Causes and Consequences of A Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism

 ,
,  ,
,  , , ,
, , , 
Abstract
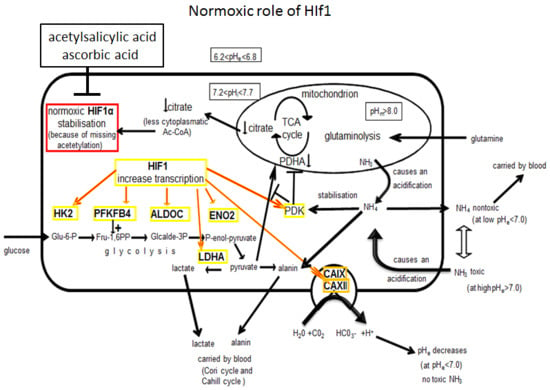
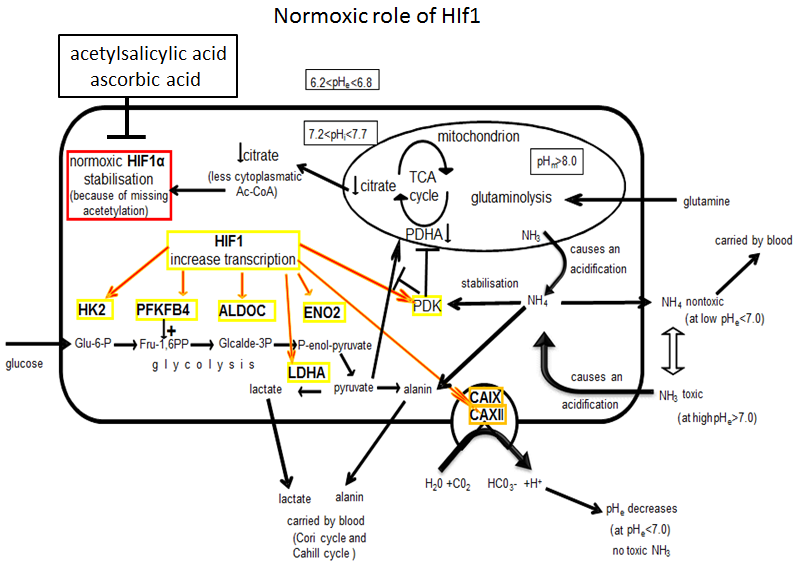
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
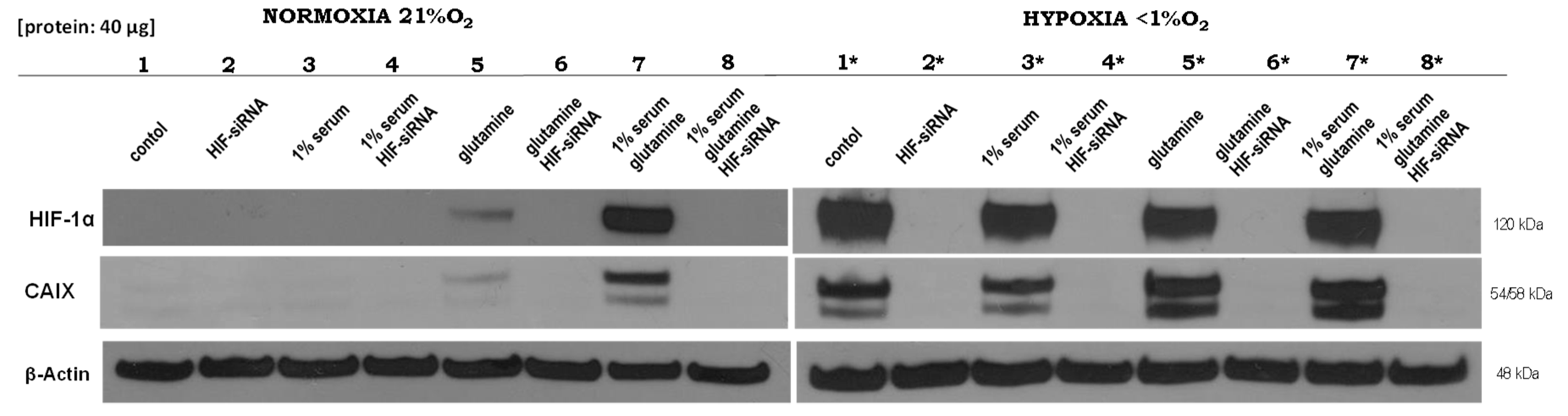
2.1. Expression of HIF1α and CAIX under Normoxic and Hypoxic Conditions
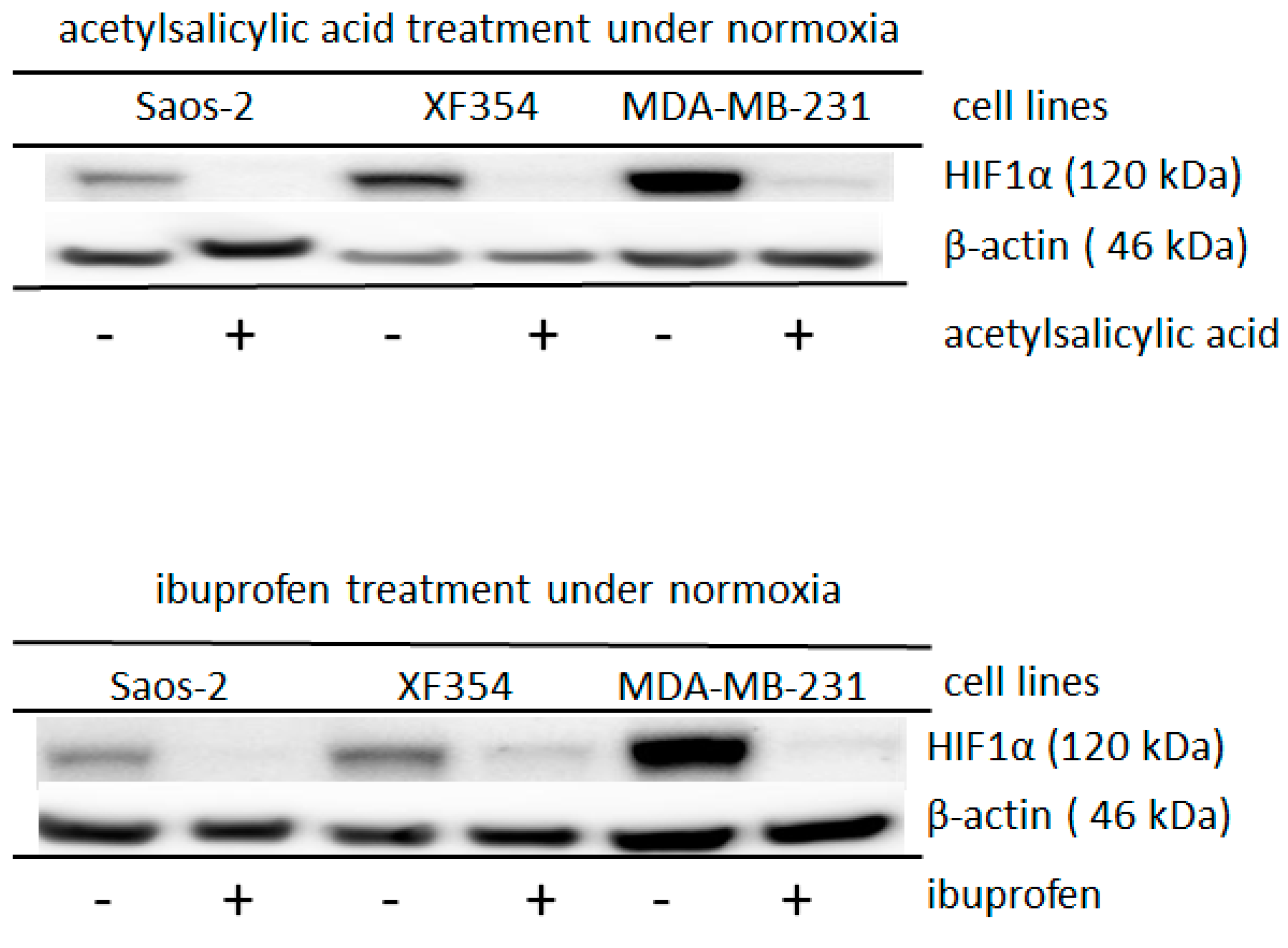
2.2. Role of Acetylation for the Normoxic and Hypoxic Stabilization of HIF1α
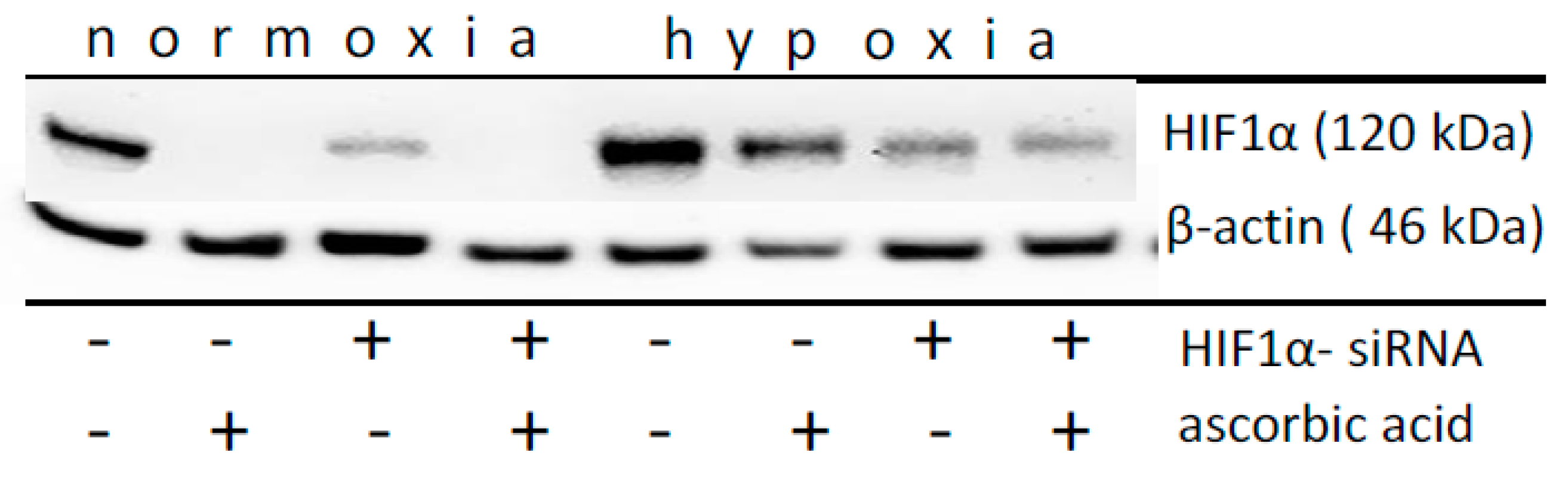
2.3. Effect of Ascorbic Acid on the Normoxic and Hypoxic Stabilization of HIF1α
2.4. Deep Sequencing Analysis of HIF1α-Positive or HIF1α-Negative Cells under Normoxic or Hypoxic Conditions
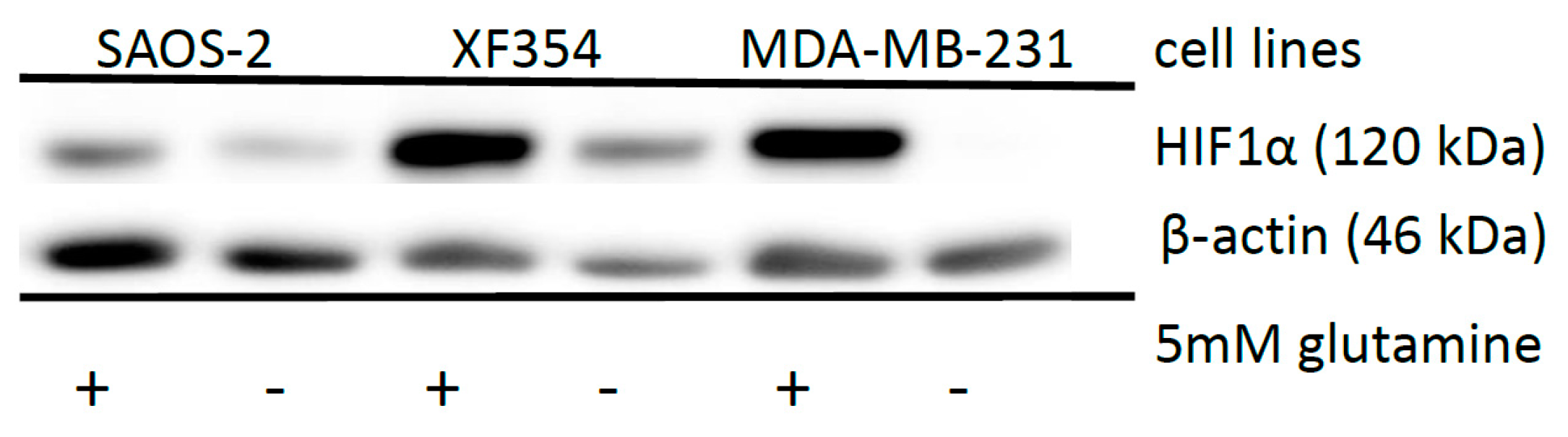
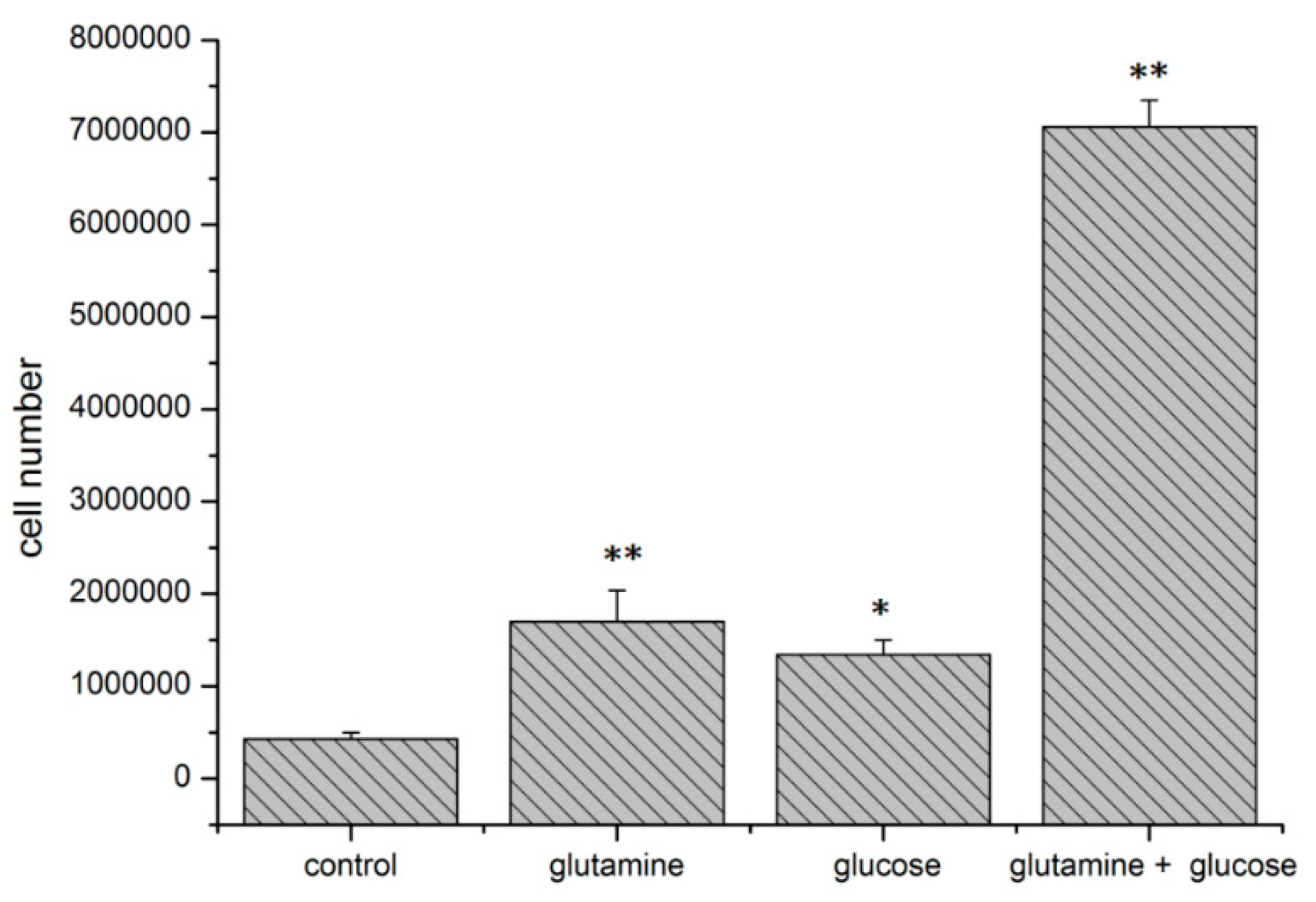
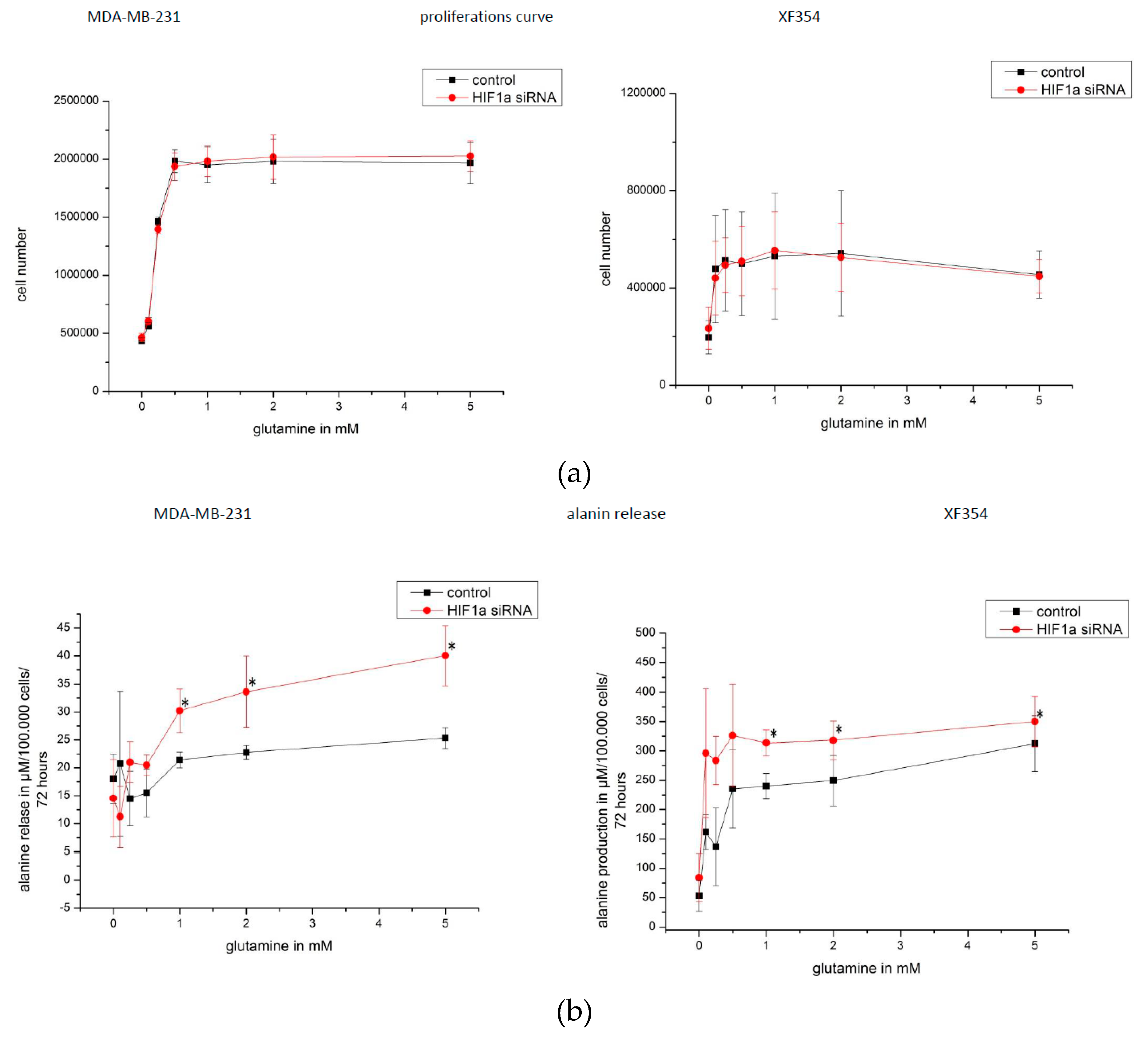
2.5. Effect of HIF1α and Different Glutamine Levels on Selected Metabolic Products in Two Tumor Cell Lines
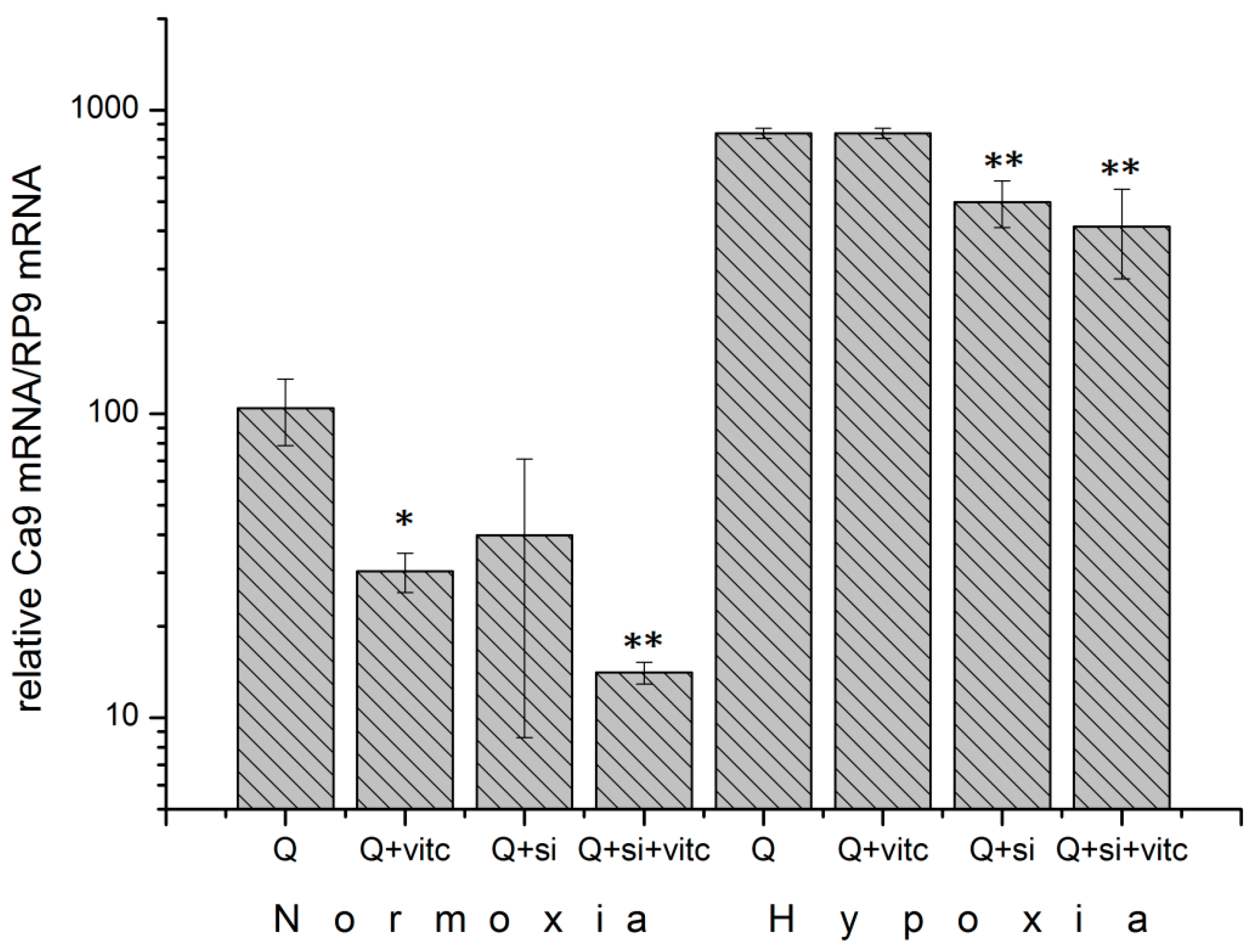
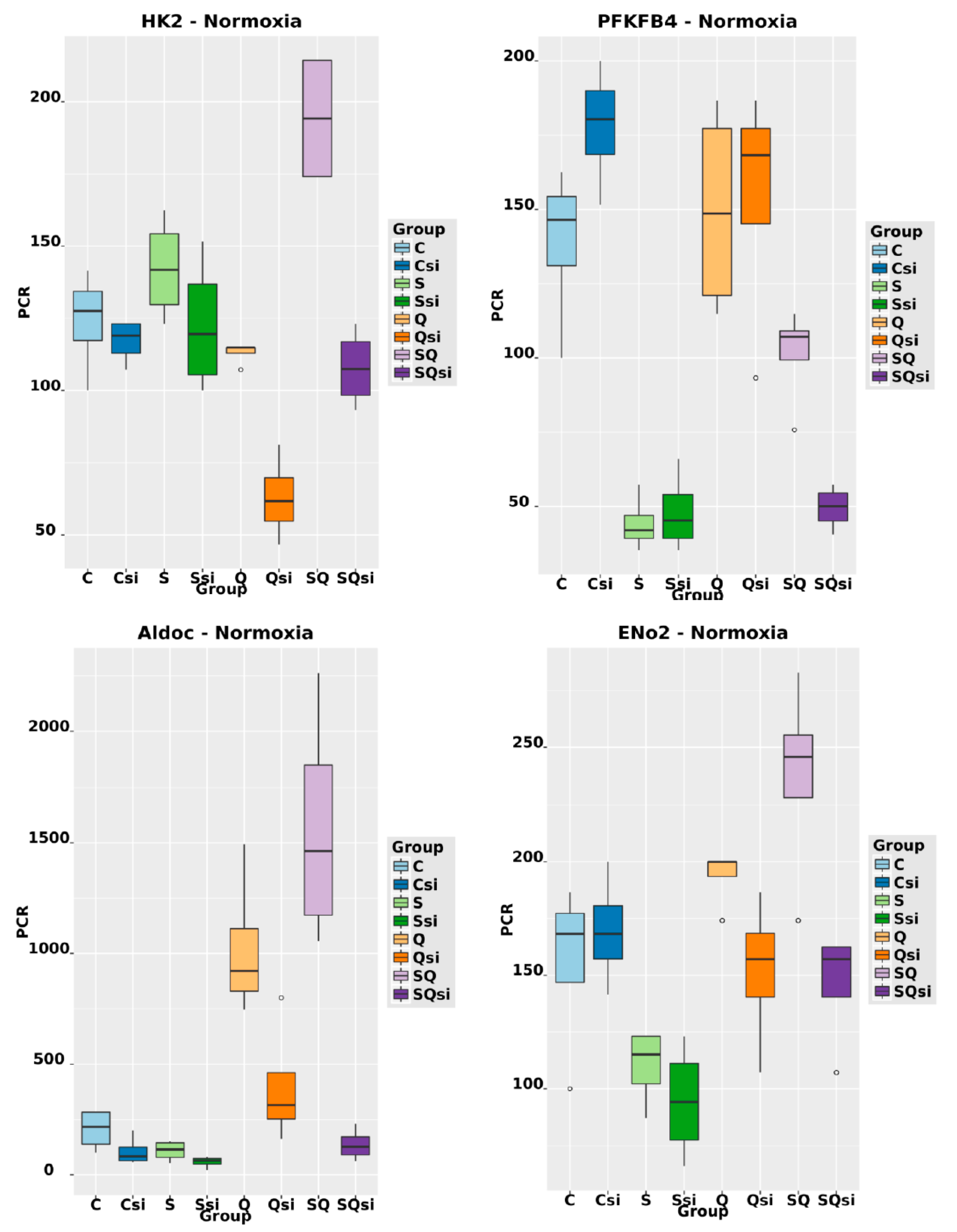
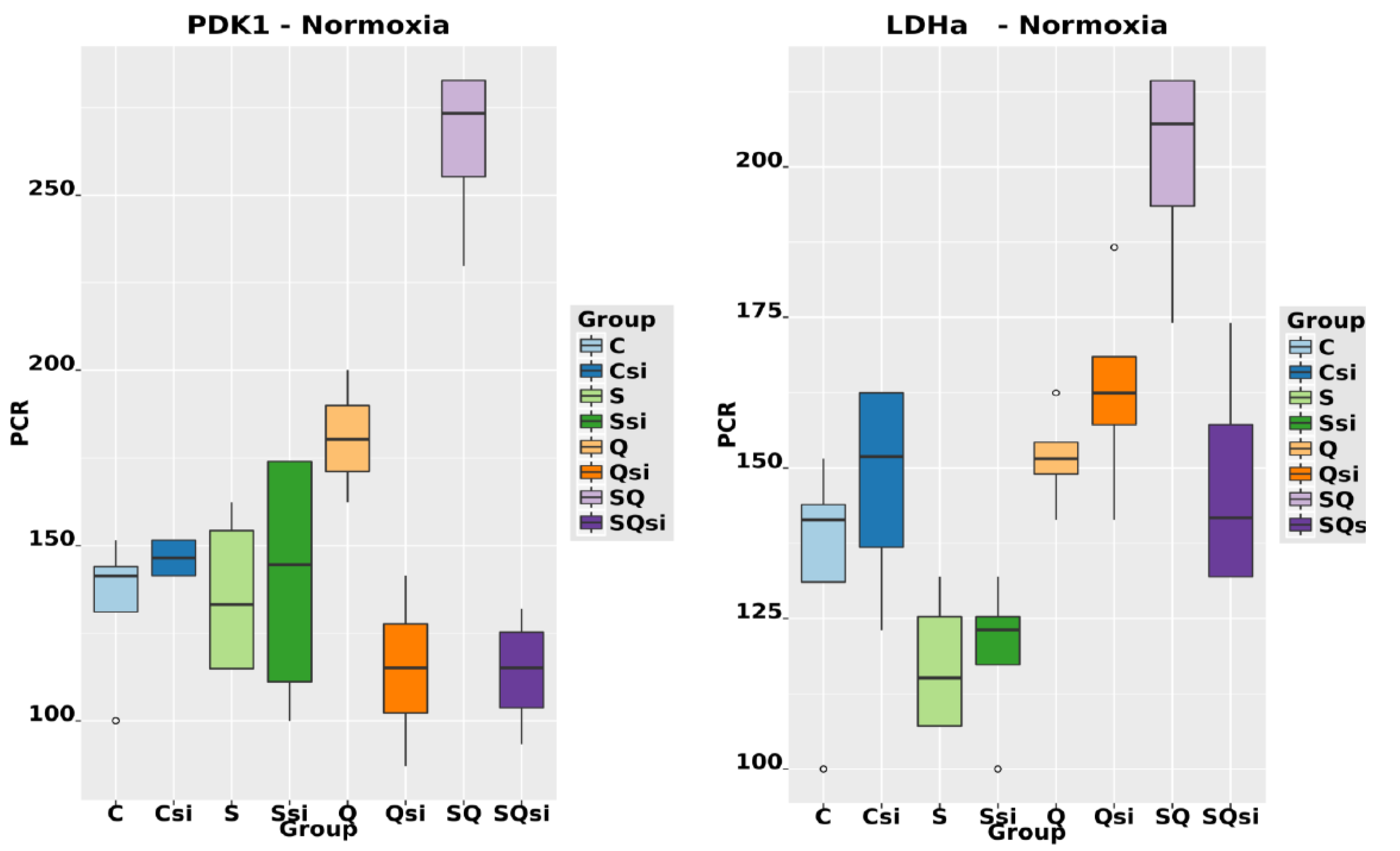
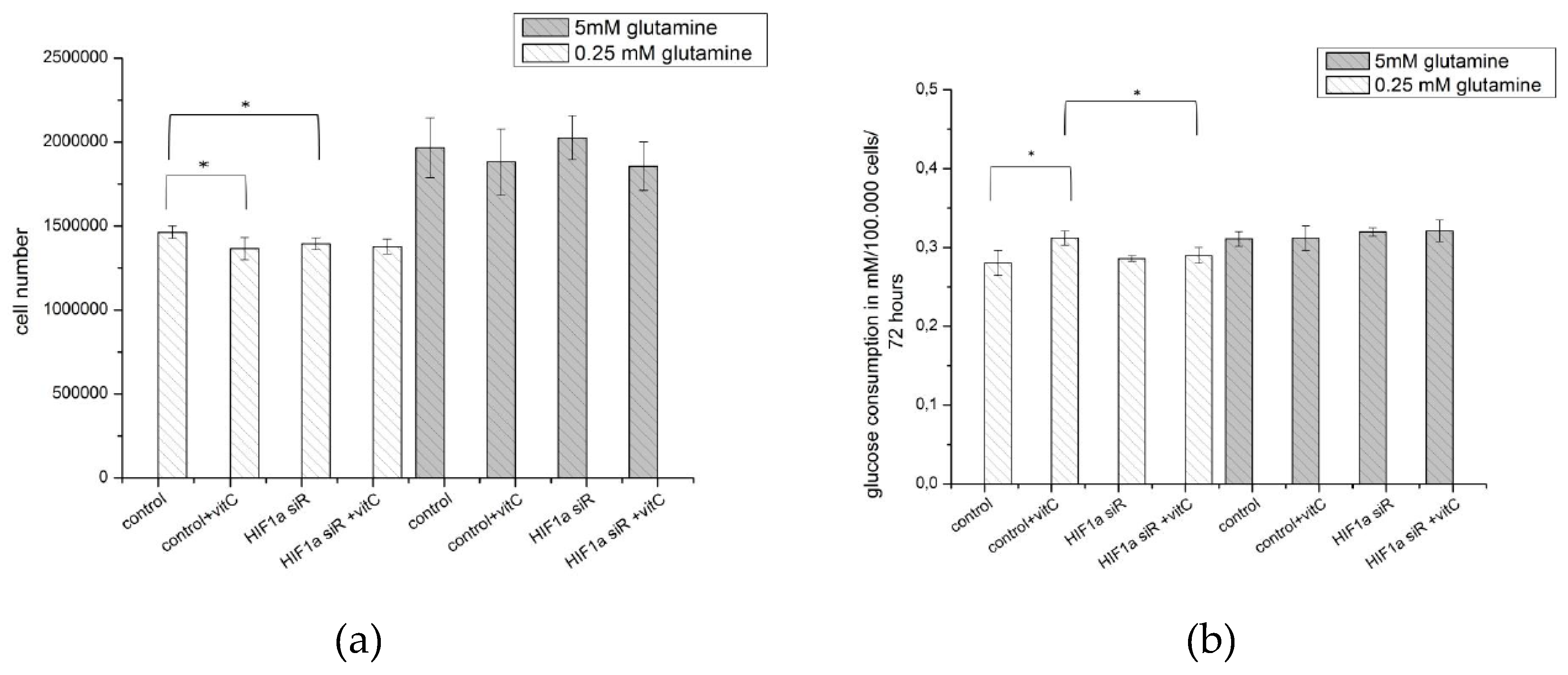
2.6. Effect of Ascorbic Acid on HIF1α and Select Metabolic Products under Normoxia at Different HIF1α Statuses and with the Addition of Two Different Concentrations of Glutamine
