALS | [122] | |||||
| 32 disease controls | |||||||
| American | 134 sporadic ALS | [123] | |||||
| 15 familial ALS | |||||||
| 101 disease controls | |||||||
| Germany | 124 sporadic ALS | [124] | |||||
| 109 disease controls | |||||||
| 50 control subjects | |||||||
| Prognostic | European | 176 sporadic ALS | High (correlation with survival time and disease duration) | [121] | |||
| 63 disease controls | |||||||
| American | 134 sporadic ALS | [123] | |||||
| 15 familial ALS | |||||||
| 101 disease controls | |||||||
| Germany | 124 sporadic ALS | [124] | |||||
| 109 disease controls | |||||||
| 50 control subjects | |||||||
| p75ECD | Increase | Urine | Diagnostic | Australian | 28 sporadic ALS | Low (not disease-specific) | [126] |
| 19 disease controls | |||||||
| 12 control subjects | |||||||
| Australian | 54 sporadic ALS | [127] | |||||
| 45 control subjects | |||||||
| Chinese | 101 sporadic ALS | [128] | |||||
| 108 disease controls | |||||||
| 97 control subjects | |||||||
| Prognostic | Australian | 28 sporadic ALS | High (correlation with survival time) | [126,127,128] | |||
| 19 disease controls | |||||||
| 12 control subjects | |||||||
| Australian | 54 sporadic ALS | [127] | |||||
| 45 control subjects | |||||||
| Chinese | 101 sporadic ALS | [128] | |||||
| 108 disease controls | |||||||
| 97 control subjects | |||||||
| Disease progression | Australian | 54 sporadic ALS | High (correlation with ALSFS-R) | [127] | |||
| 45 control subjects | |||||||
| mRNA | Changes in Levels | Kinds of Body Fluids | Patients (Origin and Number) | Disease Specificity | Reference | |
|---|---|---|---|---|---|---|
| Diagnostic biomarkers | ||||||
| VEGFA mRNA | Increase | PBMCs | Indian | 50 sporadic ALS | Low (not disease-specific) | [135] |
| 50 control subjects | ||||||
| CCL2 mRNA | Increase | PBMCs | Indian | 50 sporadic ALS | Low (not disease-specific) | [135] |
| 50 control subjects | ||||||
| KIF5C mRNA | Decrease | PBMCs | Polish | 74 sporadic ALS | Low (not disease-specific) | [136] |
| 28 disease controls | ||||||
| 65 control subjects | ||||||
| DCTN1 mRNA | Increase | PBMCs | Polish | 74 sporadic ALS | Low (not disease-specific) | [136] |
| 28 disease controls | ||||||
| 65 control subjects | ||||||
| BDNF mRNA | Decrease | PBL | Dutch | 50 sporadic ALS | Low (inconsistent results, not disease-specific) | [132] |
| 50 disease control | ||||||
| 50 control subjects | ||||||
| Increase | Whole blood | Indian | 64 sporadic ALS | [138] | ||
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| NTRK2 mRNA | Decrease | PBL | Indian | 64 sporadic ALS | Low (not disease-specific) | [138] |
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| PIK3CA mRNA | Decrease | PBL | Dutch | 50 sporadic ALS | Low (inconsistent results, not disease-specific) | [132] |
| 50 disease control | ||||||
| 50 control subjects | ||||||
| Increase | Whole blood | Indian | 64 sporadic ALS | [138] | ||
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| AKT1 mRNA | Decrease | PBL | Dutch | 50 sporadic ALS | Low (inconsistent results, not disease-specific) | [132] |
| 50 disease control | ||||||
| 50 control subjects | ||||||
| Increase | Whole blood | Indian | 64 sporadic ALS | [138] | ||
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| GSK3β mRNA | Decrease | PBL | Indian | 64 sporadic ALS | Low (not disease-specific) | [138] |
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| NFκB mRNA | Decrease | PBL | American | 123 sporadic ALS | Low (inconsistent results, not disease-specific) | [133] |
