More than just Stem Cells: Functional Roles of the Transcription Factor Sox2 in Differentiated Glia and Neurons

Abstract
:
1. Introduction
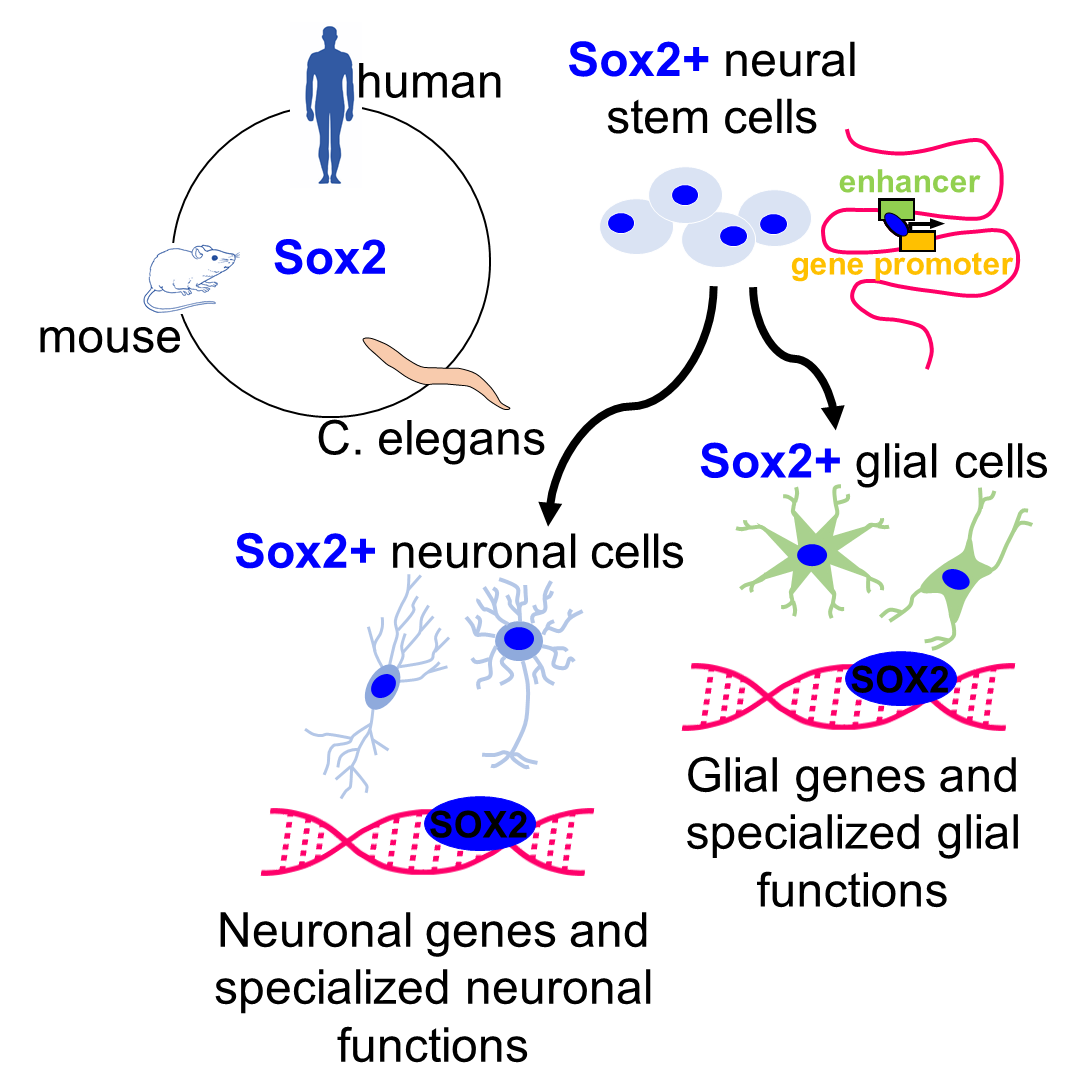
1.1. Sox2 as a “Stem Cell Gene”
1.2. SOX2 and Neurodevelopmental Genetic Disease
1.3. Recent Perspectives on SOX2 Molecular Mechanisms of Action