3. Discussion
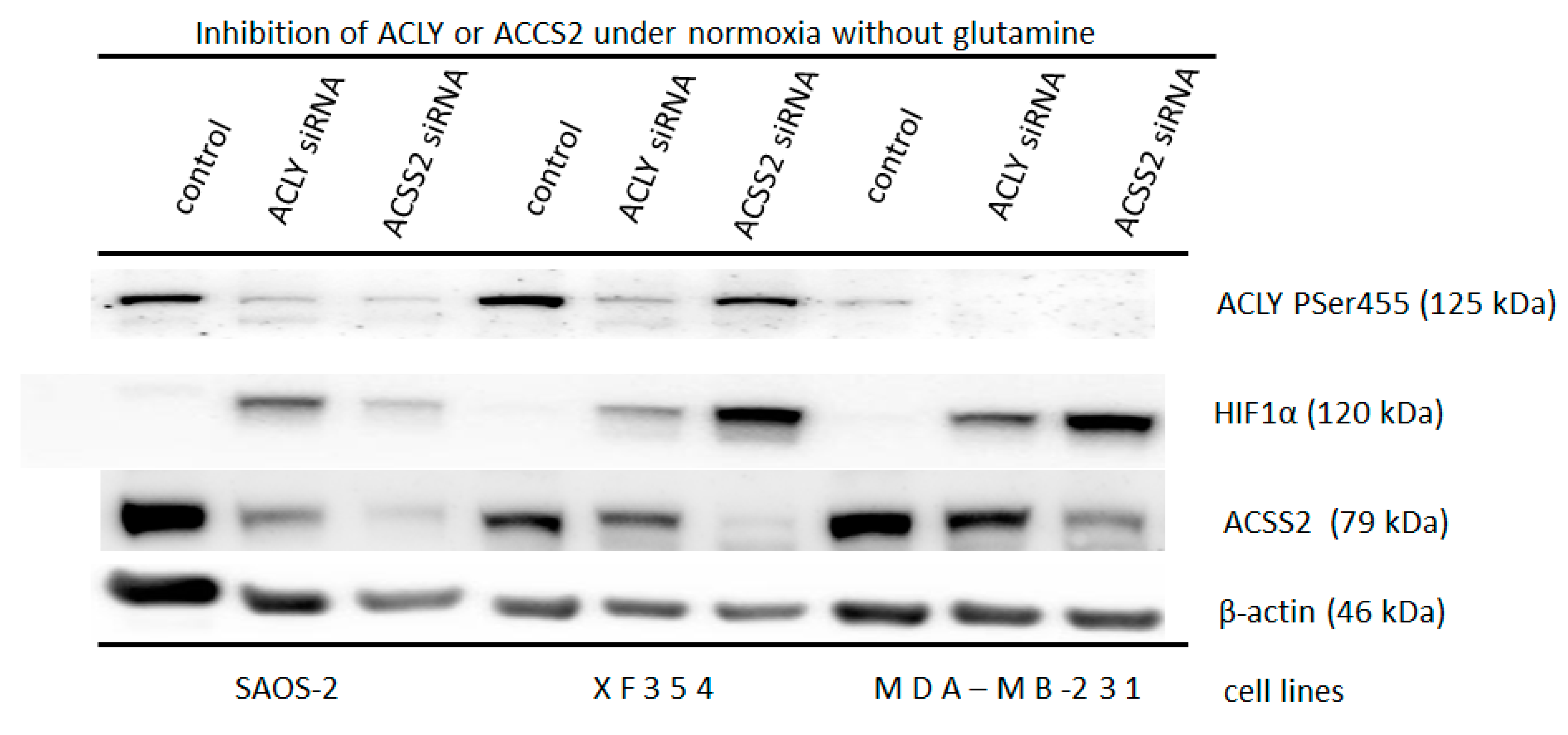
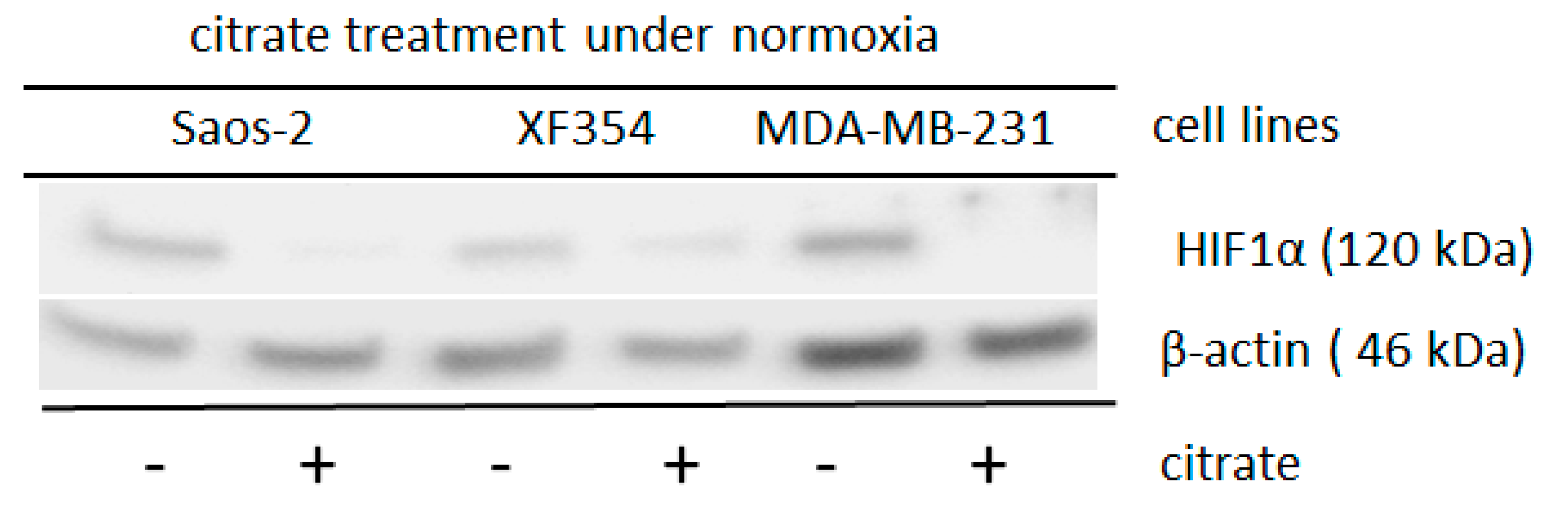
3.1. Molecular and Biochemical Reasons for the Normoxic HIF1 Activation in Tumor Cells
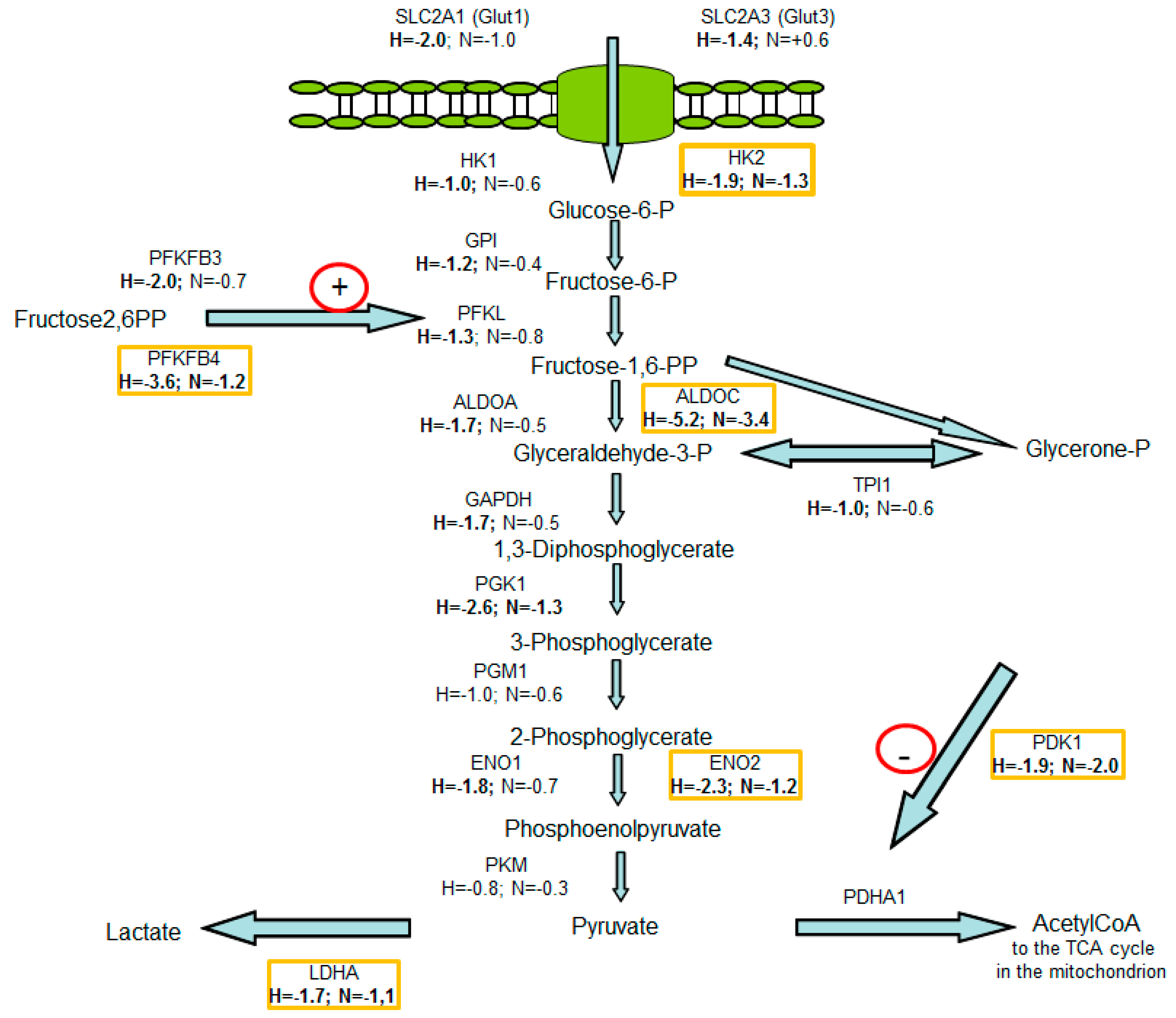
3.2. Consequences of Glutamine-Induced Normoxic HIF1 Activity for the Tumor Glycolysis/Metabolism
3.3. Consequences for A New Interpretation of the Warburg Effect
3.4. Therapeutic Consequences
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Transfection
4.3. Cell Proliferation Experiments
4.4. Metabolic Assay
4.5. Western Blot Analysis
4.6. RNA Preparation, cDNA Synthesis and Quantitative Real-Time PCR (qRT-PCR)
4.7. Deep Sequencing
4.8. Intracellular pH Measurement
4.9. Bioinformatics Approach and Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Dang, C.V.; Hamaker, M.; Sun, P.; Le, A.; Gao, P. Therapeutic targeting of cancer cell metabolism. J. Mol. Med. 2011, 89, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L. Therapeutic considerations of L-glutamine: A review of the literature. Altern. Med. Rev. 1999, 4, 239–248. [Google Scholar] [PubMed]
