Oxidation of Human Copper Chaperone Atox1 and Disulfide Bond Cleavage by Cisplatin and Glutathione

 , , ,
, , ,  ,
,  and
and 
Abstract
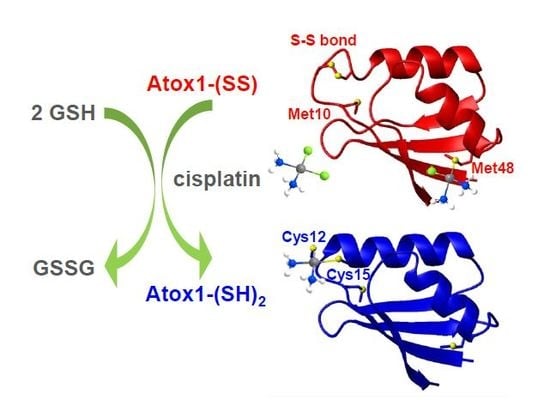
:
1. Introduction
2. Results
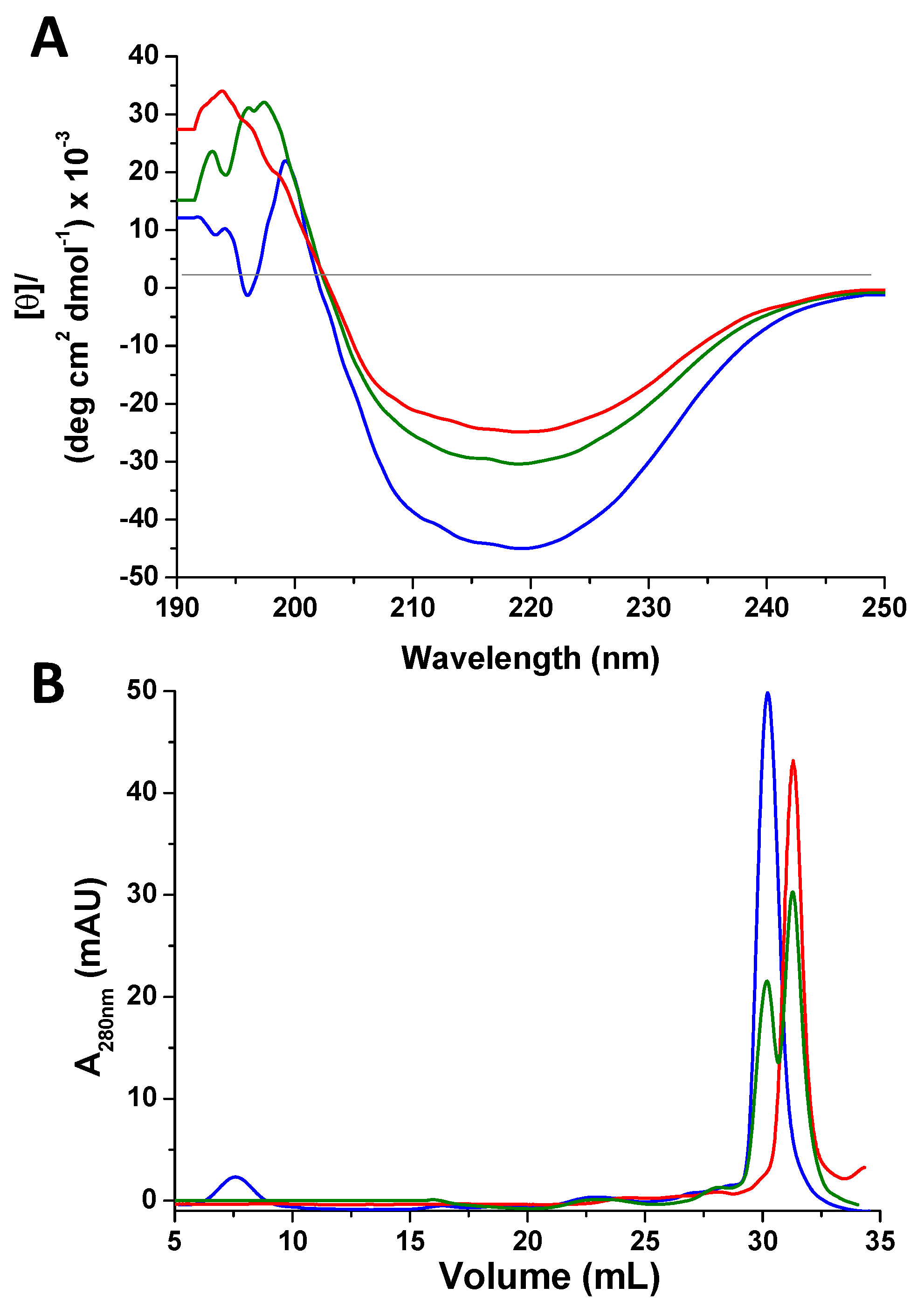
2.1. Atox1 Oxidation
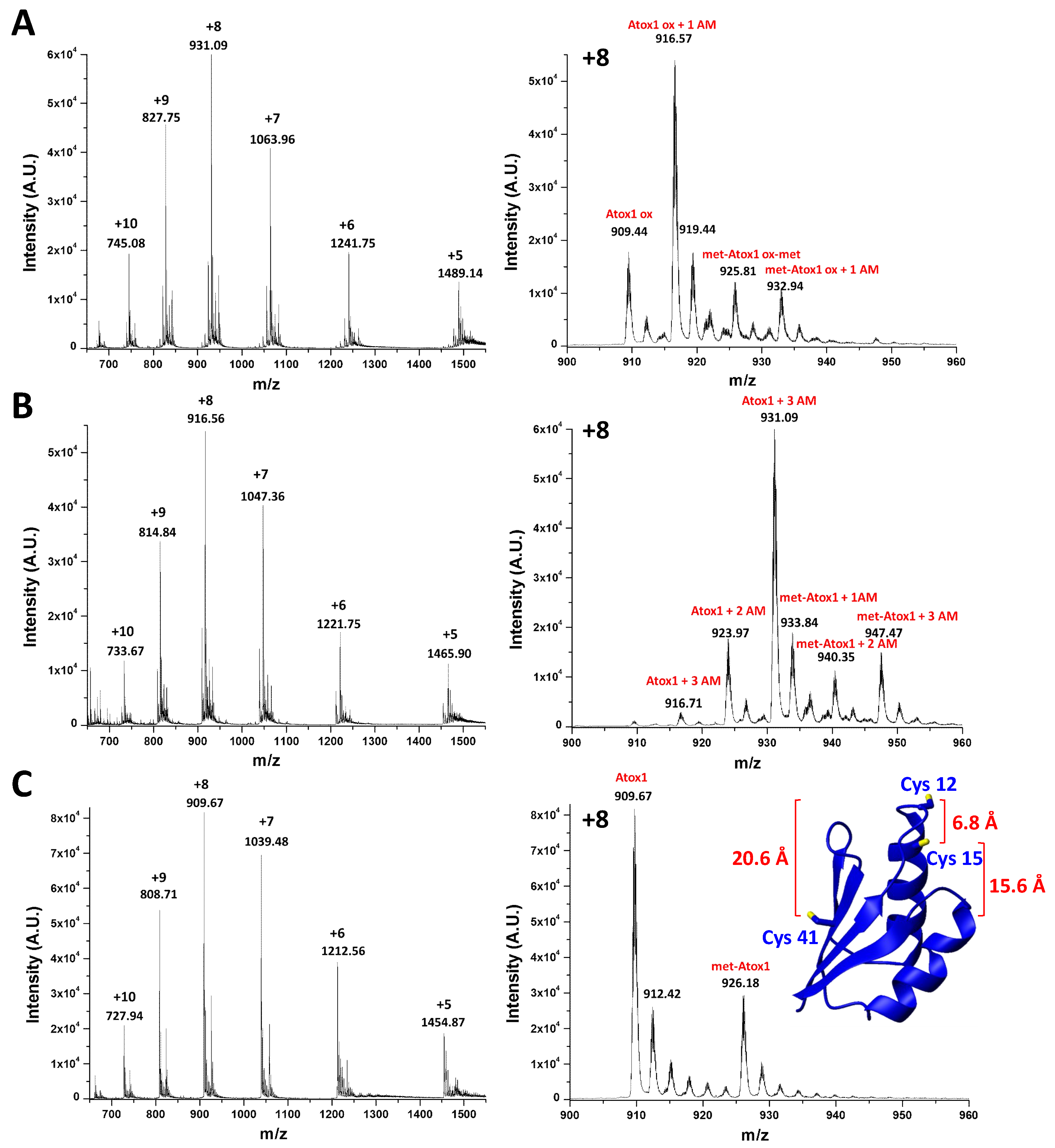
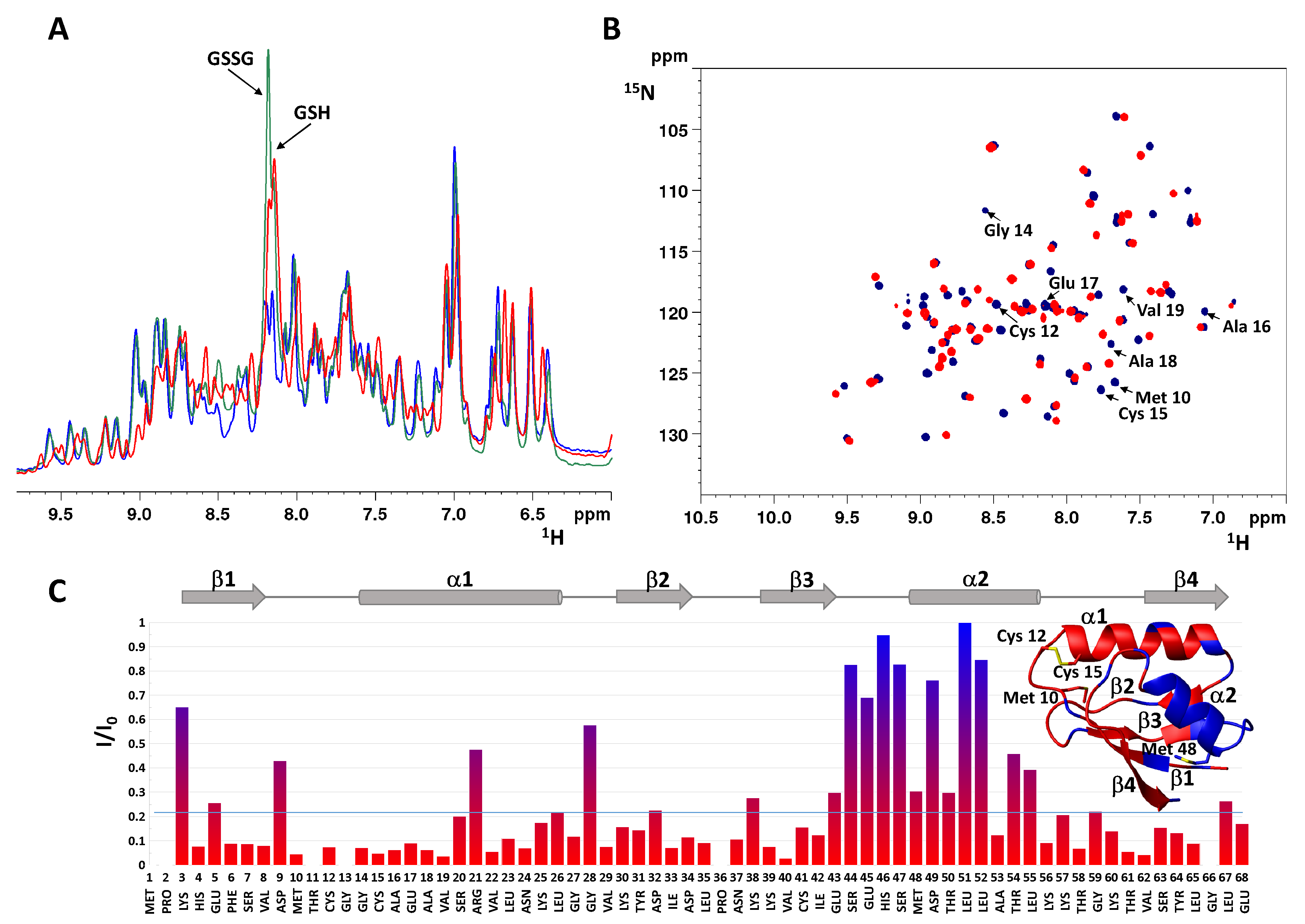