2. Sox2 Functions in Differentiated Glia and Neurons
2.1. Sox2 in Glial Cells
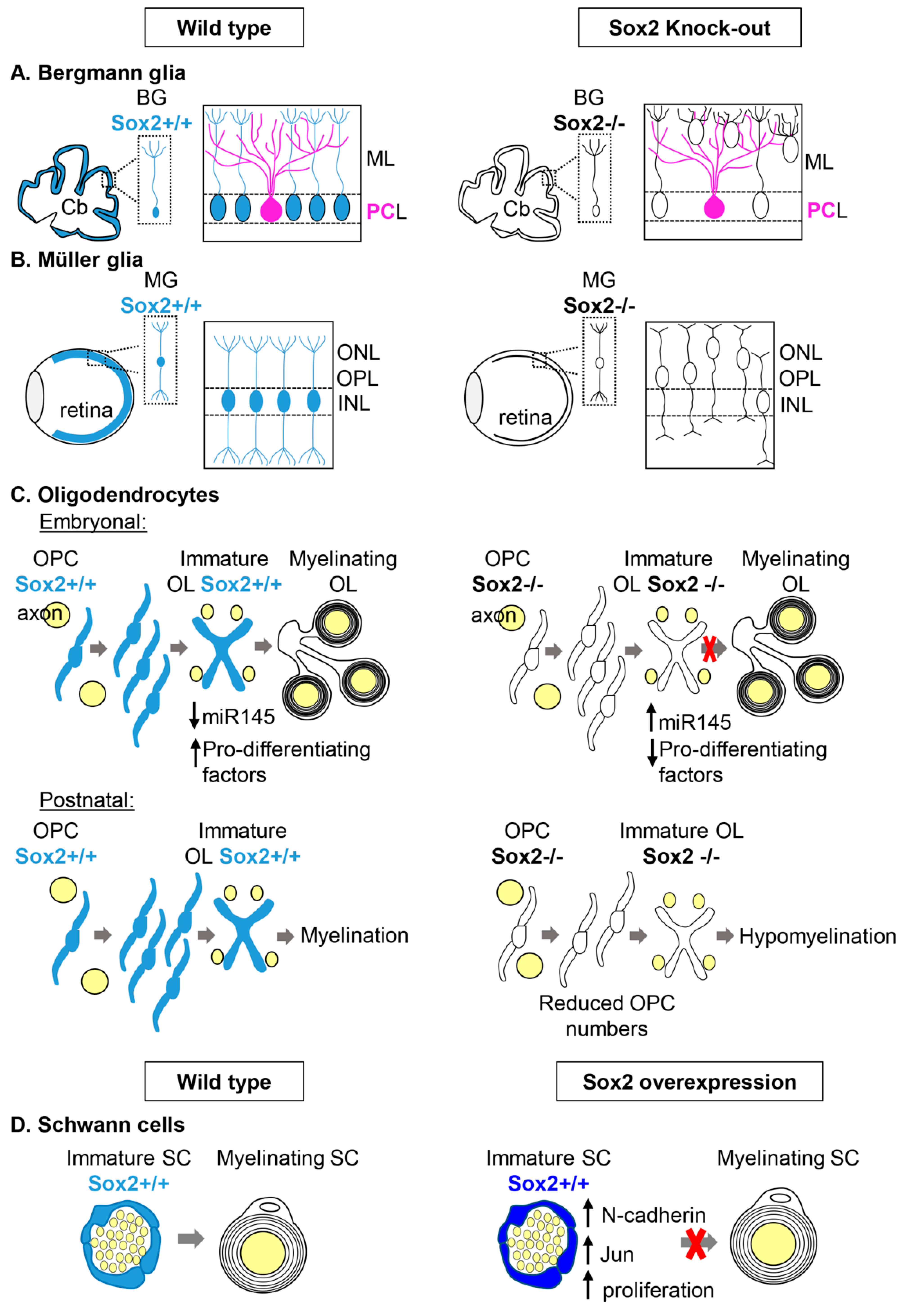
2.1.1. Bergmann Glia and Müller Glia
Bergmann Glia in the cerebellum
Müller Glia in the Retina
Can Sox2 Expressing BG and MG Include Cells with Stem Cell Features?