- Meng, M.; Chen, S.; Lao, T.; Liang, D.; Sang, N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle 2010, 9, 3921–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappler, M.; Pabst, U.; Rot, S.; Taubert, H.; Wichmann, H.; Schubert, J.; Bache, M.; Weinholdt, C.; Immel, U.-D.; Grosse, I.; et al. Normoxic accumulation of HIF1alpha is associated with glutaminolysis. Clin. Oral Investig. 2017, 21, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.R.; McSheehy, P.M.J.; Robinson, S.P.; Troy, H.; Chung, Y.-L.; Leek, R.D.; Williams, K.J.; Stratford, I.J.; Harris, A.L.; Stubbs, M. Metabolic changes detected by in vivo magnetic resonance studies of HEPA-1 wild-type tumors and tumors deficient in hypoxia-inducible factor-1beta (HIF-1beta): Evidence of an anabolic role for the HIF-1 pathway. Cancer Res. 2002, 62, 688–695. [Google Scholar] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Vordermark, D.; Kraft, P.; Katzer, A.; Bölling, T.; Willner, J.; Flentje, M. Glucose requirement for hypoxic accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha). Cancer Lett. 2005, 230, 122–133. [Google Scholar] [CrossRef]
- Peng, X.-H.; Karna, P.; Cao, Z.; Jiang, B.-H.; Zhou, M.; Yang, L. Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J. Biol. Chem. 2006, 281, 25903–25914. [Google Scholar] [CrossRef] [PubMed]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3’-kinase/Akt signaling pathway. Cancer Res. 2001, 61, 7349–7355. [Google Scholar]
