Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression



 ,
,  and
and
Abstract
:1. Introduction
2. Results
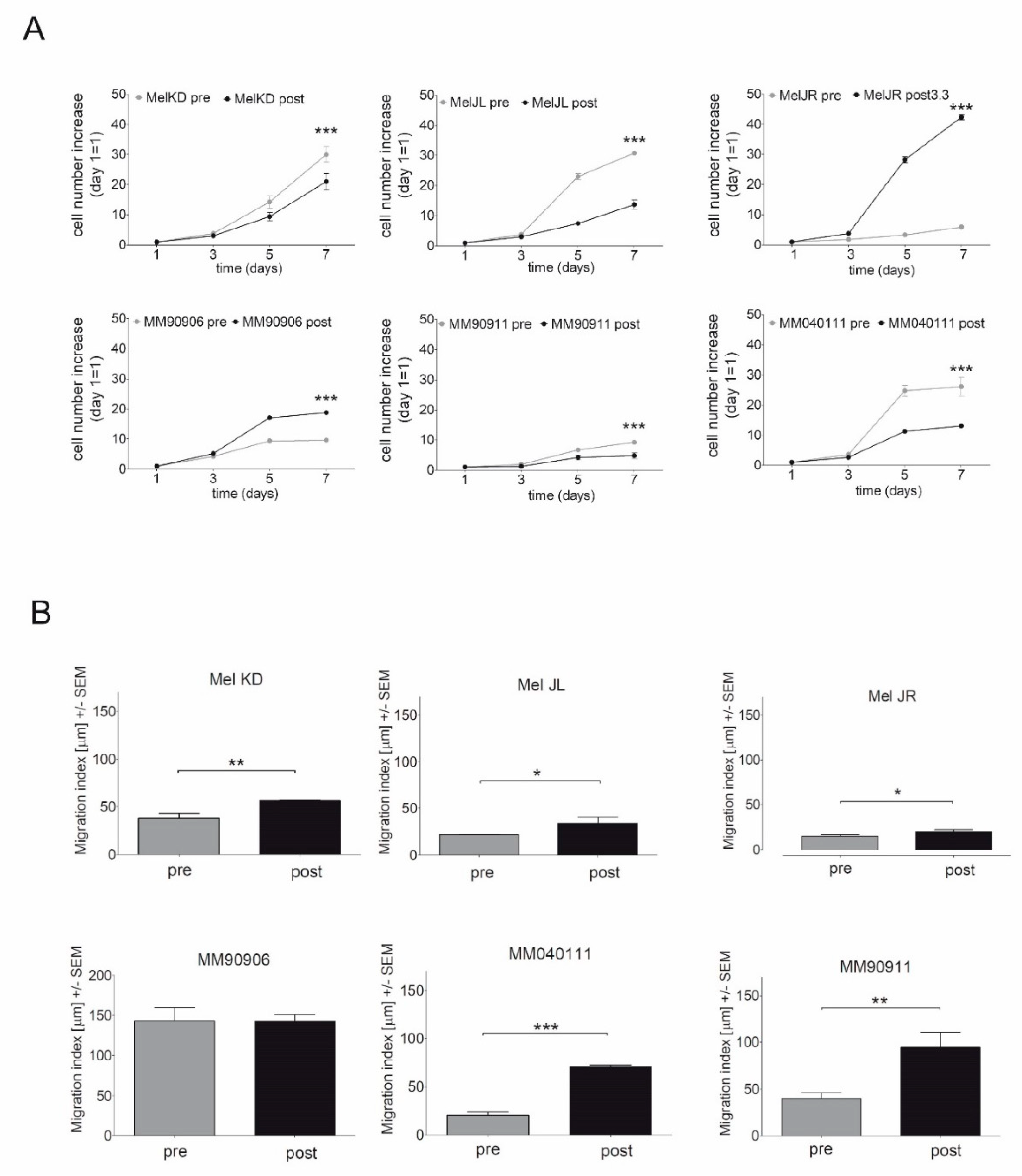
2.1. Proliferation and Migration of the Melanoma Cell Line Pairs
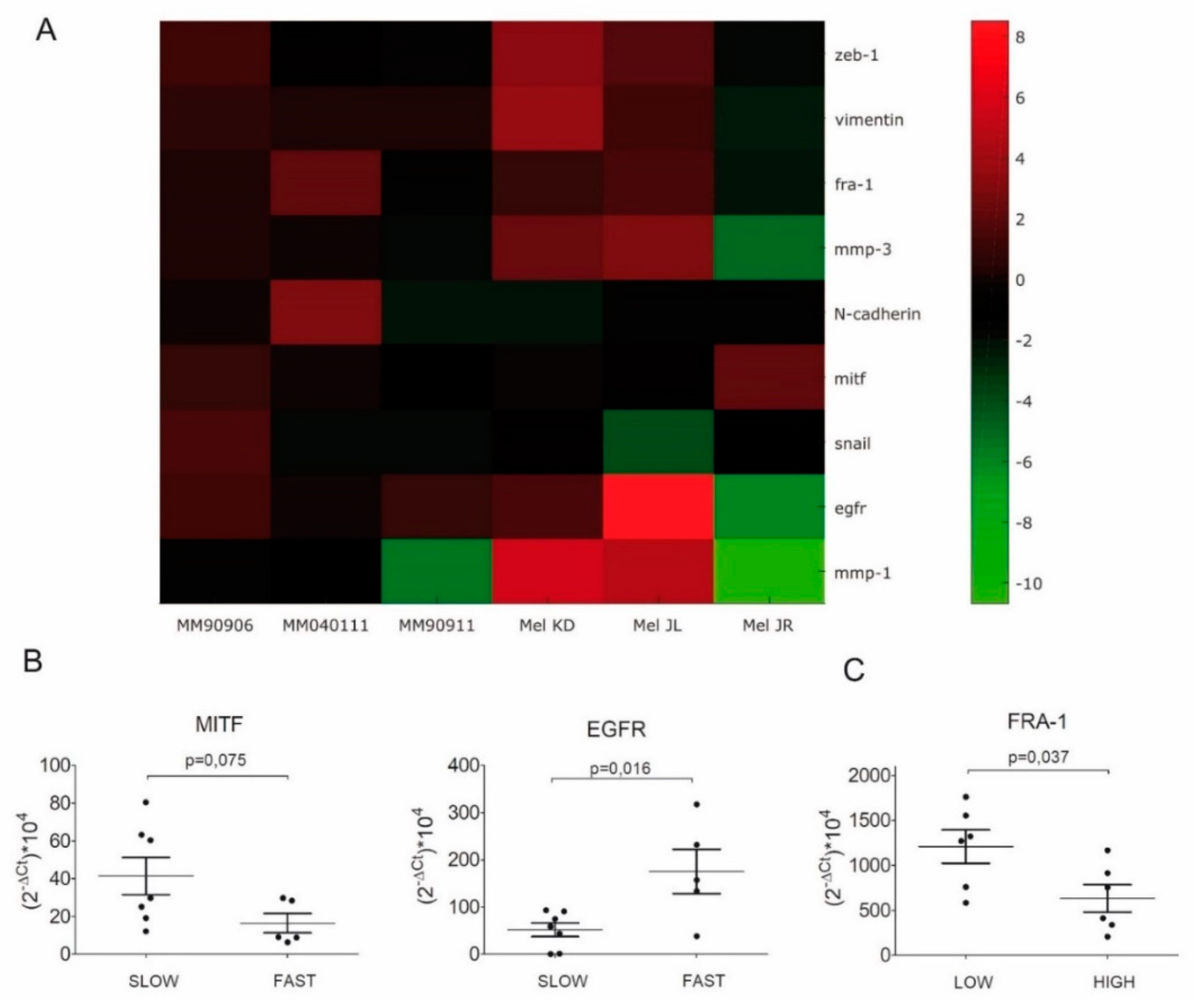
2.2. Evaluation of EMT Marker Expression by q-RT-PCR
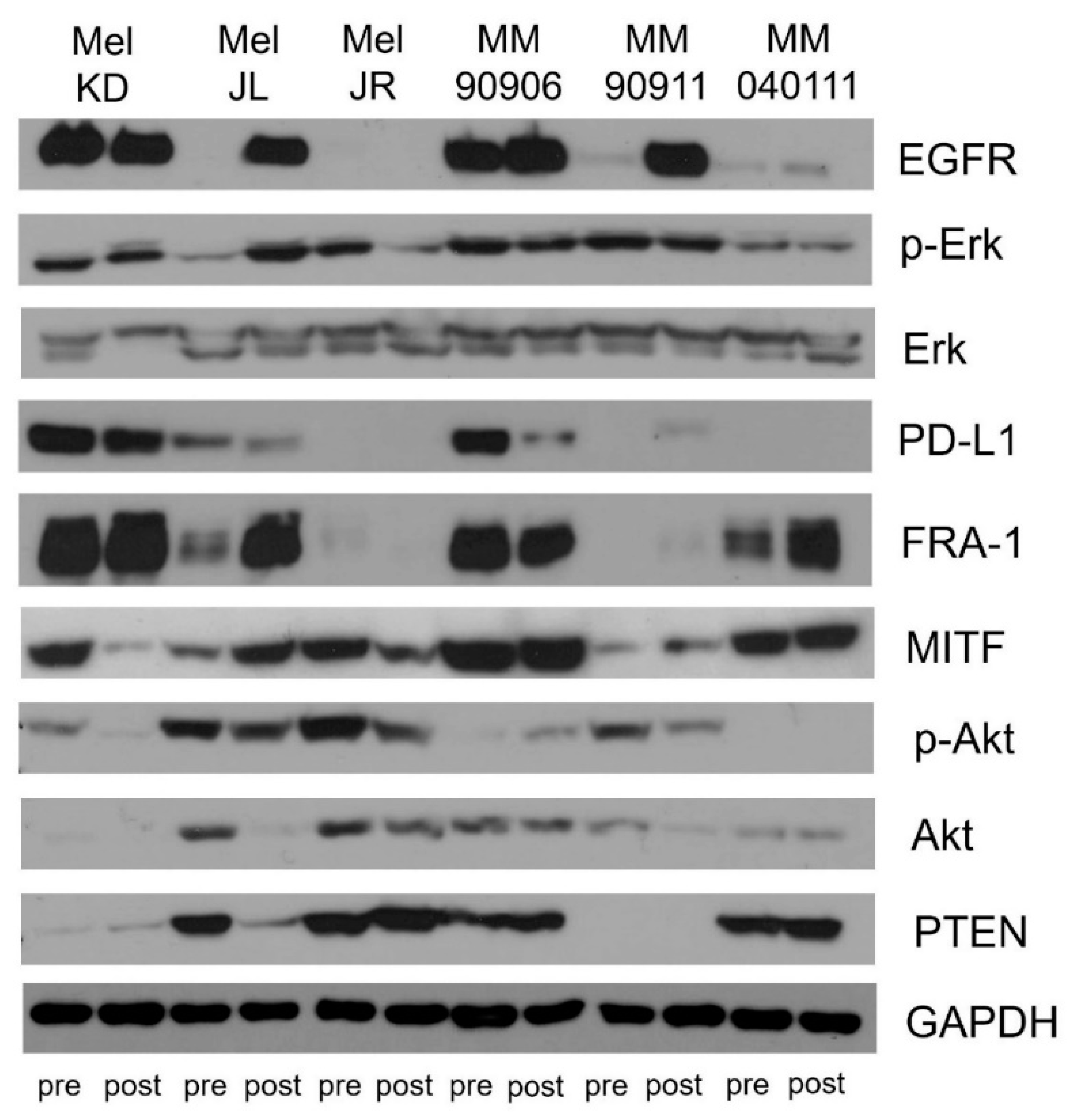
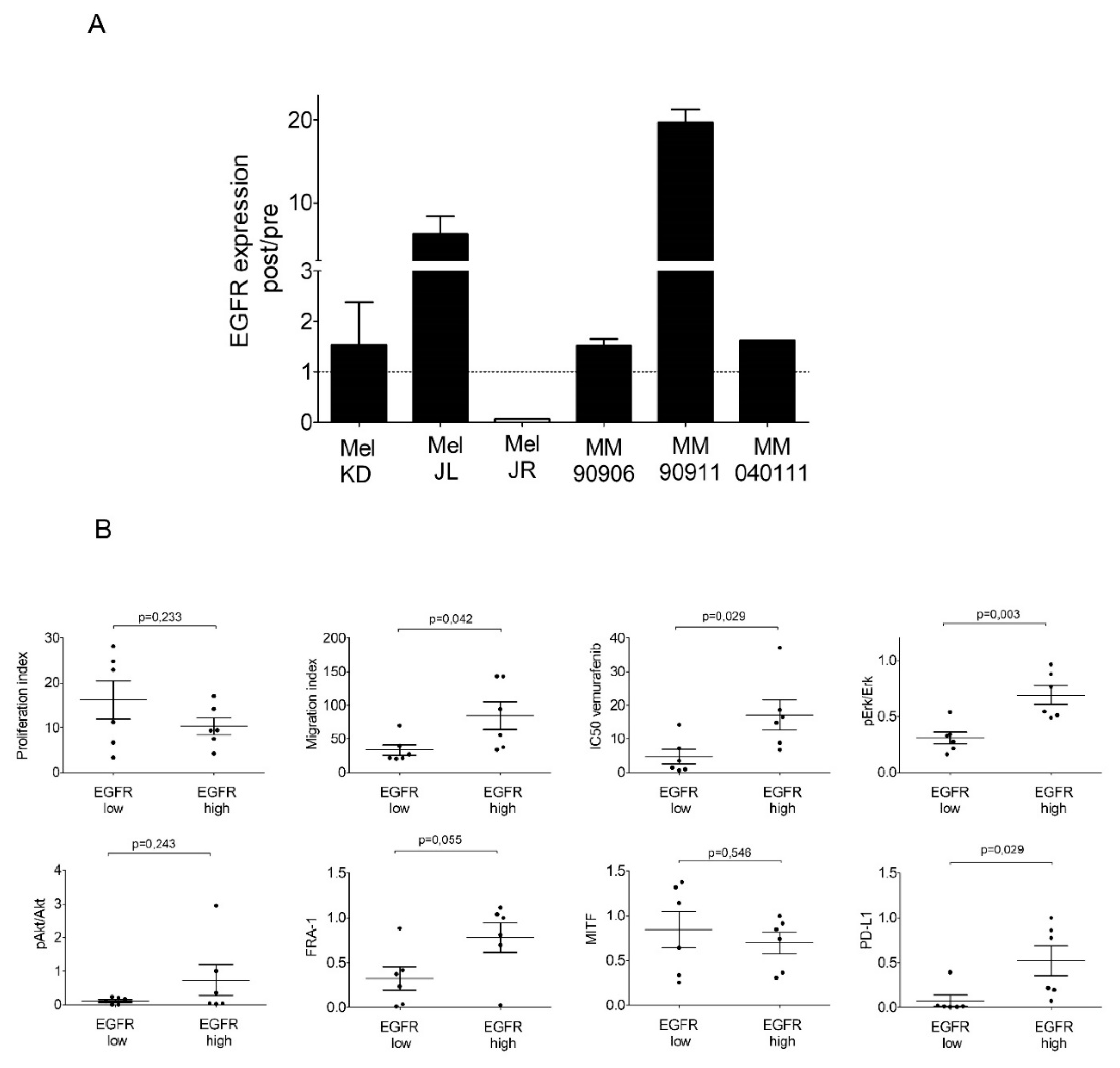
2.3. Signaling Pathway Activation and EGFR, PTEN, MITF, FRA-1, and PD-L1 Expression of the Cell Line Pairs
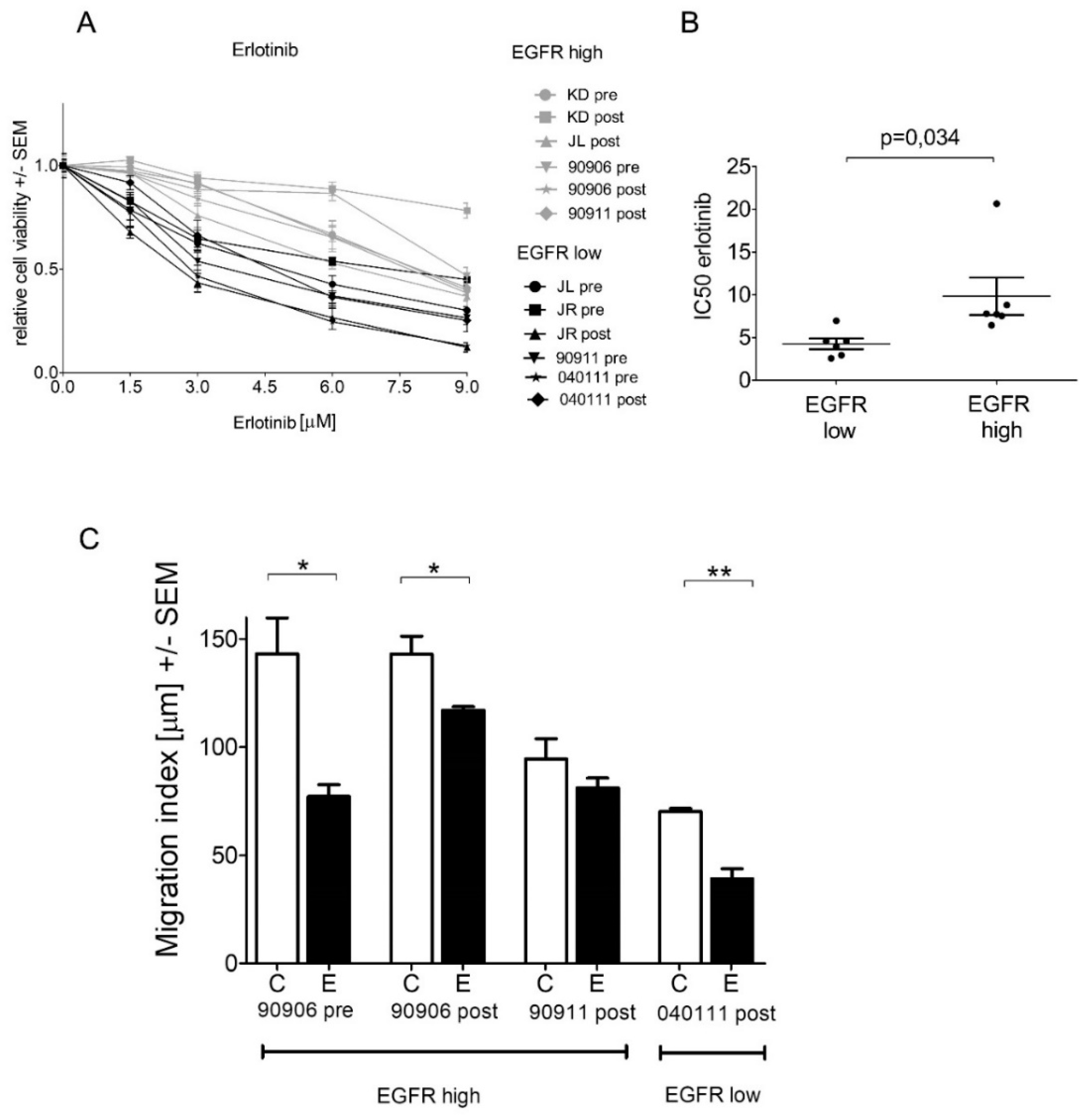
2.4. High-EGFR-Expressing Cells Are More Resistant to Erlotinib Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Proliferation Assay
4.3. Time-Lapse Microscopy
4.4. Gene Expression Analysis
4.5. Immunoblot
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corrie, P.; Hategan, M.; Fife, K.; Parkinson, C. Management of melanoma. Br. Med. Bull. 2014, 111, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Sosman, J.A. Therapeutic advances and treatment options in metastatic melanoma. JAMA Oncol. 2015, 1, 380–386. [Google Scholar] [CrossRef]