2.1.2. Myelinating Glial Cells: Oligodendrocytes and Schwann Cells
Oligodendrocytes
Schwann Cells
Could Sox2 Have a Role in Remyelination?
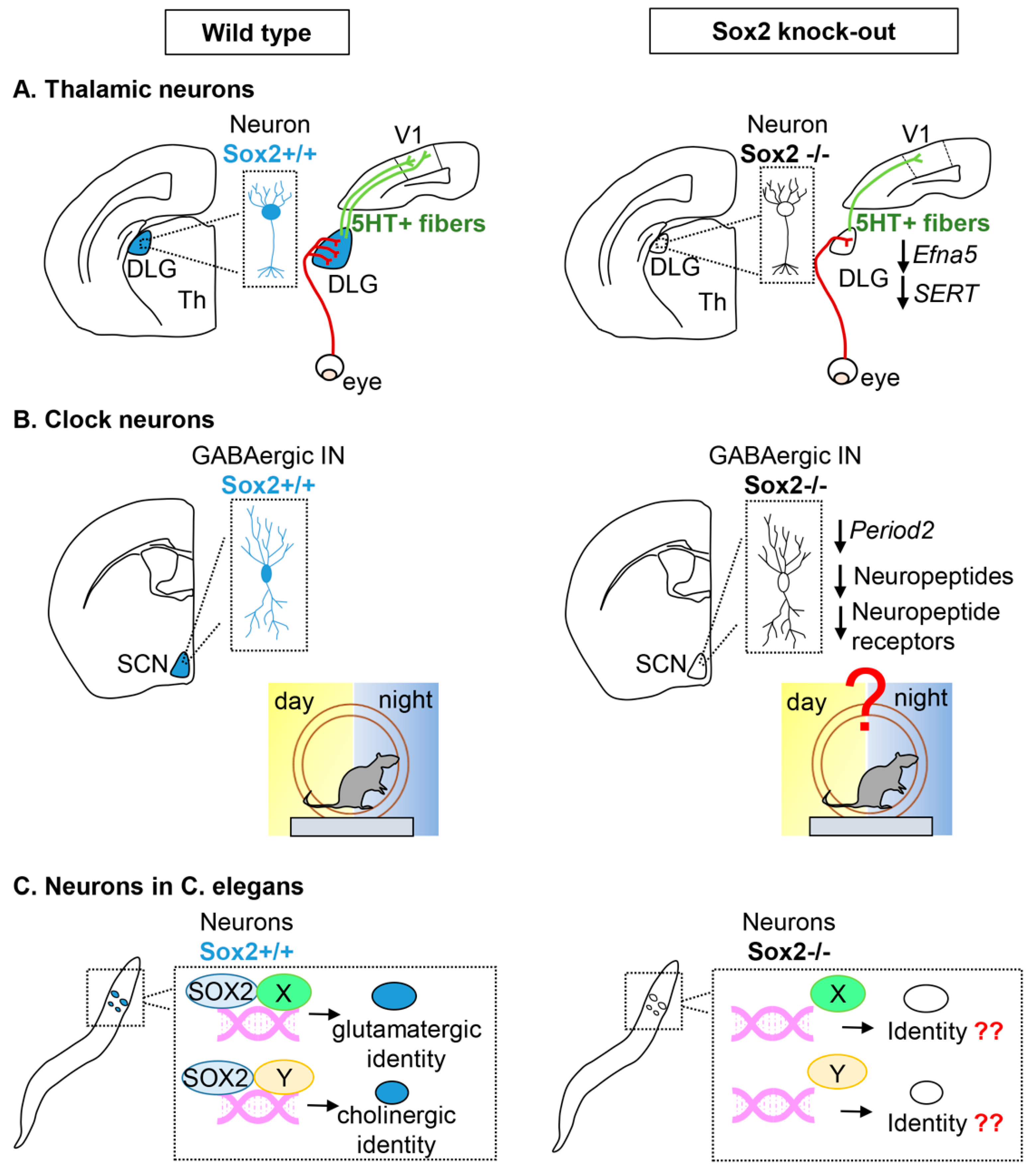
2.2. Sox2 in Neurons
Thalamic Projection Neurons
Clock Neurons in the Suprachiasmatic Thalamic Nucleus
Neurons in C. elegans
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, J.; Mercurio, S.; Favaro, R.; Mariani, J.; Ottolenghi, S.; Nicolis, S.K. Sox2-dependent regulation of neural stem cells and CNS development. In Sox2, Biology and Role in Development and Disease; Kondoh, H., Lovell-Badge, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Kondoh, H.; Lovell-Badge, R. Sox2, Biology and Role in Development and Disease; Elsevier: Amsterdam, The Netherlands; Associated Press: New York, NY, USA, 2016; ISBN 978-0-12-800352-7. [Google Scholar]
- Pevny, L.H.; Nicolis, S.K. Sox2 roles in neural stem cells. Int. J. Biochem. Cell Biol. 2010, 42, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Zappone, M.V.; Galli, R.; Catena, R.; Meani, N.; De Biasi, S.; Mattei, E.; Tiveron, C.; Vescovi, A.L.; Lovell-Badge, R.; Ottolenghi, S.; et al. Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development 2000, 127, 2367–2382. [Google Scholar]
- Favaro, R.; Valotta, M.; Ferri, A.L.; Latorre, E.; Mariani, J.; Giachino, C.; Lancini, C.; Tosetti, V.; Ottolenghi, S.; Taylor, V.; et al. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat. Neurosci. 2009, 12, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Favaro, R.; Beccari, L.; Bertolini, J.; Mercurio, S.; Nieto-Lopez, F.; Verzeroli, C.; La Regina, F.; De Pietri Tonelli, D.; Ottolenghi, S.; et al. Sox2 is required for embryonic development of the ventral telencephalon through the activation of the ventral determinants Nkx2.1 and Shh. Development 2013, 140, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Panaliappan, T.K.; Wittmann, W.; Jidigam, V.K.; Mercurio, S.; Bertolini, J.A.; Sghari, S.; Bose, R.; Patthey, C.; Nicolis, S.K.; Gunhaga, L. Sox2 is required for olfactory pit formation and olfactory neurogenesis through BMP restriction and Hes5 upregulation. Development 2018, 145. [Google Scholar] [CrossRef]
- Schwob, J.E.; Jang, W.; Holbrook, E.H.; Lin, B.; Herrick, D.B.; Peterson, J.N.; Hewitt Coleman, J. Stem and progenitor cells of the mammalian olfactory epithelium: Taking poietic license. J. Comp. Neurol. 2017, 525, 1034–1054. [Google Scholar] [CrossRef]
- Fantes, J.; Ragge, N.K.; Lynch, S.A.; McGill, N.I.; Collin, J.R.; Howard-Peebles, P.N.; Hayward, C.; Vivian, A.J.; Williamson, K.; van Heyningen, V.; et al. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003, 33, 461–463. [Google Scholar] [CrossRef]