- Dayan, F.; Bilton, R.L.; Laferrière, J.; Trottier, E.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J. Cell. Physiol. 2009, 218, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Höckel, M.; Vaupel, P. Endogenous hypoxia markers: Case not proven! Adv. Exp. Med. Biol. 2008, 614, 127–136. [Google Scholar] [PubMed]
- Wang, C.-W.; Purkayastha, A.; Jones, K.T.; Thaker, S.K.; Banerjee, U. In vivo genetic dissection of tumor growth and the Warburg effect. Elife 2016, 5, e18126. [Google Scholar] [CrossRef] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Parks, S.K.; Chiche, J.; Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Kwon, S.J.; Lee, Y.J. Effect of low glutamine/glucose on hypoxia-induced elevation of hypoxia-inducible factor-1alpha in human pancreatic cancer MiaPaCa-2 and human prostatic cancer DU-145 cells. Clin. Cancer Res. 2005, 11, 4694–4700. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Kappler, M.; Taubert, H.; Eckert, A.W. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 1845–1846. [Google Scholar] [PubMed]
- Kappler, M.; Taubert, H.; Schubert, J.; Vordermark, D.; Eckert, A.W. The real face of HIF1α in the tumor process. Cell Cycle 2012, 11, 3932–3936. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-W.; Bae, M.-K.; Ahn, M.-Y.; Kim, S.-H.; Sohn, T.-K.; Bae, M.-H.; Yoo, M.-A.; Song, E.J.; Lee, K.-J.; Kim, K.-W. Regulation and Destabilization of HIF-1α by ARD1-Mediated Acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Choi, S.Y.C.; Xue, H.; Wu, R.; Fazli, L.; Lin, D.; Collins, C.C.; Gleave, M.E.; Gout, P.W.; Wang, Y. The MCT4 Gene: A Novel, Potential Target for Therapy of Advanced Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Osipyants, A.I.; Poloznikov, A.A.; Smirnova, N.A.; Hushpulian, D.M.; Khristichenko, A.Y.; Chubar, T.A.; Zakhariants, A.A.; Ahuja, M.; Gaisina, I.N.; Thomas, B.; et al. L-ascorbic acid: A true substrate for HIF prolyl hydroxylase? Biochimie 2018, 147, 46–54. [Google Scholar] [CrossRef] [PubMed]
- KC, S.; Cárcamo, J.M.; Golde, D.W. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. Off. Publ. Fed. Am. Societies Exp. Biol. 2005, 19, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, R. Vitamin C, a Multi-Tasking Molecule, Finds a Molecular Target in Killing Cancer Cells. React. Oxyg. Species (Apex, N.C.) 2016, 1, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szarka, A.; Balogh, T. In silico aided thoughts on mitochondrial vitamin C transport. J. Theor. Biol. 2015, 365, 181–189. [Google Scholar] [CrossRef]
