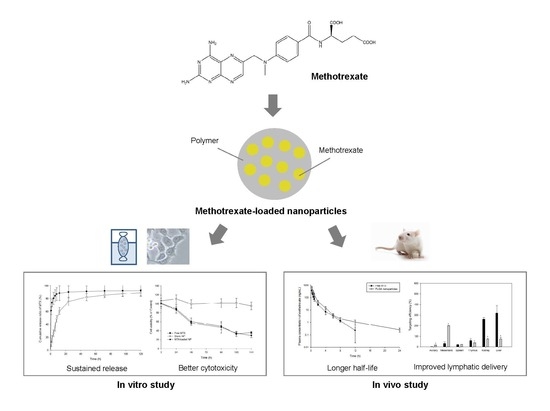
Preparation and In Vitro/In Vivo Characterization of Polymeric Nanoparticles Containing Methotrexate to Improve Lymphatic Delivery


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Preparation of MTX-Loaded NPs
2.1.1. Determination of Optimal Concentration of PLGA in Internal Oil Phase
2.1.2. Determination of Optimal Concentration of PVA in External Aqueous Phase
2.1.3. Determination of Optimal Oil–Water Phase Volume Ratio
2.2. Characterization and Evaluation of NPs
2.2.1. Particle Size, Zeta Potential, Drug Encapsulation Efficiency and Drug Loading
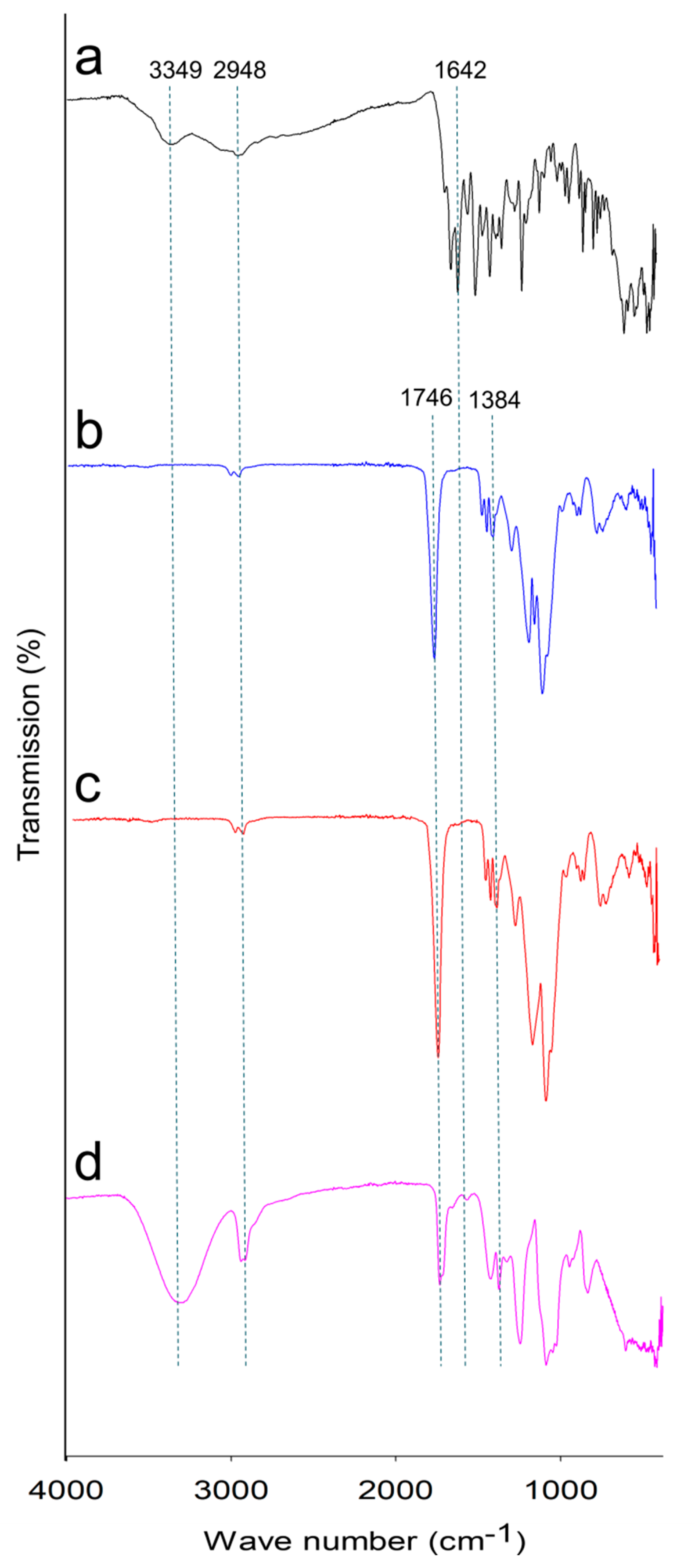
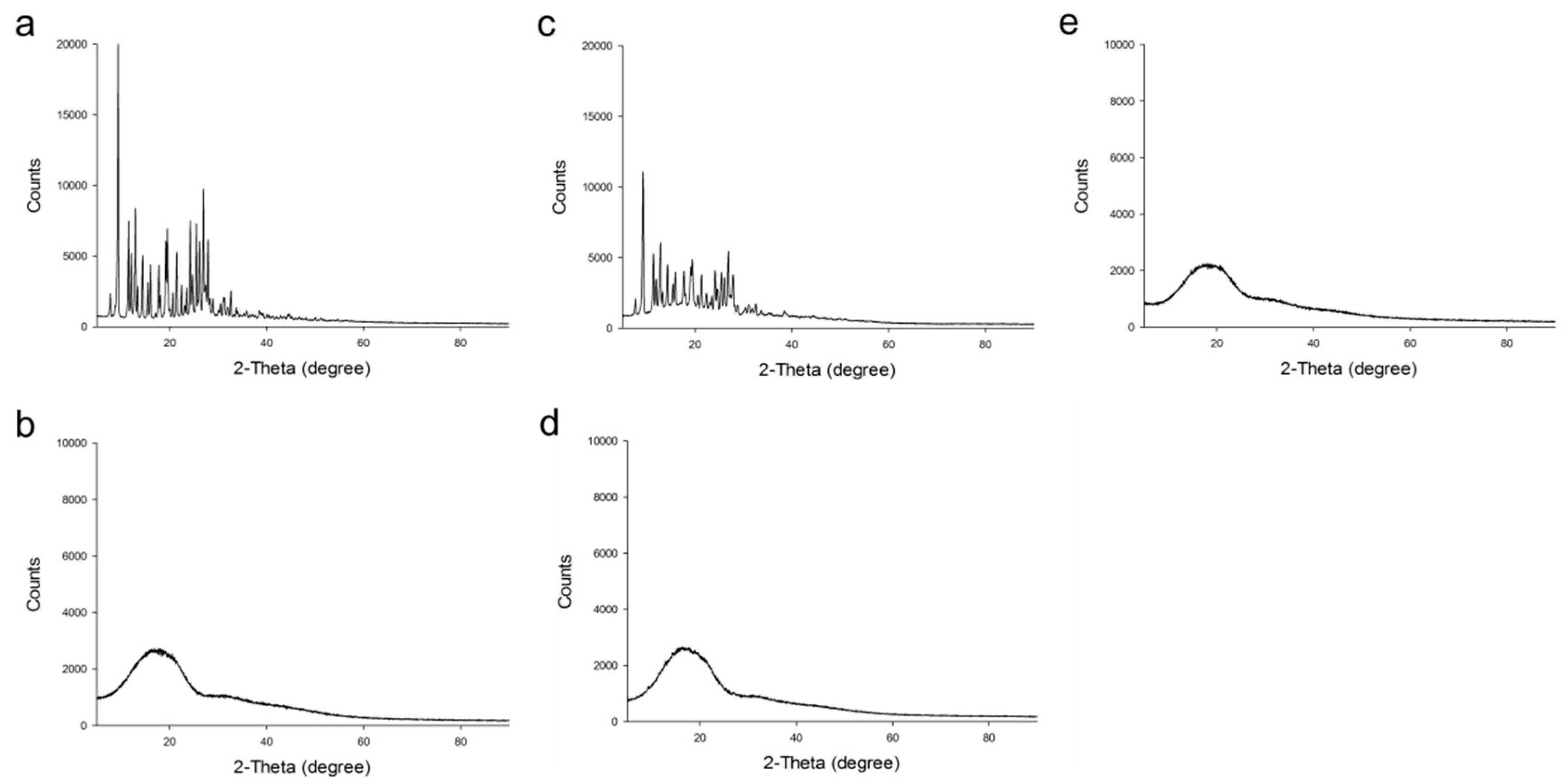
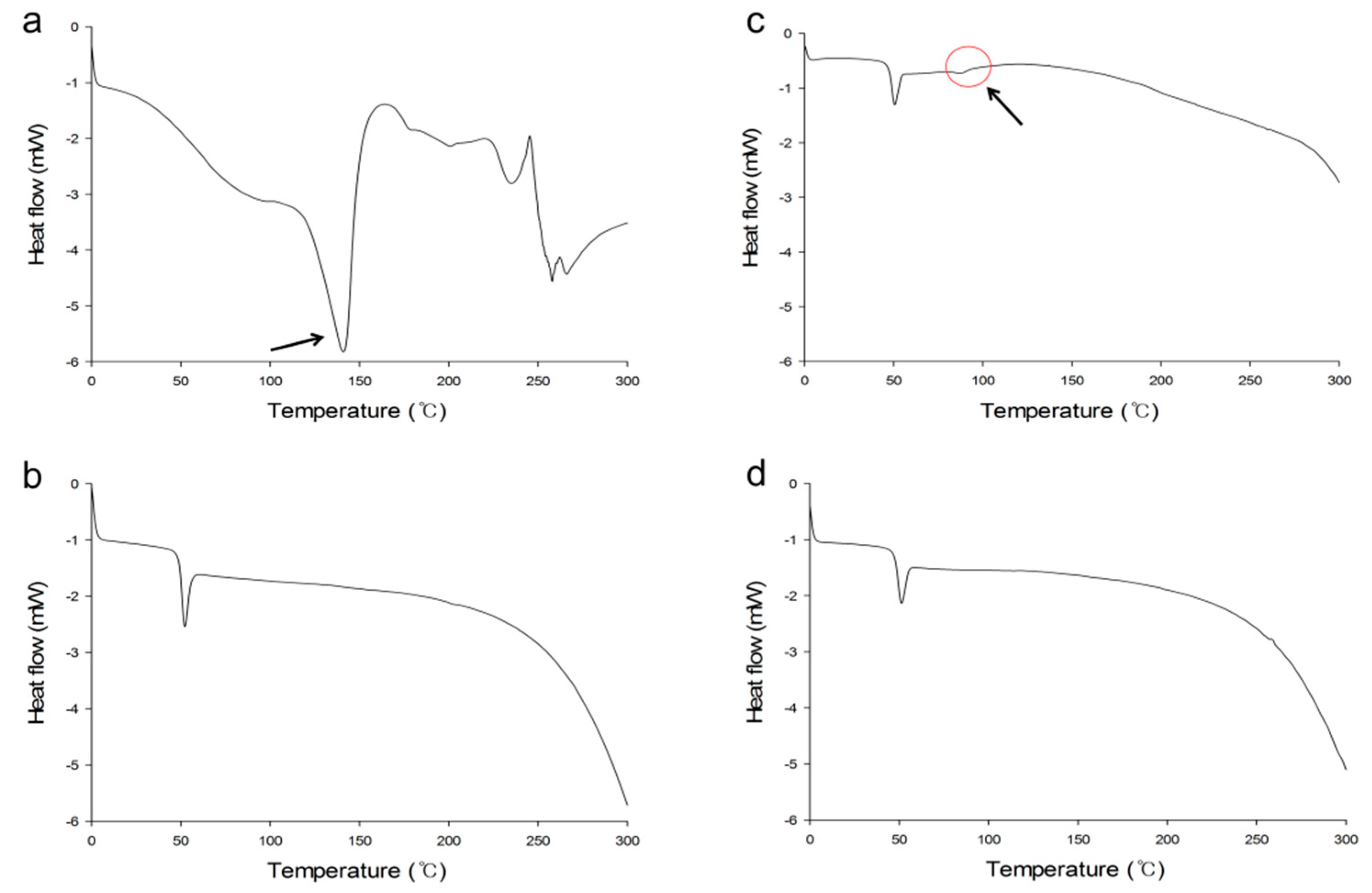
2.2.2. Fourier-Transform Infrared (FTIR) Spectroscopy, X-ray Diffraction (XRD) Analysis and Thermal Analysis
2.2.3. Morphological Studies
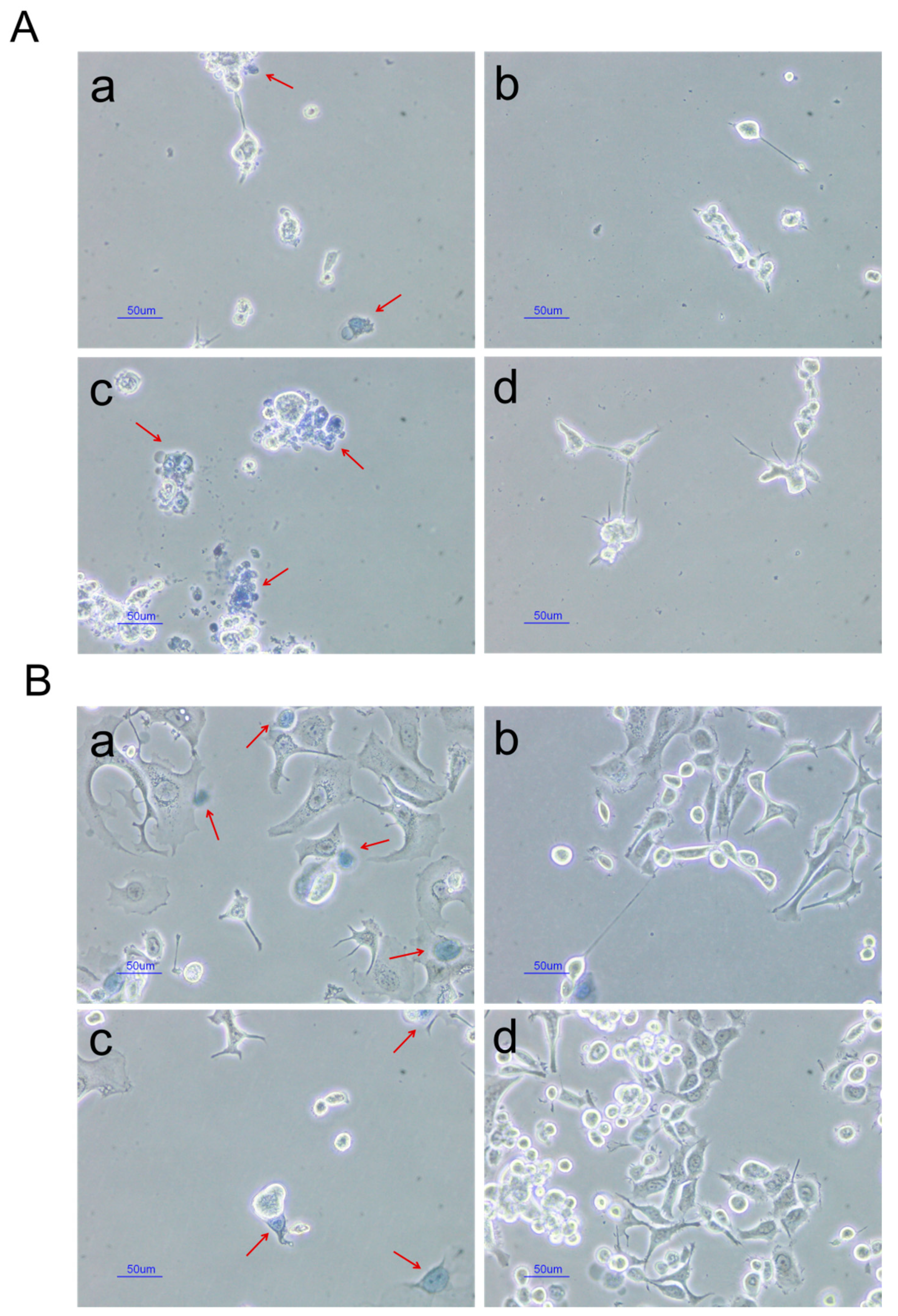
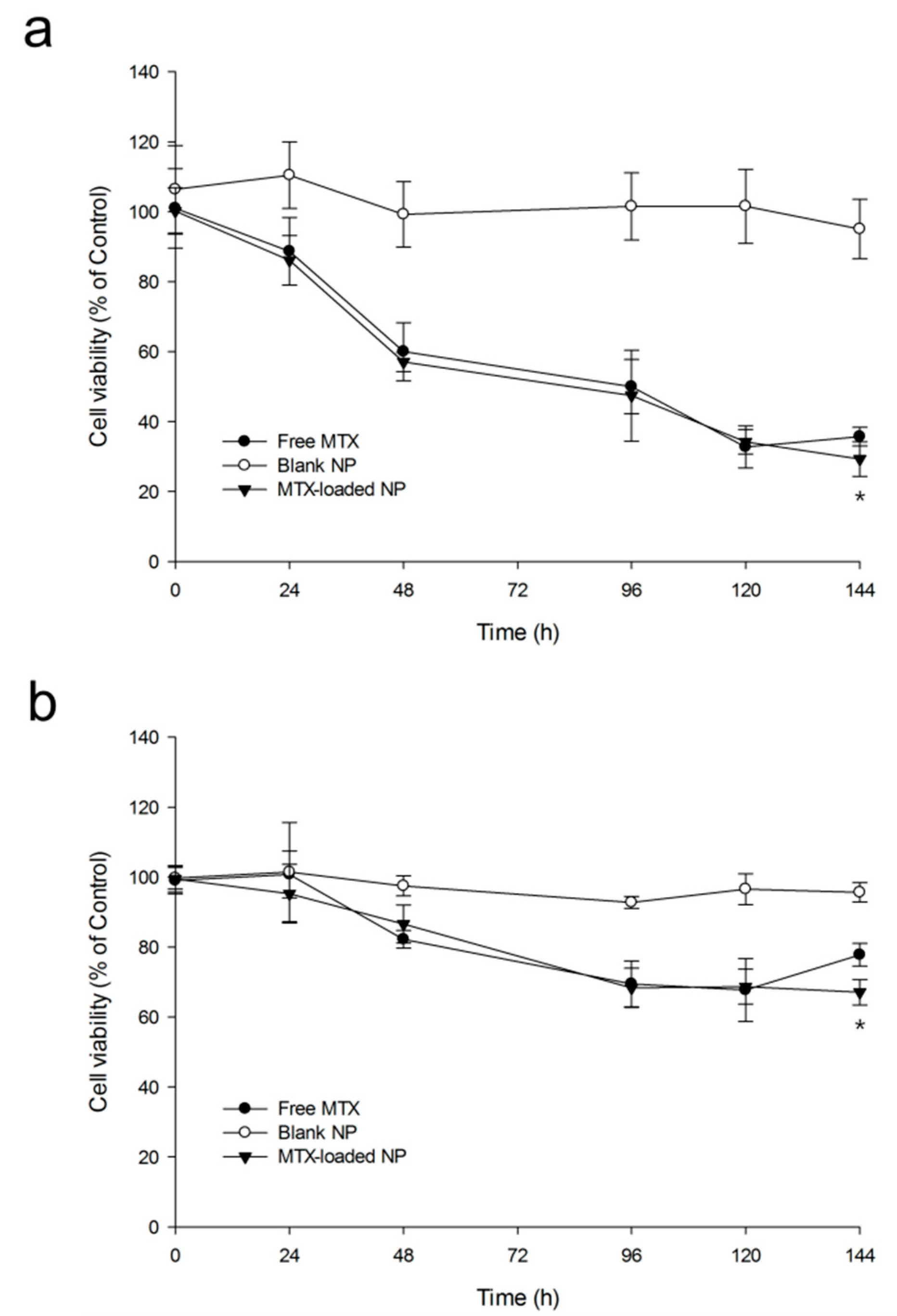
2.2.4. In Vitro Cytotoxic Studies
2.2.5. In Vitro Release Studies
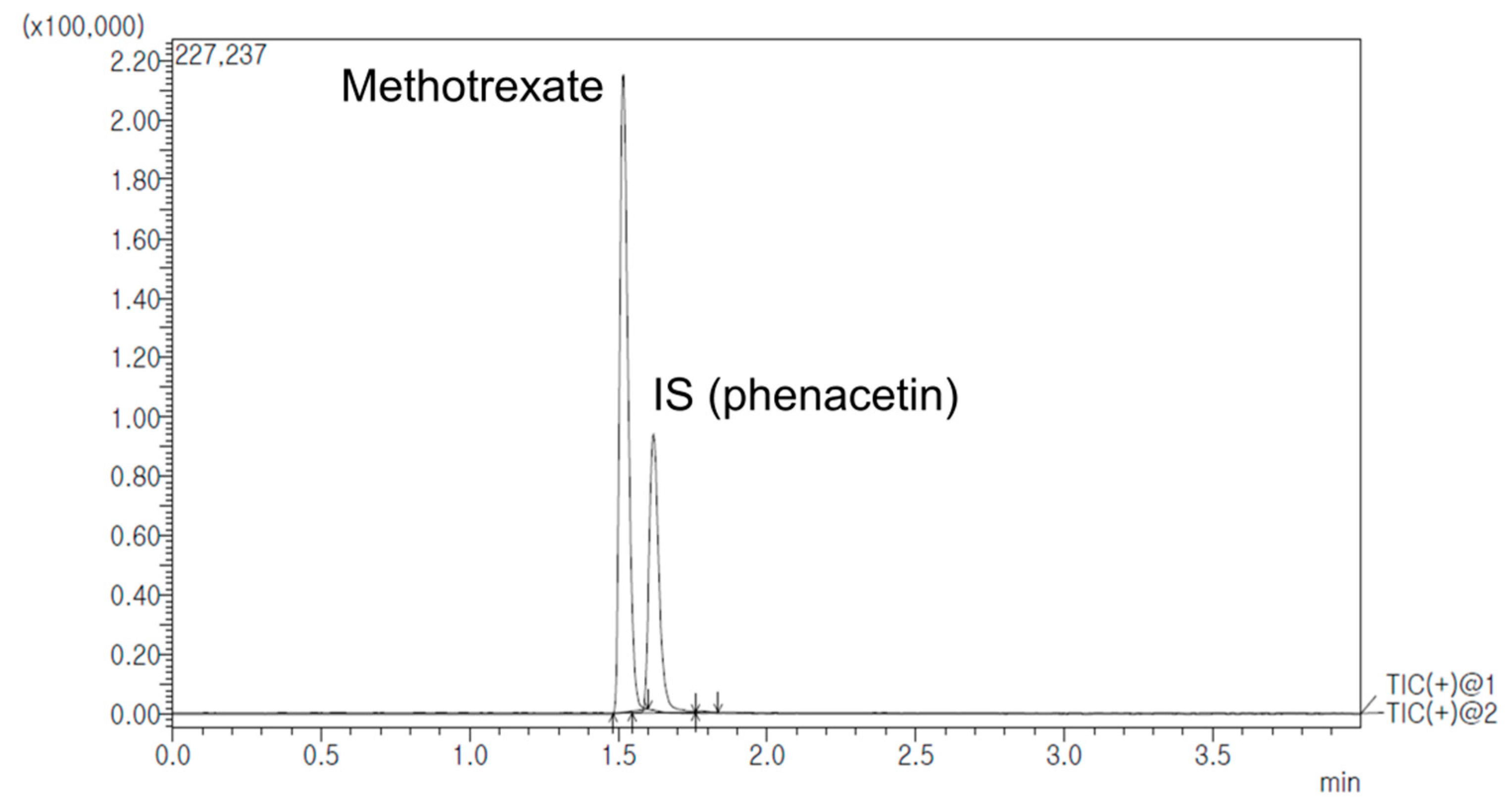
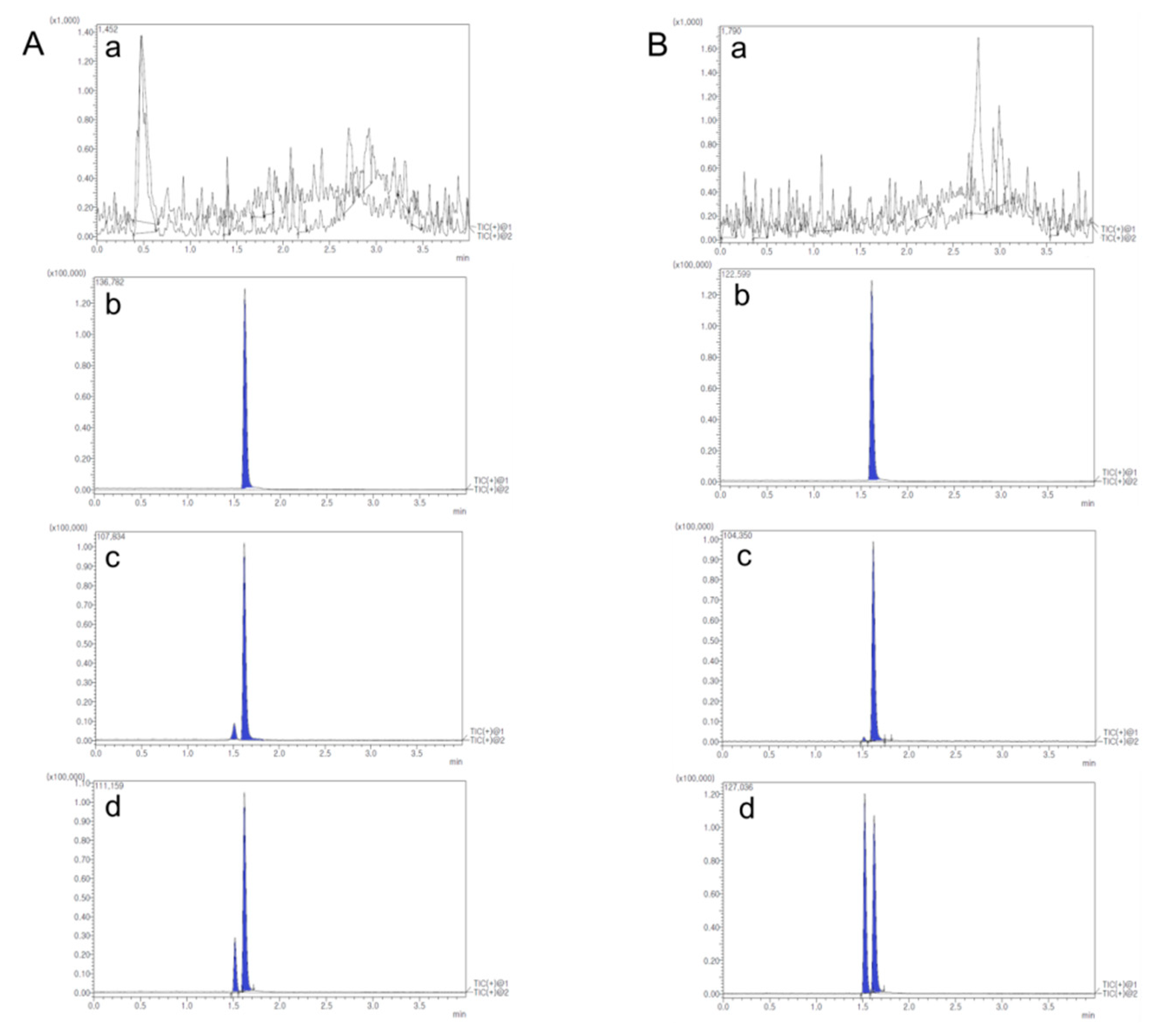
2.2.6. Determination of MTX in Rat Plasma, Lymph Node and Tissues
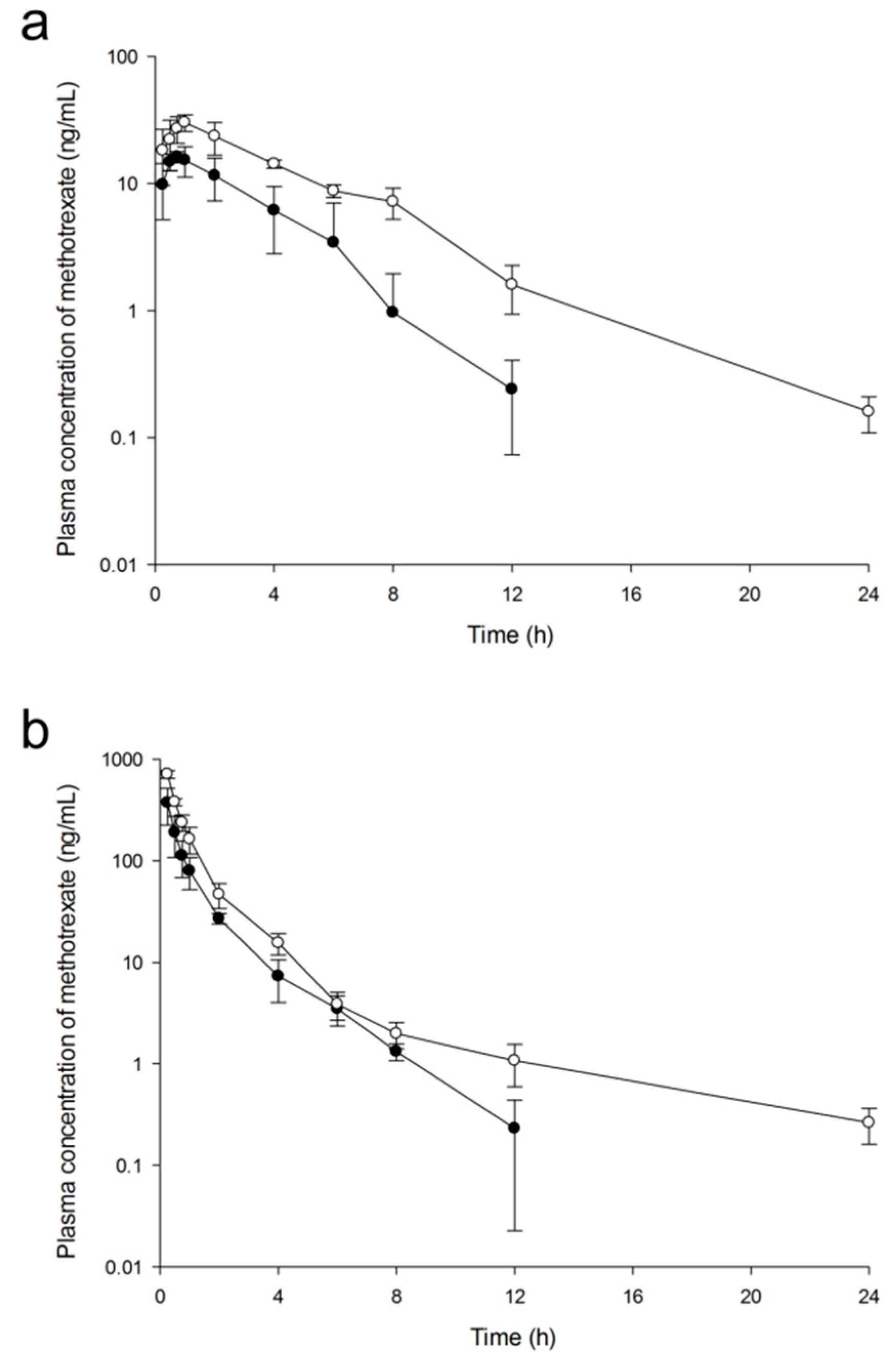
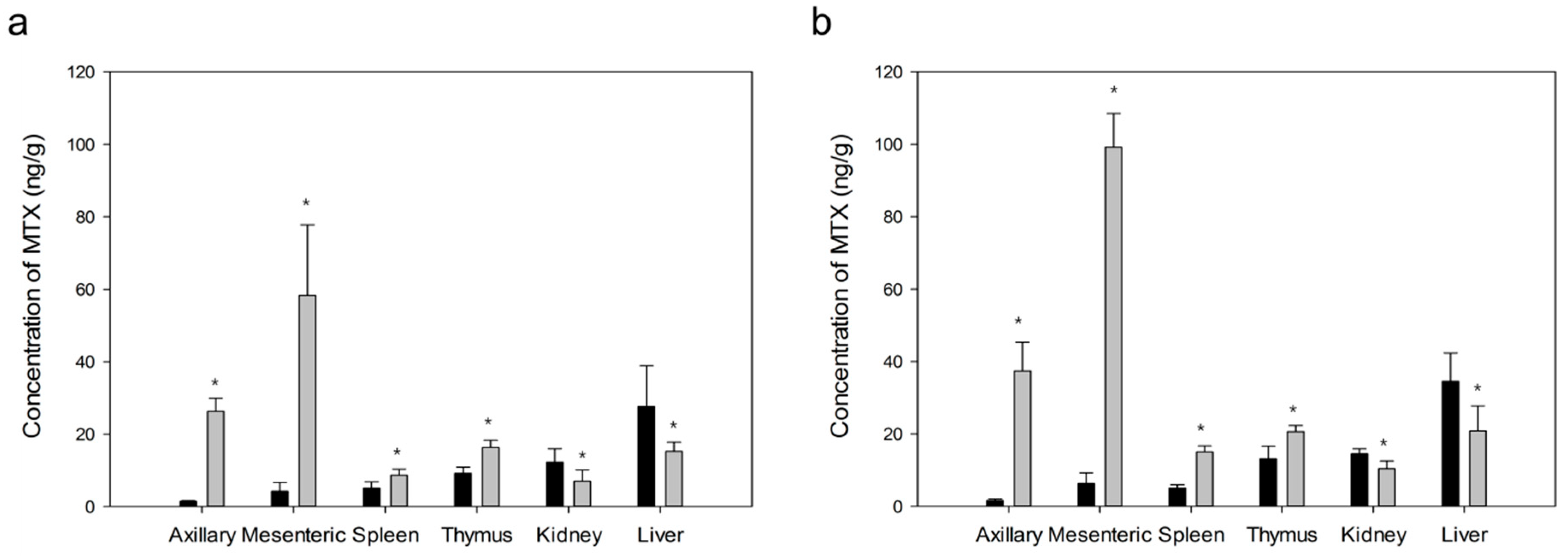
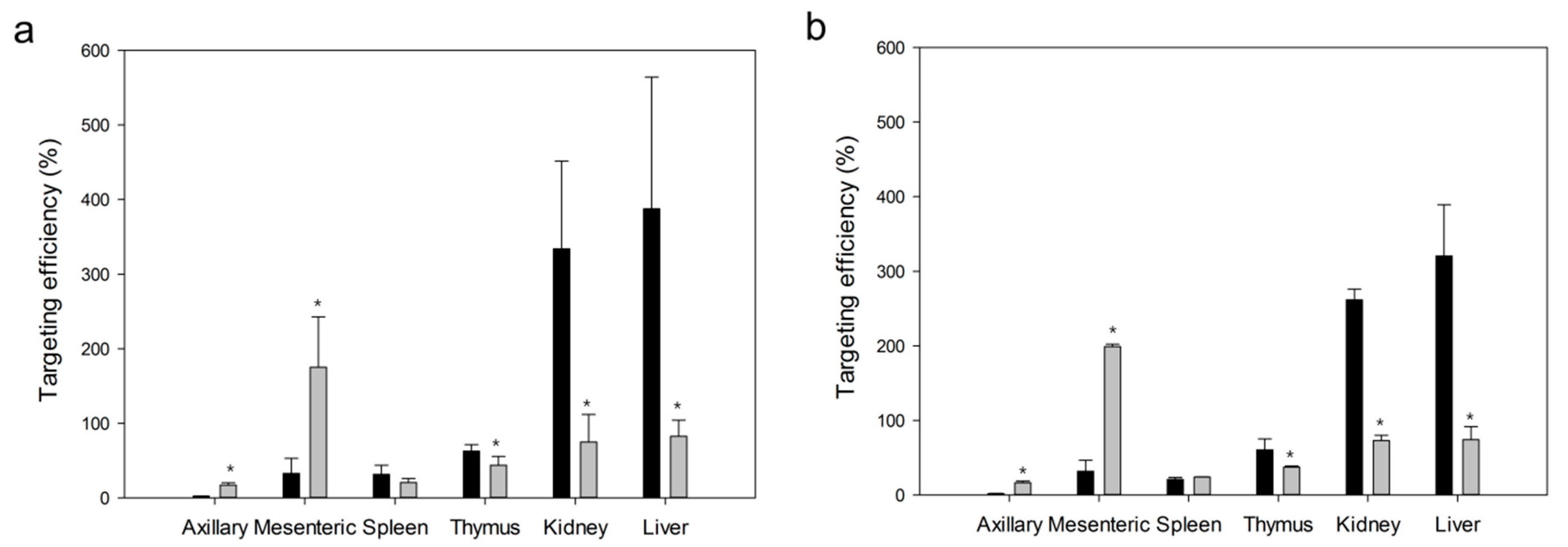
2.2.7. Pharmacokinetic Analysis and Targeting Delivery Evaluation
3. Materials and Methods
3.1. Materials
3.2. Preparation of MTX-loaded NPs
3.2.1. PLGA Concentration in the Internal Oil Phase
3.2.2. PVA Concentration in the External Aqueous Phase
3.2.3. The Oil−water Phase Volume Ratio
3.3. Characterization and Evaluation of NPs
3.3.1. Particle Size and Zeta Potential
3.3.2. Drug Encapsulation Efficiency and Drug Loading
3.3.3. Fourier-Transform Infrared (FTIR) Spectroscopy, X-ray Diffraction (XRD) Analysis and Thermal Analysis
3.3.4. Morphological Studies
3.3.5. In Vitro Release Studies
3.3.6. In Vitro Cytotoxicity Studies
Trypan Blue Assay
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) Assay
Cellular Folates Analysis
3.3.7. In Vivo Studies
3.3.8. Determination of MTX in Rat Plasma, Lymph Node and Tissues
3.3.9. Pharmacokinetic Analysis and Targeting Delivery Evaluation
3.3.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nath, B.; Barbhuiya, T. Studies on the density and surface area of nanoparticles from Camellia sinensis, A natural source. J. Chem. Pharm. Res. 2014, 11, 608–610. [Google Scholar]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: A review on formulation technology, types and applications toward targeted drug delivery. Nanomed. Nanotechnol. 2010, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Purcell, W.T.; Ettinger, D.S. Novel antifolate drugs. Curr. Oncol. Rep. 2003, 5, 114–125. [Google Scholar] [CrossRef]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef]