- Ragge, N.K.; Lorenz, B.; Schneider, A.; Bushby, K.; de Sanctis, L.; de Sanctis, U.; Salt, A.; Collin, J.R.; Vivian, A.J.; Free, S.L.; et al. SOX2 anophthalmia syndrome. Am. J. Med. Genet. A 2005, 135, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M.; Ragge, N.K.; Cavalleri, G.L.; Hever, A.; Lorenz, B.; Schneider, A.; Williamson, K.A.; Stevens, J.M.; Free, S.L.; Thompson, P.J.; et al. Role of SOX2 mutations in human hippocampal malformations and epilepsy. Epilepsia 2006, 47, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Taranova, O.V.; Magness, S.T.; Fagan, B.M.; Wu, Y.; Surzenko, N.; Hutton, S.R.; Pevny, L.H. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006, 20, 1187–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Nakamura, A.; Urata, M.; Shirae-Kurabayashi, M.; Kuraku, S.; Russell, S.; Ohtsuka, S. The evolutionally-conserved function of group B1 Sox family members confers the unique role of Sox2 in mouse ES cells. BMC Evol. Biol. 2016, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Wen, J. Overview of the roles of Sox2 in stem cell and development. Biol. Chem. 2015, 396, 883–891. [Google Scholar] [CrossRef]
- Bertolini, J.A.; Favaro, R.; Zhu, Y.; Pagin, M.; Ngan, C.Y.; Wong, C.H.; Tjong, H.; Vermunt, M.W.; Martynoga, B.; Barone, C.; et al. Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell Stem Cell 2019, 24, 462–476. [Google Scholar] [CrossRef]
- Amador-Arjona, A.; Cimadamore, F.; Huang, C.T.; Wright, R.; Lewis, S.; Gage, F.H.; Terskikh, A.V. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E1936–E1945. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Hsu, J.Y.; Linker, S.B.; Hu, L.; Schafer, S.T.; Mertens, J.; Jacinto, F.V.; Hetzer, M.W.; Gage, F.H. Nup153 Interacts with Sox2 to Enable Bimodal Gene Regulation and Maintenance of Neural Progenitor Cells. Cell Stem Cell 2017, 21, 618–634. [Google Scholar] [CrossRef]
- Kautzman, A.G.; Keeley, P.W.; Nahmou, M.M.; Luna, G.; Fisher, S.K.; Reese, B.E. Sox2 regulates astrocytic and vascular development in the retina. Glia 2018, 66, 623–636. [Google Scholar] [CrossRef]
- Hoffmann, S.A.; Hos, D.; Kuspert, M.; Lang, R.A.; Lovell-Badge, R.; Wegner, M.; Reiprich, S. Stem cell factor Sox2 and its close relative Sox3 have differentiation functions in oligodendrocytes. Development 2014, 141, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Rasai, A.; Wang, Y.; Xu, J.; Bannerman, P.; Erol, D.; Tsegaye, D.; Wang, A.; Soulika, A.; Zhan, X.; et al. The Stem Cell Factor Sox2 Is a Positive Timer of Oligodendrocyte Development in the Postnatal Murine Spinal Cord. Mol. Neurobiol. 2018, 55, 9001–9015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, X.; Gui, X.; Croteau, C.; Song, L.; Xu, J.; Wang, A.; Bannerman, P.; Guo, F. Sox2 Is Essential for Oligodendroglial Proliferation and Differentiation during Postnatal Brain Myelination and CNS Remyelination. J. Neurosci. 2018, 38, 1802–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Ma, D.; Zawadzka, M.; Fancy, S.P.; Elis-Williams, L.; Bouvier, G.; Stockley, J.H.; de Castro, G.M.; Wang, B.; Jacobs, S.; et al. Sox2 Sustains Recruitment of Oligodendrocyte Progenitor Cells following CNS Demyelination and Primes Them for Differentiation during Remyelination. J. Neurosci. 2015, 35, 11482–11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, A.L.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [PubMed]