- Volpe, V.O.; Klufas, D.M.; Hegde, U.; Grant-Kels, J.M. The new paradigm of systemic therapies for metastatic melanoma. J. Am. Acad. Derm. 2017, 77, 356–368. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Menzies, A.M.; Long, G.V. Systemic treatment for BRAF-mutant melanoma: Where do we go next? Lancet Oncol. 2014, 15, e371–e381. [Google Scholar] [CrossRef]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Derm. 2015, 135, 532–541. [Google Scholar] [CrossRef]
- Glitza, I.C.; Davies, M.A. Genotyping of cutaneous melanoma. Chin. Clin. Oncol. 2014, 3, 27. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Serra, S.; Stingi, A.; Orso, F.; Gaudino, F.; Bologna, C.; Neri, F.; Garaffo, G.; Nassini, R.; Baroni, G.; et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 2017, 8, 15894–15911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaro, F.G.; De Presbiteris, A.L.; Camerlingo, R.; Mozzillo, N.; Pirozzi, G.; Cavalcanti, E.; Manca, A.; Palmieri, G.; Cossu, A.; Ciliberto, G.; et al. Phenotype characterization of human melanoma cells resistant to dabrafenib. Oncol. Rep. 2017, 38, 2741–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef]
- Paulitschke, V.; Berger, W.; Paulitschke, P.; Hofstatter, E.; Knapp, B.; Dingelmaier-Hovorka, R.; Fodinger, D.; Jager, W.; Szekeres, T.; Meshcheryakova, A.; et al. Vemurafenib resistance signature by proteome analysis offers new strategies and rational therapeutic concepts. Mol. Cancer 2015, 14, 757–768. [Google Scholar] [CrossRef]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Pringle, J.H.; Caramel, J.; Ansieau, S. Plasticity of melanoma and EMT-TF reprogramming. Oncotarget 2014, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.C.; Mijatov, B.; Pupo, G.M.; Tran, S.L.; Gowrishankar, K.; Shaw, H.M.; Goding, C.R.; Scolyer, R.A.; Mann, G.J.; Kefford, R.F.; et al. Oncogenic B-RAF(V600E) signaling induces the T-Box3 transcriptional repressor to repress E-cadherin and enhance melanoma cell invasion. J. Investig. Derm. 2013, 133, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Fagone, P.; Nicoletti, F.; Andersen, R.S.; Høgdall, E.; Straten, P.T.; Andersen, M.H.; Svane, I.M. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012, 1, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Jiang, C.C.; Farrelly, M.L.; Zhang, X.D.; Hersey, P. Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res. 2012, 22, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Eccles, M.R.; He, S.; Ahn, A.; Slobbe, L.J.; Jeffs, A.R.; Yoon, H.S.; Baguley, B.C. MITF