- Braun, J.; Rau, R. An update on methotrexate. Curr. Opin. Rheumatol. 2009, 21, 216–223. [Google Scholar] [CrossRef]
- Setoguchi, S.; Solomon, D.H.; Weinblatt, M.E.; Katz, J.N.; Avorn, J.; Glynn, R.J.; Cook, E.F.; Carney, G.; Schneeweiss, S. Tumor necrosis factor α antagonist use and cancer in patients with rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2757–2764. [Google Scholar] [CrossRef]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernäs, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Genestier, L.; Paillot, R.; Quemeneur, L.; Izeradjene, K.; Revillard, J.P. Mechanisms of action of methotrexate. Immunopharmacology 2000, 47, 247–257. [Google Scholar] [CrossRef]
- Oh, J.M.; Park, M.; Kim, S.T.; Jung, J.Y.; Kang, Y.G.; Choy, J.H. Efficient delivery of anticancer drug MTX through MTX-LDH nanohybrid system. J. Phys. Chem. Solids 2006, 67, 1024–1027. [Google Scholar] [CrossRef]
- Bailey, L.B.; Gregory, I.J.F. Folate metabolism and requirements. J. Nutr. 1999, 129, 779–782. [Google Scholar] [CrossRef] [PubMed]
- van Roon, E.; van de Laar, M. Methotrexate bioavailability. Clin. Exp. Rheumatol. 2010, 28, S27–S32. [Google Scholar] [PubMed]
- Iqbal, M.P. Accumulation of methotrexate in human tissues following high-dose methotrexate therapy. J. Pak. Med. Assoc. 1998, 48, 341–342. [Google Scholar] [PubMed]
- Visser, K.; Van der Heijde, D. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: A systematic review of the literature. Clin. Exp. Rheumatol. 2009, 27, 1017–1025. [Google Scholar] [PubMed]
- Jadhav, P.; Bothiraja, C.; Pawar, A. Methotrexate-loaded nanomixed micelles: Formulation, characterization, bioavailability, safety, and in vitro anticancer study. J. Pharm. Innov. 2018, 13, 213–225. [Google Scholar] [CrossRef]
- Pignatello, R.; Puleo, A.; Puglisi, G.; Vicari, L.; Messina, A. Effect of liposomal delivery on in vitro antitumor activity of lipophilic conjugates of methotrexate with lipoamino acids. Drug Deliv. 2003, 10, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.I.; Seo, D.H.; Kim, D.G.; Choi, C.; Jang, M.K.; Nah, J.W.; Park, Y. Methotrexate-incorporated polymeric micelles composed of methoxy poly(ethylene glycol)-grafted chitosan. Macromol. Res. 2009, 17, 538–543. [Google Scholar] [CrossRef]