- Bachleda, A.R.; Pevny, L.H.; Weiss, E.R. Sox2-Deficient Muller Glia Disrupt the Structural and Functional Maturation of the Mammalian Retina. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1488–1499. [Google Scholar] [CrossRef]
- Surzenko, N.; Crowl, T.; Bachleda, A.; Langer, L.; Pevny, L. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Muller glia. Development 2013, 140, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, V.; Mercurio, S.; Leto, K.; Fuca, E.; Hoxha, E.; Bottes, S.; Pagin, M.; Milanese, M.; Ngan, C.Y.; Concina, G.; et al. Sox2 conditional mutation in mouse causes ataxic symptoms, cerebellar vermis hypoplasia, and postnatal defects of Bergmann glia. Glia 2018, 66, 1929–1946. [Google Scholar] [CrossRef]
- Le, N.; Nagarajan, R.; Wang, J.Y.; Araki, T.; Schmidt, R.E.; Milbrandt, J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc. Natl. Acad. Sci. USA 2005, 102, 2596–2601. [Google Scholar] [CrossRef]
- Roberts, S.L.; Dun, X.P.; Doddrell, R.D.S.; Mindos, T.; Drake, L.K.; Onaitis, M.W.; Florio, F.; Quattrini, A.; Lloyd, A.C.; D’Antonio, M.; et al. Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 2017, 144, 3114–3125. [Google Scholar] [CrossRef] [Green Version]
- Koike, T.; Wakabayashi, T.; Mori, T.; Hirahara, Y.; Yamada, H. Sox2 promotes survival of satellite glial cells in vitro. Biochem. Biophys. Res. Commun. 2015, 464, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Koike, T.; Wakabayashi, T.; Mori, T.; Takamori, Y.; Hirahara, Y.; Yamada, H. Sox2 in the adult rat sensory nervous system. Histochem. Cell Biol. 2014, 141, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.H.; Bouchard-Cannon, P.; Hegazi, S.; Lowden, C.; Fung, S.W.; Chiang, C.K.; Ness, R.W.; Cheng, H.M. SOX2-Dependent Transcription in Clock Neurons Promotes the Robustness of the Central Circadian Pacemaker. Cell Rep. 2019, 26, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, S.; Serra, L.; Motta, A.; Gesuita, L.; Sanchez-Arrones, L.; Inverardi, F.; Foglio, B.; Barone, C.; Kaimakis, P.; Martynoga, B.; et al. Sox2 Acts in Thalamic Neurons to Control the Development of Retina-Thalamus-Cortex Connectivity. iScience 2019, 15, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqadah, A.; Hsieh, Y.W.; Vidal, B.; Chang, C.; Hobert, O.; Chuang, C.F. Post-mitotic diversification of olfactory neuron types is mediated by differential activities of the HMG-box transcription factor SOX-2. EMBO J. 2015, 34, 2574–2589. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Santella, A.; Serrano-Saiz, E.; Bao, Z.; Chuang, C.F.; Hobert, O.C. elegans SoxB genes are dispensable for embryonic neurogenesis but required for terminal differentiation of specific neuron types. Development 2015, 142, 2464–2477. [Google Scholar] [CrossRef] [PubMed]