and PAX3 Play Distinct Roles in Melanoma Cell Migration; Outline of a “Genetic Switch” Theory Involving MITF and PAX3 in Proliferative and Invasive Phenotypes of Melanoma. Front. Oncol. 2013, 3, 229. [Google Scholar] [CrossRef]
- Agnarsdóttir, M.; Ponten, F.; Garmo, H.; Wagenius, G.; Mucci, L.; Magnusson, K.; Holmberg, L.; Eaker-Fält, S. MITF Expression in Cutaneous Malignant Melanoma. Mol. Biomark. Diagn. 2012, 3. [Google Scholar] [CrossRef]
- Shirsat, N.V.; Shaikh, S.A. Overexpression of the immediate early gene fra-1 inhibits proliferation, induces apoptosis, and reduces tumourigenicity of c6 glioma cells. Exp. Cell Res. 2003, 291, 91–100. [Google Scholar] [CrossRef]
- Belguise, K.; Kersual, N.; Galtier, F.; Chalbos, D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene 2005, 24, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R. A price to pay for tumor regression. Cell Res. 2015, 25, 763–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Szasz, I.; Koroknai, V.; Kiss, T.; Vizkeleti, L.; Adany, R.; Balazs, M. Molecular alterations associated with acquired resistance to BRAFV600E targeted therapy in melanoma cells. Melanoma Res. 2019, 29, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Inno, A.; Di Noia, V.; Martini, M.; D’Argento, E.; Di Salvatore, M.; Arena, V.; Schinzari, G.; Orlandi, A.; Larocca, L.M.; Cassano, A.; et al. Erlotinib for Patients with EGFR Wild-Type Metastatic NSCLC: A Retrospective Biomarkers Analysis. Pathol. Oncol. Res. 2019, 25, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol. 2005, 23, 1803–1810. [Google Scholar] [CrossRef]
- Uhlyarik, A.; Piurko, V.; Vizkeleti, L.; Papai, Z.; Raso, E.; Lahm, E.; Kiss, E.; Sikter, M.; Vachaja, J.; Kenessey, I.; et al. EGFR Protein Expression of KRAS Wild-Type Colorectal Cancer: Predictive Value of the Sidedness for Efficacy of Anti-EGFR Therapy. Pathol. Oncol. Res. 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.-J.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients With BRAF V600E–Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. J. Clin. Oncol. 2019, 37, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, I.; Kramer, Z.; Istvan, L.; Cserepes, M.T.; Garay, T.; Hegedus, B.; Dobos, J.; Timar, J.; Tovari, J. Inhibition of epidermal growth factor receptor improves antitumor efficacy of vemurafenib in BRAF-mutant human melanoma in preclinical model. Melanoma