- de Oliveira, E.G.; Machado, P.R.L.; Farias, K.J.S.; da Costa, T.R.; Melo, D.M.A.; Lacerda, A.F.; de Freitas Fernandes-Pedrosa, M.; Cornélio, A.M.; da Silva-Junior, A.A. Tailoring structural properties of spray-dried methotrexate-loaded poly (lactic acid)/poloxamer microparticle blends. J. Mater. Sci. Mater. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.M.; Rao, K.P. Preparation and characterization of pH-sensitive proteinoid microspheres for the oral delivery of methotrexate. Biomaterials 1998, 19, 725–732. [Google Scholar] [CrossRef]
- Dhanka, M.; Shetty, C.; Srivastava, R. Injectable methotrexate loaded polycaprolactone microspheres: Physicochemical characterization, biocompatibility, and hemocompatibility evaluation. Mater. Sci. Eng. C 2017, 81, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Karasulu, H.Y.; Karabulut, B.; Göker, E.; Güneri, T.; Gabor, F. Controlled release of methotrexate from W/O microemulsion and its in vitro antitumor activity. Drug Deliv. 2007, 14, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.E.; Rial-Hermida, M.I.; Ramirez, J.C.; Pazos, A.; Concheiro, A.; Alvarez-Lorenzo, C.; Peralta, R.D. Microemulsions for colorectal cancer treatments. General considerations and formulation of methotrexate. Mini-Rev. Med. Chem. 2016, 16, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Rai, S.; Vaidya, B.; Khatri, K.; Goyal, A.K.; Mishra, N.; Mehta, A.; Vyas, S.P. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine 2009, 5, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Barth, R.F.; Yang, W.; Kawabata, S.; Zhang, L.; Green-Church, K. Targeted delivery of methotrexate to epidermal growth factor receptor–positive brain tumors by means of cetuximab (IMC-C225) dendrimer bioconjugates. Mol. Cancer Ther. 2006, 5, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, H.; Adinehvand, R.; Manjili, H.K.; Rostamizadeh, K.; Danafar, H. Synthesis, characterization, and kinetic release study of methotrexate loaded mPEG–PCL polymersomes for inhibition of MCF-7 breast cancer cell line. Pharm. Dev. Technol. 2019, 24, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.R.; Jones, D.; Jung, K.; Padera, T.P. The lymph node microenvironment and its role in the progression of metastatic cancer. Semin. Cell Dev. Biol. 2015, 38, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.Y.; Lee, Y.B. Nano-sized drug delivery systems for lymphatic delivery. J. Nanosci. Nanotechnol. 2014, 14, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Tanis, P.J.; Nieweg, O.E.; Valdés Olmos, R.A.; Kroon, B.B.R. Anatomy and physiology of lymphatic drainage of the breast from the perspective of sentinel node biopsy. J. Am. Coll. Surg. 2001, 192, 399–409. [Google Scholar] [CrossRef]
- Khan, A.A.; Mudassir, J.; Mohtar, N.; Darwis, Y. Advanced drug delivery to the lymphatic system: Lipid-based nanoformulations. Int. J. Nanomedicine 2013, 8, 2733–2744. [Google Scholar]