- Koivunen, P.; Fell, S.M.; Lu, W.; Rabinowitz, J.D.; Kung, A.L.; Schlisio, S. The 2-oxoglutarate analog 3-oxoglutarate decreases normoxic hypoxia-inducible factor-1alpha in cancer cells, induces cell death, and reduces tumor xenograft growth. Hypoxia (Auckland, N.Z.) 2016, 4, 15–27. [Google Scholar]
- Zhang, Y.; Ren, Y.-J.; Guo, L.-C.; Ji, C.; Hu, J.; Zhang, H.-H.; Xu, Q.-H.; Zhu, W.-D.; Ming, Z.-J.; Yuan, Y.-S.; et al. Nucleus accumbens-associated protein-1 promotes glycolysis and survival of hypoxic tumor cells via the HDAC4-HIF-1α axis. Oncogene 2017, 36, 4171–4181. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Guan, K.-L.; Xiong, Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 2011, 36, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Huang, J.-Y.; Schwer, B.; Verdin, E. SIRT3 Regulates Mitochondrial Protein Acetylation and Intermediary Metabolism; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2011. [Google Scholar]
- Paik, W.K.; Pearson, D.; Lee, H.W.; Kim, S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochim. et Biophys. Acta (BBA)-Nucleic Acids Protein Synth. 1970, 213, 513–522. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.-L.; Xiong, Y.; Lei, Q.-Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef]
- Bae, J.M.; Kim, J.H.; Oh, H.J.; Park, H.E.; Lee, T.H.; Cho, N.-Y.; Kang, G.H. Downregulation of acetyl-CoA synthetase 2 is a metabolic hallmark of tumor progression and aggressiveness in colorectal carcinoma. Mod. Pathol. 2017, 30, 267–277. [Google Scholar] [CrossRef]
- Tatham, M.H.; Cole, C.; Scullion, P.; Wilkie, R.; Westwood, N.J.; Stark, L.A.; Hay, R.T. A Proteomic Approach to Analyze the Aspirin-mediated Lysine Acetylome. Mol. Cell. Proteom. MCP 2017, 16, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Bourseau-Guilmain, E.; Menard, J.A.; Lindqvist, E.; Indira Chandran, V.; Christianson, H.C.; Cerezo Magaña, M.; Lidfeldt, J.; Marko-Varga, G.; Welinder, C.; Belting, M. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat. Commun. 2016, 7, 11371. [Google Scholar] [CrossRef]
- Gündel, D.; Allmeroth, M.; Reime, S.; Zentel, R.; Thews, O. Endocytotic uptake of HPMA-based polymers by different cancer cells: Impact of extracellular acidosis and hypoxia. Int. J. Nanomed. 2017, 12, 5571–5584. [Google Scholar] [CrossRef]