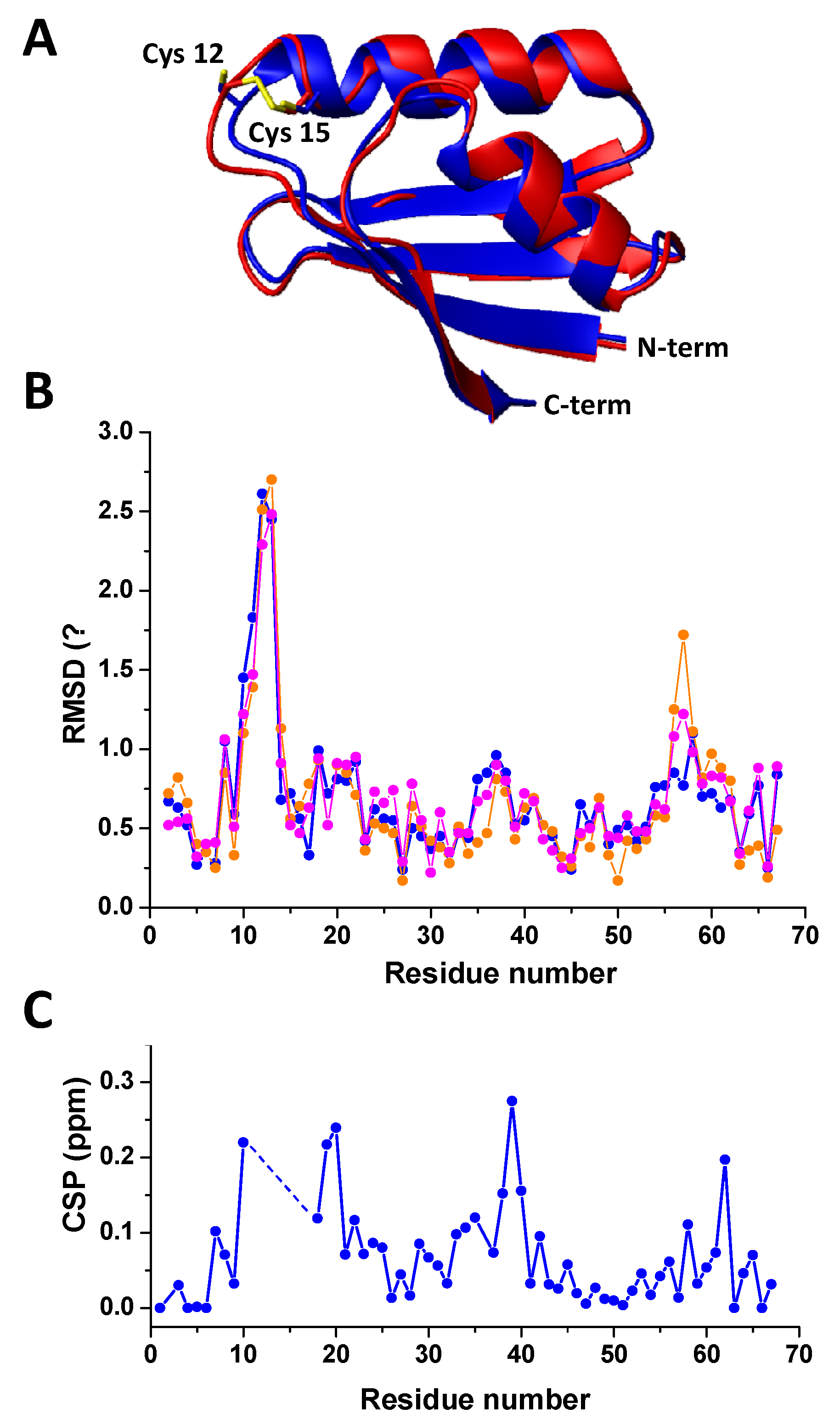
2.2. Characterization of Oxidized Atox1 in Solution
2.3. Preliminary Crystal Structure Characterization of Oxidized Atox1
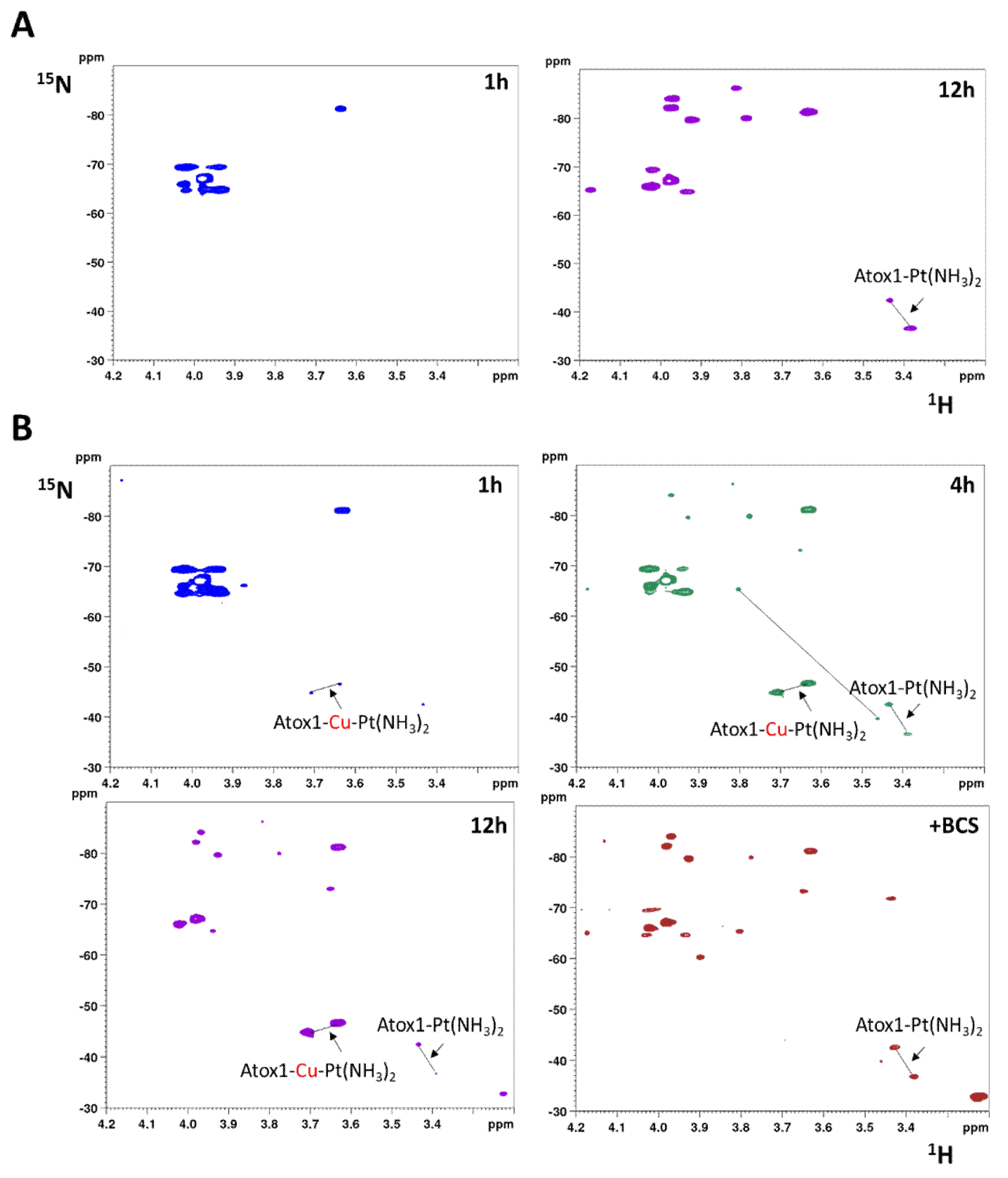
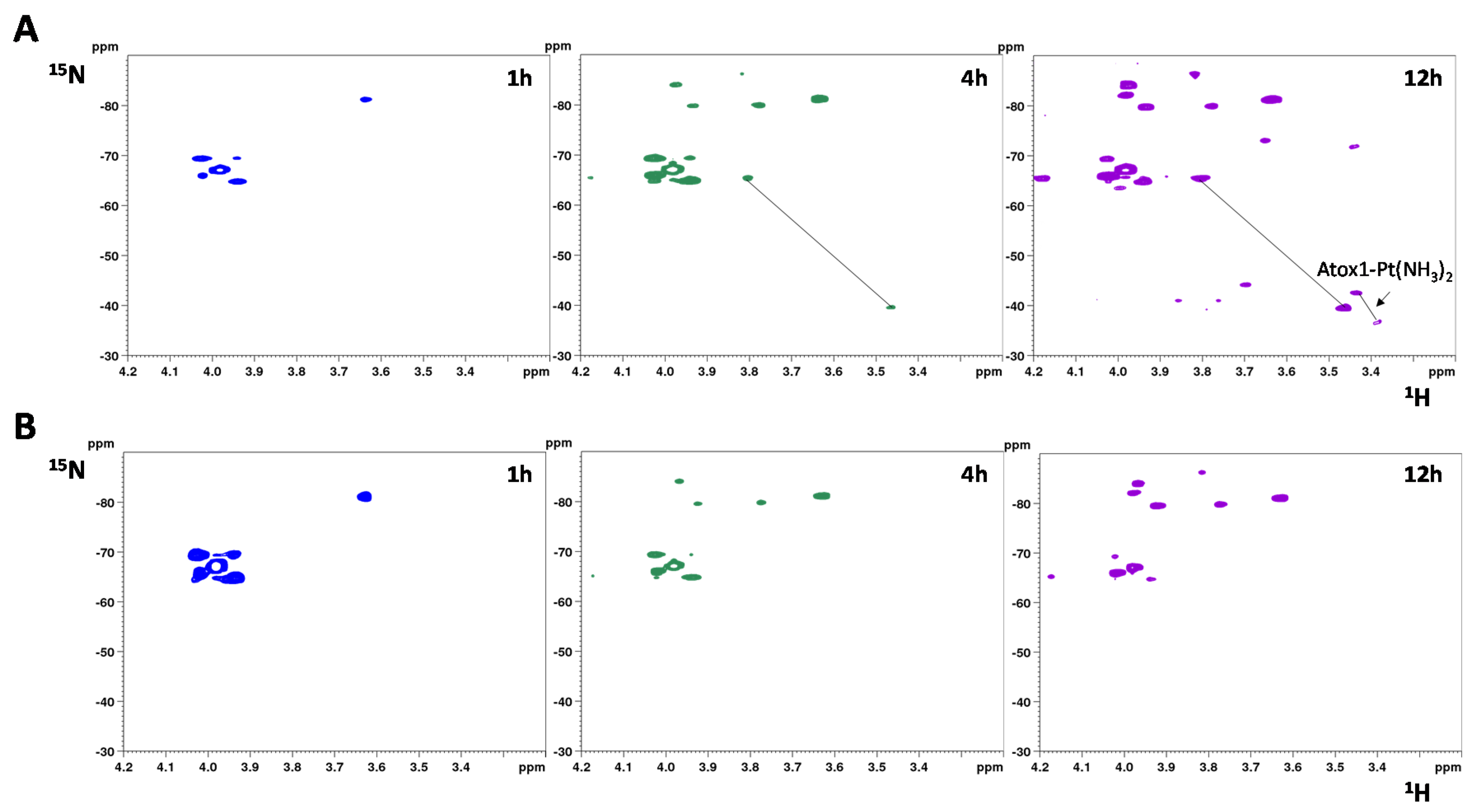
2.4. Interaction with Cisplatin
3. Discussion
- Direct attack of Pt on the disulfide (RSSR acting as monodentate ligand), followed by cleavage of the disulfide bond through formation of sulfinic acid (RSO2H) [39]. In at least one case, it was proposed that a divalent metal ion, such as Ni(II), can bind to a protein disulfide in the absence of thiol reduction [43], but in the case of Atox1 there is no evidence of formation of Pt(II) adduct with oxidized cysteines which would have produced 15NH3 chemical shifts different from those of the adduct of cisplatin with reduced apoAtox1. Instead, all minor peaks in HSQC spectra belong to cisplatin hydrolyzed forms or adducts with GSH that are removed by filtration using a 3 kDa cutoff.