- Jeong, S.H.; Jang, J.H.; Cho, H.Y.; Lee, Y.B. Soft- and hard-lipid nanoparticles: A novel approach to lymphatic drug delivery. Arch. Pharm. Res. 2018, 41, 797–814. [Google Scholar] [CrossRef] [PubMed]
- Afshari, M.; Derakhshandeh, K.; Hosseinzadeh, L. Characterisation, cytotoxicity and apoptosis studies of methotrexate-loaded PLGA and PLGA-PEG nanoparticles. J. Microencapsul. 2014, 31, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Musmade, K.P.; Deshpande, P.B.; Musmade, P.B.; Maliyakkal, M.N.; Kumar, A.R.; Reddy, M.S.; Udupa, N. Methotrexate-loaded biodegradable nanoparticles: Preparation, characterization and evaluation of its cytotoxic potential against U-343 MGa human neuronal glioblastoma cells. Bull. Mater. Sci. 2014, 37, 945–951. [Google Scholar] [CrossRef]
- Gorjikhah, F.; Azizi Jalalian, F.; Salehi, R.; Panahi, Y.; Hasanzadeh, A.; Alizadeh, E.; Akbarzadeh, A.; Davaran, S. Preparation and characterization of PLGA-β-CD polymeric nanoparticles containing methotrexate and evaluation of their effects on T47D cell line. Artif. Cells Nanomed. Biotechnol. 2017, 45, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Maincent, P.; Dellacherie, E.; Alonso, M.J.; Labrude, P.; Vigneron, C. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J. Control. Release 1998, 50, 31–40. [Google Scholar] [CrossRef]
- Murakami, H.; Kawashima, Y.; Niwa, T.; Hino, T.; Takeuchi, H.; Kobayashi, M. Influence of the degrees of hydrolyzation and polymerization of poly(vinylalcohol) on the preparation and properties of poly(dl-lactide-co-glycolide) nanoparticle. Int. J. Pharm. 1997, 149, 43–49. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Bugno, J.; Lantvit, D.; Burdette, J.E.; Hong, S. Prolonged blood circulation and enhanced tumor accumulation of folate-targeted dendrimer-polymer hybrid nanoparticles. J. Control. Release 2014, 191, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzer, C.; Dirin, M.; Winkler, A.-M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kaszuba, M.; Corbett, J.; Watson, F.M.; Jones, A. High-concentration zeta potential measurements using light-scattering techniques. Phil. Trans. R. Soc. A 2010, 368, 4439–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, D.H. Basic Principles of Colloid Science; Royal Society of Chemistry: London, UK, 2007. [Google Scholar]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Avasatthi, V.; Pawar, H.; Dora, C.P.; Bansod, P.; Gill, M.S.; Suresh, S. A novel nanogel formulation of methotrexate for topical treatment of psoriasis: Optimization, in vitro and in vivo evaluation. Pharm. Dev. Technol. 2016, 21, 554–562. [Google Scholar] [PubMed]