- Whitney, I.E.; Keeley, P.W.; St John, A.J.; Kautzman, A.G.; Kay, J.N.; Reese, B.E. Sox2 regulates cholinergic amacrine cell positioning and dendritic stratification in the retina. J. Neurosci. 2014, 34, 10109–10121. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, M.; Mariani, J.; Lancini, C.; Latorre, E.; Caccia, R.; Gullo, F.; Valotta, M.; DeBiasi, S.; Spinardi, L.; Ronchi, A.; et al. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development 2008, 135, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Watanabe, M. Cytodifferentiation of Bergmann glia and its relationship with Purkinje cells. Anat. Sci. Int. 2002, 77, 94–108. [Google Scholar] [CrossRef]
- Buffo, A.; Rossi, F. Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 2013, 109, 42–63. [Google Scholar] [CrossRef]
- De Zeeuw, C.I.; Hoogland, T.M. Reappraisal of Bergmann glial cells as modulators of cerebellar circuit function. Front. Cell. Neurosci. 2015, 9, 246. [Google Scholar] [CrossRef]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Ng, C.W.; Wamstad, J.A.; Cheng, A.W.; Thai, K.K.; Fraenkel, E.; Jaenisch, R.; Boyer, L.A. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet. 2013, 9, e1003288. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Imura, T.; Sofroniew, M.V.; Fushiki, S. Loss of adenomatous polyposis coli in Bergmann glia disrupts their unique architecture and leads to cell nonautonomous neurodegeneration of cerebellar Purkinje neurons. Glia 2011, 59, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoser, M.; Baader, S.L.; Bosl, M.R.; Ihmer, A.; Wegner, M.; Sock, E. Prolonged glial expression of Sox4 in the CNS leads to architectural cerebellar defects and ataxia. J. Neurosci. 2007, 27, 5495–5505. [Google Scholar] [CrossRef] [PubMed]
- Dooves, S.; Bugiani, M.; Wisse, L.E.; Abbink, T.E.M.; van der Knaap, M.S.; Heine, V.M. Bergmann glia translocation: A new disease marker for vanishing white matter identifies therapeutic effects of Guanabenz treatment. Neuropathol. Appl. Neurobiol. 2018, 44, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Livesey, F.J.; Cepko, C.L. Vertebrate neural cell-fate determination: Lessons from the retina. Nat. Rev. Neurosci. 2001, 2, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Willbold, E.; Berger, J.; Reinicke, M.; Wolburg, H. On the role of Muller glia cells in histogenesis: Only retinal spheroids, but not tectal, telencephalic and cerebellar spheroids develop histotypical patterns. J. Hirnforsch. 1997, 38, 383–396. [Google Scholar]
- Furukawa, T.; Mukherjee, S.; Bao, Z.Z.; Morrow, E.M.; Cepko, C.L. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 2000, 26, 383–394. [Google Scholar] [CrossRef]
- Jadhav, A.P.; Roesch, K.; Cepko, C.L. Development and neurogenic potential of Muller glial cells in the vertebrate retina. Prog. Retin. Eye Res. 2009, 28, 249–262. [Google Scholar] [CrossRef]