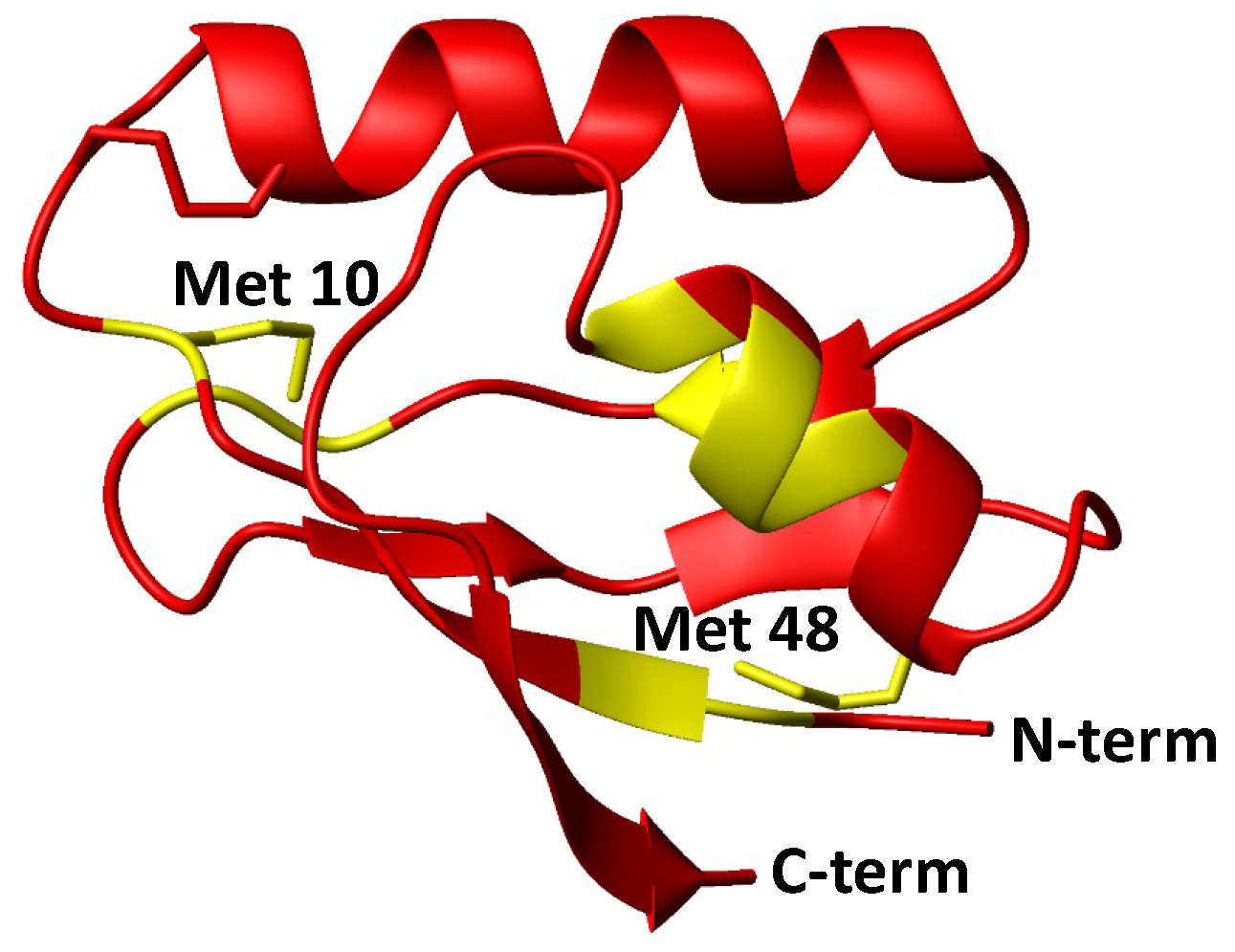
- Initial hydrolysis of disulfide bond to give thiol and sulfenic acid (RSSR + H2O → RSH + RSOH) followed by cisplatin coordination to one thiol (as proposed for Ag+) [44], but in the case of Atox1 there is no binding of cisplatin to thiol(s) in the absence of GSH/GSSG. In a condition where no binding of 15N-labeled cisplatin is detected, the perturbation of intensity of 1H,15N protein cross-peaks can be informative about changes in the chemical environment at the protein surface owing to weak interactions with the drug. The result of this analysis indicates that cisplatin perturbs the protein region around Met48, which is far from the CxxC motif and is not affected by disulfide formation. Slight variations in intensity are also detected at the Met10 site, involving residues in close contact with its buried side-chain [45]. The poor accessibility of Met10 would hamper the binding of cisplatin; however, a conformational change induced by disulfide formation may project the side-chain of Met10 into the solvent, similarly to what occurs with the homologous residue Met12 in the oxidized form of the mercury-binding protein MerP [46]. Analogously to Atox1, cisplatin does not bind to cysteines in fully oxidized Cox17 (the copper chaperone for cytochrome c oxidase) and results indicate that a methionine residue could be the binding site [47].
- Involvement of glutathione in the reaction mechanism. In the present case, the monofunctional adduct [PtCl(NH3)2GS] is formed in the presence of GSH/GSSG, which could also mediate the interaction with oxidized Atox1. The NMR spectra did not show intermediate species that could encompass the protein and the Pt moiety (e.g., monofunctional adducts of cisplatin with the protein), therefore Pt(II) chelate binding appears to be very efficient once a good template structure is obtained. In this regard, the reaction of Cu(I)-Atox1 with cisplatin is indicative, since it is found that when GSH is present in slight excess, the bidentate adduct contains a Cu(I) ion, located in the vicinity of Pt(II), whose role is to keep the two cysteines very close to one another, thus allowing the chelate binding of cisplatin (and possibly of other cis compounds). When a disulfide bond is present between the two cysteines, the S-S distance is lower compared to Cu(I)-Atox1 (2.0–2.1 vs. 3.6–3.7 Å) and Pt binding is only possible provided that a source of reducing equivalents is found nearby. It might be argued that some residual reduced apoprotein present in the GSH/GSSG solution can bind cisplatin instead of oxidized Atox1. However, it has been shown that reduced apoAtox1 is not able to bind cisplatin in the presence of excess GSH [34], thus it is inferred that formation of Atox1-Pt(NH3)2 in GSH/GSSG should occur through a concerted mechanism that involves the oxidized protein and GSH. Pt-thioether type adducts (especially with Met10) may serve as a drug reservoir for subsequent delivery to CxxC once the disulfide is reduced.