- El-Houssiny, A.S.; Ward, A.A.; Mostafa, D.M.; Abd-El-Messieh, S.L.; Abdel-Nour, K.N.; Darwish, M.M.; Khalil, W.A. Drug–polymer interaction between glucosamine sulfate and alginate nanoparticles: FTIR, DSC and dielectric spectroscopy studies. Adv. Nat. Sci. Nanosci. 2016, 7. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Avelar-Freitas, B.A.; Almeida, V.G.; Pinto, M.C.X.; Mourão, F.A.G.; Massensini, A.R.; Martins-Filho, O.A.; Rocha-Vieira, E.; Brito-Melo, G.E.A. Trypan blue exclusion assay by flow cytometry. Braz. J. Med. Biol. Res. 2014, 47, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Tennant, J.R. Evaluation of the trypan blue technique for determination of cell viability. Transplantation 1964, 2, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Mitchell, J.B. Viability measurements in mammalian cell systems. Anal. Biochem. 1989, 179, 1–7. [Google Scholar] [CrossRef]
- Wessels, J.A.M.; Huizinga, T.W.J.; Guchelaar, H.-J. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology 2007, 47, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Shibata, H.; Izutsu, K.-I.; Yomota, C.; Okuda, H.; Goda, Y. Investigation of factors affecting in vitro doxorubicin release from PEGylated liposomal doxorubicin for the development of in vitro release testing conditions. Drug Dev. Ind. Pharm. 2015, 41, 1376–1386. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Mei, S.; Zhu, L.; Li, X.; Wang, J.; Jiang, X.; Chen, H.; Huo, J.; Yang, L.; Lin, S.; Zhao, Z. UPLC-MS/MS analysis of methotrexate in human plasma and comparison with the fluorescence polarization immunoassay. Anal. Sci. 2017, 33, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Giri, K.; Dhiman, V.; Dixit, A.; Zainuddin, M.; Mullangi, R. A validated LC-MS/MS assay for simultaneous quantification of methotrexate and tofacitinib in rat plasma: Application to a pharmacokinetic study. Biomed. Chromatogr. 2015, 29, 722–732. [Google Scholar] [CrossRef] [PubMed]
- FDA. Bioanalytical Method Validation Guidance for Industry; U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM): Rockville, MD, USA, 2018.
- Kuroda, T.; Namba, K.; Torimaru, T.; Yamamoto, S.; Akahori, M. Variability of oral bioavailability for low dose methotrexate in rats. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 227–234. [Google Scholar] [CrossRef]
- Kuroda, T.; Namba, K.; Torimaru, T.; Kawashima, K.; Hayashi, M. Species differences in oral bioavailability of methotrexate between rats and monkeys. Biol. Pharm. Bull. 2000, 23, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Parmar, A.; Kori, S.; Sandhir, R. PLGA-based nanoparticles: A new paradigm in biomedical applications. Trends Analyt. Chem. 2016, 80, 30–40. [Google Scholar] [CrossRef]