- Matsushima, D.; Heavner, W.; Pevny, L.H. Combinatorial regulation of optic cup progenitor cell fate by SOX2 and PAX6. Development 2011, 138, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlfeld, J.; Filser, S.; Schmidt, F.; Wefers, A.K.; Merk, D.J.; Glass, R.; Herms, J.; Schuller, U. Neurogenesis from Sox2 expressing cells in the adult cerebellar cortex. Sci. Rep. 2017, 7, 6137. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Cepko, C.L. Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 2000, 3, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Gorsuch, R.A.; Lahne, M.; Yarka, C.E.; Petravick, M.E.; Li, J.; Hyde, D.R. Sox2 regulates Muller glia reprogramming and proliferation in the regenerating zebrafish retina via Lin28 and Ascl1a. Exp. Eye Res. 2017, 161, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Raff, M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004, 18, 2963–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsland, M.; Ramsköld, D.; Zaouter, C.; Klum, S.; Sandberg, R.; Muhr, J. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev. 2011, 25, 2453–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, M. SOX after SOX: SOXession regulates neurogenesis. Genes Dev. 2011, 25, 2423–2428. [Google Scholar] [CrossRef]
- Lu, Y.; Futtner, C.; Rock, J.R.; Xu, X.; Whitworth, W.; Hogan, B.L.; Onaitis, M.W. Evidence that SOX2 overexpression is oncogenic in the lung. PLoS ONE 2010, 5, e11022. [Google Scholar] [CrossRef]
- Feltri, M.L.; D’Antonio, M.; Previtali, S.; Fasolini, M.; Messing, A.; Wrabetz, L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann. N. Y. Acad. Sci. 1999, 883, 116–123. [Google Scholar] [CrossRef]
- Oliver-De La Cruz, J.; Carrion-Navarro, J.; Garcia-Romero, N.; Gutierrez-Martin, A.; Lazaro-Ibanez, E.; Escobedo-Lucea, C.; Perona, R.; Belda-Iniesta, C.; Ayuso-Sacido, A. SOX2+ cell population from normal human brain white matter is able to generate mature oligodendrocytes. PLoS ONE 2014, 9, e99253. [Google Scholar] [CrossRef]
- Chou, S.J.; Babot, Z.; Leingartner, A.; Studer, M.; Nakagawa, Y.; O’Leary, D.D. Geniculocortical input drives genetic distinctions between primary and higher-order visual areas. Science 2013, 340, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ye, R.; Gargus, J.J.; Blakely, R.D.; Dobrenis, K.; Sze, J.Y. Disruption of Transient Serotonin Accumulation by Non-Serotonin-Producing Neurons Impairs Cortical Map Development. Cell Rep. 2015, 10, 346–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, P.; Cases, O.; Maroteaux, L. The developmental role of serotonin: News from mouse molecular genetics. Nat. Rev. Neurosci. 2003, 4, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Mengual, E.; Moessner, R.; Hall, F.S.; Revay, R.S.; Sora, I.; Arellano, J.; DeFelipe, J.; Gimenez-Amaya, J.M.; Conciatori, M.; et al. Barrel pattern formation requires serotonin uptake by thalamocortical afferents, and not vesicular monoamine release. J. Neurosci. 2001, 21, 6862–6873. [Google Scholar] [CrossRef] [PubMed]