4. Materials and Methods
4.1. Preparation of GSH and Cisplatin Solutions
4.2. Protein Expression and Purification
4.3. Size-Exclusion Chromatography
4.4. Electrospray Mass Spectrometry
4.5. Nuclear Magnetic Resonance Spectroscopy
4.6. Circular Dichroism
4.7. Protein Crystallization
4.8. X-ray Diffraction Data Collection and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AM | Carbamidomethyl |
| Atox1 | Antioxidant 1 Copper Chaperone |
| BCS | Bathocuproine Disulfonate |
| CD | Circular Dichroism |
| CSP | Chemical Shift Perturbation |
| EDTA | Ethylenediaminetetraacetic Acid |
| ESI-MS | Electrospray Mass Spectrometry |
| Grx1 | Glutaredoxin 1 |
| GSH | Reduced Glutathione |
| GSSG | Oxidized Glutathione dimer |
| NADPH | Reduced Nicotinamide Adenine Dinucleotide Phosphate |
| HSQC | Heteronuclear Single Quantum Coherence |
| IAM | 2-Iodoacetamide |
| MR | Molecular Replacement |
| NADPH | Reduced Nicotinamide Adenine Dinucleotide Phosphate |
| NMR | Nuclear Magnetic Resonance |
| PDB | Protein Data Bank |
| RMSD | Root-Mean-Square Deviation |
| ROS | Reactive Oxygen Species |
| RV | Retention Volume |
| SEC | Size-Exclusion Chromatography |
References
- Cairns, R.; Harris, I.; Mak, T. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Brabec, V. DNA Modifications by Antitumor Platinum and Ruthenium Compounds: Their Recognition and Repair. Prog. Nucleic Acid Res. Mol. Biol. 2004, 71, 1–68. [Google Scholar]
- Cepeda, V.; Fuertes, M.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J. Biochemical Mechanisms of Cisplatin Cytotoxicity. Anti Cancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; Prieto-García, L.; Prieto, M.; López-Novoa, J.M.; López-Hernández, F.J. Subcellular Targets of Cisplatin Cytotoxicity: An Integrated View. Pharmacol. Ther. 2012, 136, 35–55. [Google Scholar] [CrossRef]
- Corte-Rodríguez, M.; Espina, M.; Sierra, L.M.; Blanco, E.; Ames, T.; Montes-Bayón, M.; Sanz-Medel, A. Quantitative Evaluation of Cellular Uptake, DNA Incorporation and Adduct Formation in Cisplatin Sensitive and Resistant Cell Lines: Comparison of Different Pt-Containing Drugs. Biochem. Pharmacol. 2015, 98, 69–77. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. New Insights into the Molecular and Epigenetic Effects of Antitumor Pt(IV)-Valproic Acid Conjugates in Human Ovarian Cancer Cells. Biochem. Pharmacol. 2015, 95, 133–144. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zanellato, I.; Marzano, C.; Pracharova, J.; Kasparkova, J.; Gibson, D.; Gandin, V.; Osella, D.; Brabec, V. Epigenetic and Antitumor Effects of Platinum(IV)-Octanoato Conjugates. Sci. Rep. 2017, 7, 3751. [Google Scholar] [CrossRef]
- Hall, M.D.; Okabe, M.; Shen, D.-W.; Liang, X.-J.; Gottesman, M.M. The Role of Cellular Accumulation in Determining Sensitivity to Platinum-Based Chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535. [Google Scholar] [CrossRef]
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against Novel Anticancer Metal Compounds: Differences and Similarities. Drug Resist. Updates 2008, 11, 1–16. [Google Scholar] [CrossRef]
- Palm, M.E.; Weise, C.F.; Lundin, C.; Wingsle, G.; Nygren, Y.; Bjorn, E.; Naredi, P.; Wolf-Watz, M.; Wittung-Stafshede, P. Cisplatin Binds Human Copper Chaperone Atox1 and Promotes Unfolding in Vitro. Proc. Natl. Acad. Sci. USA 2011, 108, 6951–6956. [Google Scholar] [CrossRef]
- Pufahl, R.A.; Singer, C.P.; Peariso, K.L.; Lin, S.J.; Schmidt, P.J.; Fahrni, C.J.; Cizewski Culotta, V.; Penner-Hahn, J.E.; O’Halloran, T.V. Metal Ion Chaperone Function of the Soluble Cu(I) Receptor Atx1. Science 1997, 278, 853–856. [Google Scholar] [CrossRef]
- Hung, I.H.; Casareno, R.L.B.; Labesse, G.; Mathews, F.S.; Gitlin, J.D. HAH1 Is a Copper-Binding Protein with Distinct Amino Acid Residues Mediating Copper Homeostasis and Antioxidant Defense. J. Biol. Chem. 1998, 273, 1749–1754. [Google Scholar] [CrossRef]
- Hamza, I.; Prohaska, J.; Gitlin, J.D. Essential Role for Atox1 in the Copper-Mediated Intracellular Trafficking of the Menkes ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 1215–1220. [Google Scholar] [CrossRef]
- Anastassopoulou, I.; Banci, L.; Bertini, I.; Cantini, F.; Katsari, E.; Rosato, A. Solution Structure of the Apo and Copper(I)-Loaded Human Metallochaperone HAH1. Biochemistry 2004, 43, 13046–13053. [Google Scholar] [CrossRef]
- Arnesano, F.; Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Molteni, E.; Huffman, D.L.; O’Halloran, T.V. Metallochaperones and Metal-Transporting ATPases: A Comparative Analysis of Sequences and Structures. Genome Res. 2002, 12, 255–271. [Google Scholar] [CrossRef] [Green Version]
- Arnesano, F.; Banci, L.; Bertini, I.; Felli, I.C.; Losacco, M.; Natile, G. Probing the Interaction of Cisplatin with the Human Copper Chaperone Atox1 by Solution and In-Cell NMR Spectroscopy. J. Am. Chem. Soc. 2011, 133, 18361–18369. [Google Scholar] [CrossRef]