- Porter, C.J. Drug delivery to the lymphatic system. Crit. Rev. Ther. Drug Carrier Syst. 1997, 14, 333–393. [Google Scholar] [PubMed]
- Singh, I.; Swami, R.; Khan, W.; Sistla, R. Lymphatic system: A prospective area for advanced targeting of particulate drug carriers. Expert Opin. Drug Deliv. 2014, 11, 211–229. [Google Scholar] [CrossRef]
- Moghimi, S. Nanoparticle engineering for the lymphatic system and lymph node targeting. In Polymer-Based Nanostructures: Medical Applications; Broz, P., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2010; pp. 81–97. [Google Scholar]
- Madani, F.; Goodarzi, A.; Hashemi, M.; Mujokoro, B.; Khosravani, M.; Adabi, M. Preparation of methotrexate loaded PLGA nanoparticles coated with PVA and Poloxamer188. Nanomed. Res. J. 2018, 3, 19–24. [Google Scholar]
- Tahir, N.; Madni, A.; Balasubramanian, V.; Rehman, M.; Correia, A.; Kashif, P.M.; Mäkilä, E.; Salonen, J.; Santos, H.A. Development and optimization of methotrexate-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery applications. Int. J. Pharm. 2017, 533, 156–168. [Google Scholar] [CrossRef]
- Vakilinezhad, M.A.; Alipour, S.; Montaseri, H. Fabrication and in vitro evaluation of magnetic PLGA nanoparticles as a potential Methotrexate delivery system for breast cancer. J. Drug Deliv. Sci. Technol. 2018, 44, 467–474. [Google Scholar] [CrossRef]
- Maleki, H.; Farid, D.; Mahdi, A.; Masood, K.; Arzani, H.; Morteza, K. Methotrexate-loaded PLGA nanoparticles: Preparation, characterization and their cytotoxicity effect on human glioblastoma U87MG cells. Int. J. Med. Nano Res. 2017. [Google Scholar] [CrossRef]
- Muntoni, E.; Martina, K.; Marini, E.; Giorgis, M.; Lazzarato, L.; Salaroglio, I.C.; Riganti, C.; Lanotte, M.; Battaglia, L. Methotrexate-loaded solid lipid nanoparticles: Protein functionalization to improve brain biodistribution. Pharmaceutics 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Jain, A.; Jain, A.; Jain, A.K.; Garg, N.K.; Tekade, R.K.; Raj Singh, T.R.; Iyer, A.K. Cationic bovine serum albumin (CBA) conjugated poly lactic-co-glycolic acid (PLGA) nanoparticles for extended delivery of methotrexate into brain tumors. RSC Adv. 2016, 6, 89040–89050. [Google Scholar] [CrossRef] [Green Version]
- Parmar, A.; Jain, A.; Uppal, S.; Mehta, S.K.; Kaur, K.; Singh, B.; Sandhir, R.; Sharma, S. Anti-proliferate and apoptosis triggering potential of methotrexate-transferrin conjugate encapsulated PLGA nanoparticles with enhanced cellular uptake by high-affinity folate receptors. Artif. Cells Nanomed. Biotechnol. 2018, 46, 704–719. [Google Scholar] [CrossRef] [Green Version]