| 123 control subjects | ||||||
| Increase | Whole blood | Indian | 64 sporadic ALS | [138] | ||
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| FASLG mRNA | Increase | PBL | Indian | 64 sporadic ALS | Low (not disease-specific) | [138] |
| 122 disease controls | ||||||
| 120 control subjects | ||||||
| CYFIP2 mRNA | Increase | PBL | Dutch | 50 sporadic ALS | Possible (More studies are needed) | [132] |
| 50 disease control | ||||||
| 50 control subjects | ||||||
| Whole blood | Israeli | 6 sporadic ALS | [137] | |||
| 3 non-ALS | ||||||
| RBBP9 mRNA | Increase | PBL | Israeli | 6 sporadic ALS | Possible (More studies are needed) | [137] |
| 3 non-ALS | ||||||
| Prognostic biomarker | ||||||
| COL19A1 mRNA | Increase | Whole blood | Spanish | 59 sporadic ALS | Possible (More studies are needed) | [177] |
| 24 disease controls | ||||||
| 58 controls subjects | ||||||
| Disease progression biomarkers | ||||||
| VEGFA mRNA | Increase | PBMCs | Indian | 50 sporadic ALS | High (correlation with respiratory dysfunction) | [135] |
| 50 control subjects | ||||||
| CCL2 mRNA | Increase | PBMCs | Indian | 50 sporadic ALS | High (correlation with respiratory dysfunction) | [135] |
| 50 control subjects | ||||||
| miRNA | Changes in Levels | Kinds of Body Fluids | Patients (Origin and Number) | Disease Specificity | Reference | |
|---|---|---|---|---|---|---|
| Diagnostic biomarkers | ||||||
| miR-9 | Increase | Plasma | American | 50 sporadic ALS | Low (not disease-specific) | [151] |
| 50 AD | ||||||
| 50 PD | ||||||
| 50 FTLD | ||||||
| 50 control subjects | ||||||
| CSF | British | 32 sporadic ALS | [152] | |||
| 6 MS | ||||||
| 10 control subjects | ||||||
| PBL | Slovenian | 77 sporadic ALS | [153] | |||
| 7 familial ALS | ||||||
| 27 control subjects | ||||||
| miR-124 | Increase | CSF | British | 32 sporadic ALS | Low (not disease-specific) | [152] |
| 6 MS | ||||||
| 10 control subjects | ||||||
| miR-146a | Decrease | CSF | British | 32 sporadic ALS | Low (inconsistent results, not disease-specific) | [152] |
| 6 MS | ||||||
| 10 control subjects | ||||||
| Increase | PBMCs | American | 22 sporadic ALS | [154] | ||
| 4 familial ALS | ||||||
| 24 control subjects | ||||||
| Decrease | Serum | Italian | 14 sporadic ALS | [155] | ||
| 8 control subjects | ||||||
| miR-128 | Decrease | Whole blood | Italian | 50 sporadic ALS | Low (not disease-specific) | [156] |
| 15 control subjects | ||||||
| miR-183 | Decrease | Whole blood | Italian | 50 sporadic ALS | Low (not disease-specific) | [156] |
| 15 control subjects | ||||||
| PBL | Chinese | 83 sporadic ALS | [157] | |||
| 24 PD | ||||||
| 61 control subjects | ||||||
| miR-206 | Increase | plasma | American | 50 sporadic ALS | Low (not disease-specific) | [151] |
| 50 AD | ||||||
| 50 PD | ||||||
| 50 FTLD | ||||||
| 50 control subjects | ||||||
| PBL | Slovenian | 77 sporadic ALS | [153] | |||
| 7 familial ALS | ||||||
| 27 control subjects | ||||||
| Serum | Spanish | 12 sporadic ALS | [158] | |||
| 12 control subjects | ||||||
| Serum | British | 27 sporadic ALS | [159] | |||
| 36 disease controls | ||||||
| 25 control subjects | ||||||
| miR-338-3p | Increase | Serum | Italian | 72 sporadic ALS | Low (not disease-specific) | [172] |
| PBL | 62 control subjects | |||||
| CSF | ||||||
| PBL | Italian | 14 sporadic ALS | [173] | |||
| 14 control subjects | ||||||