- Palm-Espling, M.; Lundin, C.; Björn, E.; Naredi, P.; Wittung-Stafshede, P. Interaction between the Anticancer Drug Cisplatin and the Copper Chaperone Atox1 in Human Melanoma Cells. Protein Pept. Lett. 2013, 21, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, R.M.; DeRose, V.J. Platinum Binds Proteins in the Endoplasmic Reticulum of S. Cerevisiae and Induces Endoplasmic Reticulum Stress. ACS Chem. Biol. 2017, 12, 2737–2745. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Timm, K.N.; Hu, D.E.; Williams, M.; Wright, A.J.; Kettunen, M.I.; Kennedy, B.W.C.; Larkin, T.J.; Dzien, P.; Marco-Rius, I.; Bohndiek, S.E.; et al. Assessing Oxidative Stress in Tumors by Measuring the Rate of Hyperpolarized [1–13C] Dehydroascorbic Acid Reduction Using 13C Magnetic Resonance Spectroscopy. J. Biol. Chem. 2017, 292, 1737–1748. [Google Scholar] [CrossRef]
- Hegedűs, C.; Kovács, K.; Polgár, Z.; Regdon, Z.; Szabó, É.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virág, L. Redox Control of Cancer Cell Destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The Role of Glutathione Reductase and Related Enzymes on Cellular Redox Homoeostasis Network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hatori, Y.; Clasen, S.; Hasan, N.M.; Barry, A.N.; Lutsenko, S. Functional Partnership of the Copper Export Machinery and Glutathione Balance in Human Cells. J. Biol. Chem. 2012, 287, 26678–26687. [Google Scholar] [CrossRef] [Green Version]
- Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef]
- Brose, J.; La Fontaine, S.; Wedd, A.G.; Xiao, Z. Redox Sulfur Chemistry of the Copper Chaperone Atox1 Is Regulated by the Enzyme Glutaredoxin 1, the Reduction Potential of the Glutathione Couple GSSG/2GSH and the Availability of Cu(I). Metallomics 2014, 6, 793–808. [Google Scholar] [CrossRef]
- Zabet-Moghaddam, M.; Shaikh, A.L.; Niwayama, S. Peptide Peak Intensities Enhanced by Cysteine Modifiers and MALDI TOF MS. J. Mass Spectrom. 2012, 47, 1546–1553. [Google Scholar] [CrossRef]
- Boal, A.K.; Rosenzweig, A.C. Crystal Structures of Cisplatin Bound to a Human Copper Chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar] [CrossRef] [Green Version]
- Belviso, B.D.; Galliani, A.; Lasorsa, A.; Mirabelli, V.; Caliandro, R.; Arnesano, F.; Natile, G. Oxaliplatin Binding to Human Copper Chaperone Atox1 and Protein Dimerization. Inorg. Chem. 2016, 55, 6563–6573. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Ronconi, L.; Sadler, P.J. Insights into the Mechanism of Action of Platinum Anticancer Drugs from Multinuclear NMR Spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 65–98. [Google Scholar] [CrossRef]
- Galliani, A.; Losacco, M.; Lasorsa, A.; Natile, G.; Arnesano, F. Cisplatin Handover between Copper Transporters: The Effect of Reducing Agents. J. Biol. Inorg. Chem. 2014, 19, 705–714. [Google Scholar] [CrossRef]
- Xi, Z.; Guo, W.; Tian, C.; Wang, F.; Liu, Y. Copper Binding Promotes the Interaction of Cisplatin with Human Copper Chaperone Atox1. Chem. Commun. 2013, 49, 11197–11199. [Google Scholar] [CrossRef]
- Kasherman, Y.; Sturup, S.; Gibson, D. Is Glutathione the Major Cellular Target of Cisplatin? A Study of the Interactions of Cisplatin with Cancer Cell Extracts. J. Med. Chem. 2009, 52, 4319–4328. [Google Scholar] [CrossRef]
- Xiao, Z.; Brose, J.; Schimo, S.; Ackland, S.M.; La Fontaine, S.; Wedd, A.G. Unification of the Copper(I) Binding Affinities of the Metallo-Chaperones Atx1, Atox1, and Related Proteins: Detection Probes and Affinity Standards. J. Biol. Chem. 2011, 286, 11047–11055. [Google Scholar] [CrossRef]
- Lempers, E.L.M.; Inagaki, K.; Reedijk, J. Reactions of [PtCl(Dien)]Cl with Glutathione, Oxidized Glutathione and S-Methyl Glutathione. Formation of an S-Bridged Dinuclear Unit. Inorg. Chim. Acta 1988, 152, 201–207. [Google Scholar] [CrossRef]
- Fazlur-Rahman, A.K.; Verkade, J.G. Reactions of [Pt(Dien)Cl]Cl and [Pt(Trpy)Cl]Cl with Thiols, Thioethers, and Dialkyl Disulfides: A195Pt NMR Study. Inorg. Chem. 1992, 31, 2064–2069. [Google Scholar] [CrossRef]
- Del Socorro Murdoch, P.; Kratochwil, N.A.; Parkinson, J.A.; Patriarca, M.; Sadler, P.J. A Novel Dinuclear Diaminoplatinum(II) Glutathione Macrochelate. Angew. Chem. Int. Ed. 1999, 38, 2949–2951. [Google Scholar] [CrossRef]
- Fakih, S.; Munk, V.P.; Shipman, M.A.; Murdoch, P.d.S.; Parkinson, J.A.; Sadler, P.J. Novel Adducts of the Anticancer Drug Oxaliplatin with Glutathione and Redox Reactions with Glutathione Disulfide. Eur. J. Inorg. Chem. 2003, 2003, 1206–1214. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Blaževitš, O.; Calderone, V.; Cantini, F.; Mao, J.; Trapananti, A.; Vieru, M.; Amori, I.; Cozzolino, M.; et al. Interaction of Cisplatin with Human Superoxide Dismutase. J. Am. Chem. Soc. 2012, 134, 7009–7014. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Calderone, V.; Ciofi-Baffoni, S.; Mangani, S.; Martinelli, M.; Palumaa, P.; Wang, S. A Hint for the Function of Human Sco1 from Different Structures. Proc. Natl. Acad. Sci. USA 2006, 103, 8595–8600. [Google Scholar] [CrossRef]
- Cecil, R.; McPhee, J.R. Further Studies on the Reaction of Disulphides with Silver Nitrate. Biochem. J. 1957, 66, 538–543. [Google Scholar] [CrossRef]