- Giri, T.K.; Choudhary, C.; Ajazuddin; Alexander, A.; Badwaik, H.; Tripathi, D.K. Prospects of pharmaceuticals and biopharmaceuticals loaded microparticles prepared by double emulsion technique for controlled delivery. Saudi Pharm. J. 2013, 21, 125–141. [Google Scholar] [CrossRef]
- Shin, S.B.; Cho, H.Y.; Kim, D.D.; Choi, H.G.; Lee, Y.B. Preparation and evaluation of tacrolimus-loaded nanoparticles for lymphatic delivery. Eur. J. Pharm. Biopharm. 2010, 74, 164–171. [Google Scholar] [CrossRef]
- Washington, C. Drug release from microdisperse systems: A critical review. Int. J. Pharm. 1990, 58, 1–12. [Google Scholar] [CrossRef]
- Chen, L.; Ducker, G.S.; Lu, W.; Teng, X.; Rabinowitz, J.D. An LC-MS chemical derivatization method for the measurement of five different one-carbon states of cellular tetrahydrofolate. Anal. Bioanal. Chem. 2017, 409, 5955–5964. [Google Scholar] [CrossRef]
- Odin, E.; Wettergren, Y.; Carlsson, G.; Gustavsson, B. Determination of reduced folates in tumor and adjacent mucosa of colorectal cancer patients using LC-MS/MS. Biomed. Chromatogr. 2013, 27, 487–495. [Google Scholar] [CrossRef]
- Malaviya, A.N.; Sharma, A.; Agarwal, D.; Kapoor, S.; Garg, S.; Sawhney, S. Low-dose and high-dose methotrexate are two different drugs in practical terms. Int. J. Rheum. Dis. 2010, 13, 288–293. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA Conc. (%, w/v) | PVA Conc. (%, w/v) | Oil:Water Phase Ratio (v/v) | Nanoparticle Size (nm) |
|---|---|---|---|
| 0.5 | 10 | 1:3 | 110.5 ± 8.38 |
| 1 | 10 | 1:3 | 117.3 ± 9.54 |
| 2 | 10 | 1:3 | 152.7 ± 12.17 * |
| 4 | 10 | 1:3 | 276.7 ± 19.25 * |
| 6 | 10 | 1:3 | 456.2 ± 23.03 * |
| 8 | 10 | 1:3 | 873.8 ± 30.11 * |
| PVA Conc. (%, w/v) | PLGA Conc. (%, w/v) | Oil–Water Phase Ratio (v/v) | Nanoparticle Size (nm) |
|---|---|---|---|
| 1 | 4 | 1:3 | 135.5 ± 7.92 |
| 2.5 | 4 | 1:3 | 167.9 ± 10.33 * |
| 5 | 4 | 1:3 | 212.8 ± 13.20 * |
| 7.5 | 4 | 1:3 | 269.5 ± 15.09 * |
| 10 | 4 | 1:3 | 313.8 ± 15.86 * |
| 15 | 4 | 1:3 | 404.3 ± 20.42 * |
| Oil:Water Phase Ratio (v/v) | PLGA Conc. (%, w/v) | PVA Conc. (%, w/v) | Nanoparticle Size (nm) |
|---|---|---|---|
| 1:1 | 0.5 | 1 | 332.3 ± 15.56 |
| 1:2 | 0.5 | 1 | 201.2 ± 20.25 * |
| 1:3 | 0.5 | 1 | 103.0 ± 11.74 * |
| 1:4 | 0.5 | 1 | 199.3 ± 14.38 * |
| 1:5 | 0.5 | 1 | 254.3 ± 18.31 * |
| 1:6 | 0.5 | 1 | 315.5 ± 17.90 * |
| Methotrexate Amount (mg) | PLGA Conc. (%, w/v) | PVA Conc. (%, w/v) | Oil–Water Phase Ratio (v/v) | Nanoparticle Size (nm) | Zeta Potential (mV) | Encapsulation Efficacy (%) | Drug Loading (%) |