Res. 2018, 28, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Vanderbilt, C.M.; Cotzia, P.; Arias Stella, J.A., 3rd; Chang, J.C.; Chen, Y.; Tang, L.H.; DeLair, D.F.; Yao, J.; Ladanyi, M.; et al. JAK2, PD-L1, and PD-L2 (9p24.1) amplification in metastatic mucosal and cutaneous melanomas with durable response to immunotherapy. Hum. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology 2018, 7, e1466016. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF Mutations in Advanced Colorectal Cancer Are Associated With Poor Prognosis but Do Not Preclude Benefit From Oxaliplatin or Irinotecan: Results From the MRC FOCUS Trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Garay, T.; Juhasz, E.; Molnar, E.; Eisenbauer, M.; Czirok, A.; Dekan, B.; Laszlo, V.; Hoda, M.A.; Dome, B.; Timar, J.; et al. Cell migration or cytokinesis and proliferation?—Revisiting the “go or grow” hypothesis in cancer cells in vitro. Exp. Cell Res. 2013, 319, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Pre-TX IC50 [µM] | Post-TX IC50 [µM] | Sex | Age | Response to Vemurafenib | Reference |
|---|---|---|---|---|---|---|
| Mel KD | 6.7 | 18.6 | M | 35 | PR | Patient 1 in [27] |