| miR-133b | Increase | Serum | American | 20 sporadic ALS | Low (not disease-specific) | [176] |
| 3 familial ALS | ||||||
| 30 control subjects | ||||||
| Prognostic biomarkers | ||||||
| miR-206 | Increase | Plasma | Brazilian | 39 sporadic ALS | High (correlation with Medical Research Council Score) | [178] |
| 39 control subjects | ||||||
| miR-9 | Increase | Plasma | American | 50 sporadic ALS | Possible (More studies are needed) | [151] |
| 50 AD | ||||||
| 50 PD | ||||||
| 50 FTLD | ||||||
| 50 control subjects | ||||||
| CSF | British | 32 sporadic ALS | [152] | |||
| 6 MS | ||||||
| 10 control subjects | ||||||
| PBL | Slovenian | 77 sporadic ALS | [153] | |||
| 7 familial ALS | ||||||
| 27 control subjects | ||||||
| miR-133b | Increase | Serum | American | 20 sporadic ALS | Possible (More studies are needed) | [176] |
| 3 familial ALS | ||||||
| 30 control subjects | ||||||
| Pharmacodynamic biomarkers | ||||||
| miR-9 | Increase | Plasma | American | 50 sporadic ALS | Possible (More studies are needed) | [151] |
| 50 AD | ||||||
| 50 PD | ||||||
| 50 FTLD | ||||||
| 50 control subjects | ||||||
| CSF | British | 32 sporadic ALS | [152] | |||
| 6 MS | ||||||
| 10 control subjects | ||||||
| PBL | Slovenian | 77 sporadic ALS | [153] | |||
| 7 familial ALS | ||||||
| 27 control subjects | ||||||
| miR-206 | Increase | Plasma | American | 50 sporadic ALS | Possible (More studies are needed) | [151] |
| 50 AD | ||||||
| 50 PD | ||||||
| 50 FTLD | ||||||
| 50 control subjects | ||||||
| PBL | Slovenian | 77 sporadic ALS | [153] | |||
| 7 familial ALS | ||||||
| 27 control subjects | ||||||
| Serum | Spanish | 12 sporadic ALS | [158] | |||
| 12 control subjects | ||||||
| Serum | British | 27 sporadic ALS | [159] | |||
| 36 disease controls | ||||||
| 25 control subjects | ||||||
| Disease progression biomarker | ||||||
| miR-206 | Increase | Serum | British | 36 disease controls | Low (no correlation with disease progression) | [159] |
| 25 control subjects | ||||||
| Changes in Levels | Kinds of Body Fluids | Reliability | Reference | |
|---|---|---|---|---|
| Diagnostic biomarkers | ||||
| Glutamate | Increase | Serum | Low (not disease-specific) | [188,189,190,191] |
| Plasma | ||||
| CSF | ||||
| miR-124 | Increase | CSF | Low (not disease-specific) | [152] |
| Editing efficiencies at the ADAR2-dependent sites | Decrease | In vitro | Possible (More studies are needed) | [223] |
| Prognostic biomarkers | ||||
| Glutamate | Increase | Plasma | Low | [188] |
| Predictive biomarkers | ||||
| Glutamate | Increase | Plasma | Low | [191] |
| Editing efficiencies at the ADAR2-dependent sites | Decrease | In vitro | Possible (More studies are needed) | [223] |
| Pharmacodynamic biomarkers | ||||
| Glutamate | Increase | Plasma | Low | [191] |
| Editing efficiencies at the ADAR2-dependent sites | Decrease | In vitro | Possible (More studies are needed) | [223] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosaka, T.; Yamashita, T.; Tamaoka, A.; Kwak, S. Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3148. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133148
Hosaka T, Yamashita T, Tamaoka A, Kwak S. Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(13):3148. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133148
Chicago/Turabian StyleHosaka, Takashi, Takenari Yamashita, Akira Tamaoka, and Shin Kwak. 2019. "Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 13: 3148. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133148