|---|---|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 1:3 | 103.0 ± 11.74 * | −20.4 ± 1.54 | - | - |
| 3 | 0.5 | 1 | 1:3 | 163.7 ± 10.25 | −18.5 ± 2.28 | 93.34 ± 0.51 | 15.45 ± 0.34 |
| 6 | 0.5 | 1 | 1:3 | 206.1 ± 18.14 * | −17.3 ± 2.63 | 90.26 ± 0.63 * | 13.10 ± 0.48 * |
| 9 | 0.5 | 1 | 1:3 | 258.3 ± 15.32 * | −19.7 ± 3.10 | 91.12 ± 0.49 * | 14.33 ± 0.31 * |
| 15 | 0.5 | 1 | 1:3 | 299.2 ± 17.85 * | −18.9 ± 2.98 | 88.28 ± 1.02 * | 11.07 ± 0.85 * |
| 20 | 0.5 | 1 | 1:3 | 351.4 ± 20.10 * | −17.8 ± 3.24 | 84.31 ± 1.90 * | 9.69 ± 1.36 * |
| Intracellular Concentration (μmol/L) | ||||
|---|---|---|---|---|
| Free MTX | Blank NPs | MTX-Loaded NPs | Control | |
| CWR22Rv1 | ||||
| DHF | 4.85 ± 1.30 * | 1.05 ± 0.33 | 8.42 ± 2.17 *,# | 1.14 ± 0.38 |
| THF | 0.89 ± 0.21 * | 2.10 ± 0.64 | 0.44 ± 0.15* | 1.92 ± 0.49 |
| MTX | 1.04 ± 0.27 | - | 2.52 ± 0.61# | - |
| MCF-7 | ||||
| DHF | 2.74 ± 1.12 * | 1.50 ± 0.54 | 4.03 ± 1.76 *,# | 1.46 ± 0.60 |
| THF | 1.47 ± 0.49 * | 2.62 ± 0.79 | 1.02 ± 0.34 * | 2.59 ± 0.84 |
| MTX | 0.95 ± 0.31 | - | 2.38 ± 0.55 # | - |
| Parameters | Free MTX (5 mg/kg) | PLGA NPs (5 mg/kg as MTX) | ||
|---|---|---|---|---|
| Oral | Intravenous | Oral | Intravenous | |
| AUC0-t (ng·h/mL) | 59.73 ± 23.74 | 375.46 ± 114.61 | 152.59 ± 7.32 * | 728.17 ± 84.37 * |
| AUC0-∞ (ng·h/mL) | 60.33 ± 23.19 | 376.09 ± 115.31 | 153.29 ± 7.34 * | 730.33 ± 85.06 * |
| t1/2 (h) | 1.53 ± 0.54 | 1.56 ± 0.56 | 3.01 ± 0.28 * | 4.94 ± 0.15 * |
| Cmax (ng/mL) | 16.95 ± 2.24 | - | 31.19 ± 5.15 * | - |
| Tmax (h) | 0.75 ± 0.25 | - | 0.92 ± 0.14 | - |
| CL (L/h·kg) | - | 3.53 ± 1.03 | - | 1.73 ± 0.21 * |
| Vd (L/kg) | - | 29.84 ± 4.59 | - | 55.96 ± 9.76 * |
| Fab (%) | 16.04 | - | 20.99 * | - |
| Frb (%) | - | - | 254.09 | 194.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.-H.; Jeong, S.-H.; Lee, Y.-B. Preparation and In Vitro/In Vivo Characterization of Polymeric Nanoparticles Containing Methotrexate to Improve Lymphatic Delivery. Int. J. Mol. Sci. 2019, 20, 3312. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133312
Jang J-H, Jeong S-H, Lee Y-B. Preparation and In Vitro/In Vivo Characterization of Polymeric Nanoparticles Containing Methotrexate to Improve Lymphatic Delivery. International Journal of Molecular Sciences. 2019; 20(13):3312. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133312
Chicago/Turabian StyleJang, Ji-Hun, Seung-Hyun Jeong, and Yong-Bok Lee. 2019. "Preparation and In Vitro/In Vivo Characterization of Polymeric Nanoparticles Containing Methotrexate to Improve Lymphatic Delivery" International Journal of Molecular Sciences 20, no. 13: 3312. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133312