- Hoefflin, S.; Carter, D.A. Neuronal expression of SOX2 is enriched in specific hypothalamic cell groups. J. Chem. Neuroanat. 2014, 61–62, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Phys. 2001, 63, 647–676. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 2002, 417, 405–410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Glia | Sox2 Expression | Sox2 Function | References |
| Macroglia in the Central nervous system | |||
| Astrocytes | Yes | Cellular maturation and morphology (retina) | [21] |
| OPC and Oligodendrocytes | Yes | Proliferation, differentiation, myelination | [22,23,24,25] |
| Ependymal cells | Yes | Cellular structure (cilia reduction in Sox2 mutants) | [26] |
| Müller glia (retina) | Yes | Cellular structure and positioning | [27,28] |
| Bergmann glia (cerebellum) | Yes | Cellular morphology and positioning (in Sox2 mutants, defective motor control) | [29] |
| Macroglia in the peripheral nervous system | |||
| Schwann cells | Yes | Myelination | [30,31] |
| Satellite cells | Yes | Survival | [32,33] |
| Neurons | Sox2 Expression | Sox2 Function | References |
| Clock neurons in Suprachiasmatic nucleus | Yes | Regulation of circadian rhythms | [34] |
| Sensory thalamic nuclei (including DLG) | Yes | Signalling to incoming retinal axons; DLG growth; development of thalamic neurons projections to the visual and somatosensory cortex | [35] |
| Specific glutamatergic and cholinergic neurons in C. elegans | Yes | Differentiation of specific neuron-types | [36,37] |
| Cholinergic amacrine neurons | Yes | Cell positioning and dendritic stratification | [38] |
| GABA-ergic interneurons, rare pyramidal cells in the cortex | Yes | Morphology and maturation (impaired GABAergic neuroblast migration and differentiation; intraneuronal aggregates) | [26,39] |
| Some striatal cells | Yes | N.A. | [26] |
| Thalamic cells | Yes | N.A. | [26] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercurio, S.; Serra, L.; Nicolis, S.K. More than just Stem Cells: Functional Roles of the Transcription Factor Sox2 in Differentiated Glia and Neurons. Int. J. Mol. Sci. 2019, 20, 4540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184540
Mercurio S, Serra L, Nicolis SK. More than just Stem Cells: Functional Roles of the Transcription Factor Sox2 in Differentiated Glia and Neurons. International Journal of Molecular Sciences. 2019; 20(18):4540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184540
Chicago/Turabian StyleMercurio, Sara, Linda Serra, and Silvia K. Nicolis. 2019. "More than just Stem Cells: Functional Roles of the Transcription Factor Sox2 in Differentiated Glia and Neurons" International Journal of Molecular Sciences 20, no. 18: 4540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184540