| Mel JL | 14.2 | 16.5 | F | 53 | PR | Patient 4 in [27] |
| Mel JR | 1 | 7.1 | F | 54 | PR | Patient 3 in [27] |
| MM90906 | 8.8 | 14.8 | M | 36 | n.a. | Patient 5 in [26] |
| MM90911 | 1.5 | 37.1 | M | 41 | n.a. | Patient 4 in [26] |
| MM040111 | 0.7 | 3.5 | F | 78 | n.a. | Patient 3 in [26] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molnár, E.; Garay, T.; Donia, M.; Baranyi, M.; Rittler, D.; Berger, W.; Tímár, J.; Grusch, M.; Hegedűs, B. Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression. Int. J. Mol. Sci. 2019, 20, 4484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184484
Molnár E, Garay T, Donia M, Baranyi M, Rittler D, Berger W, Tímár J, Grusch M, Hegedűs B. Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression. International Journal of Molecular Sciences. 2019; 20(18):4484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184484
Chicago/Turabian StyleMolnár, Eszter, Tamás Garay, Marco Donia, Marcell Baranyi, Dominika Rittler, Walter Berger, József Tímár, Michael Grusch, and Balázs Hegedűs. 2019. "Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression" International Journal of Molecular Sciences 20, no. 18: 4484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184484