- Magistrato, A.; Pavlin, M.; Qasem, Z.; Ruthstein, S. Copper Trafficking in Eukaryotic Systems: Current Knowledge from Experimental and Computational Efforts. Curr. Opin. Struct. Biol. 2019, 58, 26–33. [Google Scholar] [CrossRef]
- Serre, L.; Rossy, E.; Pebay-Peyroula, E.; Cohen-Addad, C.; Covès, J. Crystal Structure of the Oxidized Form of the Periplasmic Mercury-Binding Protein MerP from Ralstonia Metallidurans CH34. J. Mol. Biol. 2004, 339, 161–171. [Google Scholar] [CrossRef]
- Zhao, L.; Cheng, Q.; Wang, Z.; Xi, Z.; Xu, D.; Liu, Y. Cisplatin Binds to Human Copper Chaperone Cox17: The Mechanistic Implication of Drug Delivery to Mitochondria. Chem. Commun. 2014, 50, 2667–2669. [Google Scholar] [CrossRef]
- Dhara, S.C. Rapid Method for the Synthesis of Cis[Pt(NH3)2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Garrett, D.S.; Seok, Y.J.; Peterkofsky, A.; Clore, G.M.; Gronenborn, A.M. Identification by NMR of the Binding Surface for the Histidine—Containing Phosphocarrier Protein HPr on the N-Terminal Domain of Enzyme I of the Escherichia Coli Phosphotransferase System. Biochemistry 1997, 36, 4393–4398. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP 4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Strong, M.; Sawaya, M.R.; Wang, S.; Phillips, M.; Cascio, D.; Eisenberg, D. Toward the Structural Genomics of Complexes: Crystal Structure of a PE/PPE Protein Complex from Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 8060–8065. [Google Scholar] [CrossRef]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mazzone, A.; Siliqi, D. Molecular Replacement: The Probabilistic Approach of the Program REMO09 and Its Applications. Acta Crystallogr. Sect. A Found. Crystallogr. 2009, 65, 512–527. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal Structure Determination and Refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Zwart, P.H. Anomalous Signal Indicators in Protein Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1437–1448. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Kosower, N.S.; Kosower, E.M. The Glutathione Status of Cells. Int. Rev. Cytol. 1978, 54, 109–160. [Google Scholar]
- Russo, A.; DeGraff, W.; Friedman, N.; Mitchell, J.B. Selective Modulation of Glutathione Levels in Human Normal versus Tumor Cells and Subsequent Differential Response to Chemotherapy Drugs. Cancer Res. 1986, 46, 2845–2848. [Google Scholar]
- Hermann, G.; Heffeter, P.; Kryeziu, K.; Berger, W.; Hann, S.; Koellensperger, G. The Study of Reduced versus Oxidized Glutathione in Cancer Cell Models Employing Isotopically Labelled Standards. Anal. Methods 2014, 6, 3086–3094. [Google Scholar] [CrossRef]
- Chisholm, C.L.; Wang, H.; Hang-Heng Wong, A.; Vazquez-Ortiz, G.; Chen, W.; Xu, X.; Deng, C.-X. Ammonium Tetrathiomolybdate Treatment Targets the Copper Transporter ATP7A and Enhances Sensitivity of Breast Cancer to Cisplatin. Oncotarget 2016, 7, 84439–84452. [Google Scholar] [CrossRef]
- Tian, Y.; Fang, T.; Yuan, S.; Zheng, Y.; Arnesano, F.; Natile, G.; Liu, Y. Tetrathiomolybdate Inhibits the Reaction of Cisplatin with Human Copper Chaperone Atox1. Metallomics 2018, 10, 745–750. [Google Scholar] [CrossRef]
- Arnesano, F.; Nardella, M.I.; Natile, G. Platinum Drugs, Copper Transporters and Copper Chelators. Coord. Chem. Rev. 2018, 374, 254–260. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
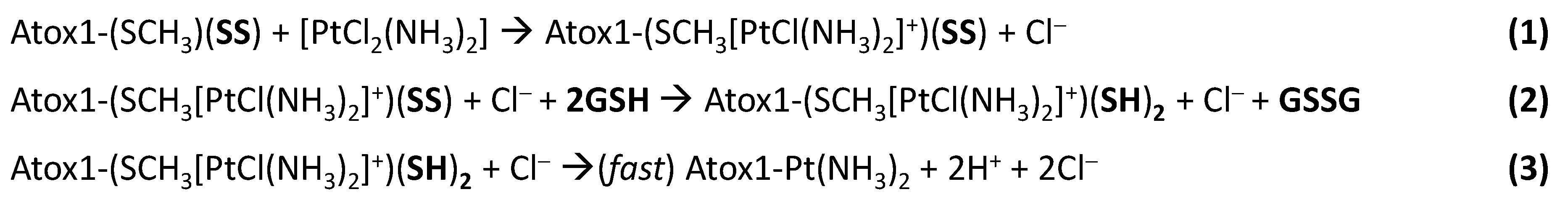{kind=link}
| Without Anisotropy Correction | With Anisotropy Correction | |
|---|---|---|
| Resolution range (Å) | 45.95–3.79 (4.24–3.79) | 45.95–3.90 (4.36–3.90) |
| Space group | P3221 | P3221 |
| Crystal Cell (Å) a,c | 53.06, 56.48 | 53.06, 56.48 |
| Total reflections | 2332 (627) | 1512 (156) |
| Unique reflections | 954 (245) | 606 (50) |
| Completeness (%) | 93.8 (91.2) | 62.7 (17.0) |
| Multiplicity | 2.4 (2.6) | 2.5 (3.1) |
| <I/σ> | 4.8 (1.8) | 6.6 (5.5) |
| Rmerge (%) | 9.9 (43.8) | 8.1 (14.4) |
| Rmeas (%) | 12.2 (53.2) | 10.0 (17.3) |
| CC1/2 | 0.995 (0.904) | 0.994 (0.996) |
| Mosaicity (°) | 0.312 | 0.312 |
| B Wilson (Å2) | 74.4 | 50.1 |
| Rwork | 0.305 | 0.247 |
| Rfree | 0.343 | 0.268 |
| Number of water molecules | 1 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardella, M.I.; Rosato, A.; Belviso, B.D.; Caliandro, R.; Natile, G.; Arnesano, F. Oxidation of Human Copper Chaperone Atox1 and Disulfide Bond Cleavage by Cisplatin and Glutathione. Int. J. Mol. Sci. 2019, 20, 4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184390
Nardella MI, Rosato A, Belviso BD, Caliandro R, Natile G, Arnesano F. Oxidation of Human Copper Chaperone Atox1 and Disulfide Bond Cleavage by Cisplatin and Glutathione. International Journal of Molecular Sciences. 2019; 20(18):4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184390
Chicago/Turabian StyleNardella, Maria I., Antonio Rosato, Benny D. Belviso, Rocco Caliandro, Giovanni Natile, and Fabio Arnesano. 2019. "Oxidation of Human Copper Chaperone Atox1 and Disulfide Bond Cleavage by Cisplatin and Glutathione" International Journal of Molecular Sciences 20, no. 18: 4390. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184390