- Simon, L.M.; Axline, S.G.; Pesanti, E.L. Adaptations of phagocytosis and pinocytosis in mouse lung macrophages after sustained in vitro hypoxia. Am. Rev. Respir. Dis. 1981, 123, 64–68. [Google Scholar]
- Hernandez, V.J.; Weng, J.; Ly, P.; Pompey, S.; Dong, H.; Mishra, L.; Schwarz, M.; Anderson, R.G.W.; Michaely, P. Cavin-3 dictates the balance between ERK and Akt signaling. Elife 2013, 2, e00905. [Google Scholar] [CrossRef]
- McQueen, A.; Bailey, J.E. Effect of ammonium ion and extracellular pH on hybridoma cell metabolism and antibody production. Biotechnol. Bioeng. 1990, 35, 1067–1077. [Google Scholar] [CrossRef]
- Schuller, K.A.; Randall, D.D. Regulation of pea mitochondrial pyruvate dehydrogenase complex: Does photorespiratory ammonium influence mitochondrial carbon metabolism? Plant Physiol. 1989, 89, 1207–1212. [Google Scholar] [CrossRef]
- Perez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; van Hee, V.F.; de Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell cycle (Georgetown, Tex.) 2016, 15, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef]
- Felig, P.; Pozefsk, T.; Marlis, E.; Cahill, G.F. Alanine: Key Role in Gluconeogenesis. Science 1970, 167, 1003–1004. [Google Scholar] [CrossRef]
- Martin, G.; Baverel, G. Lactate, alanine and glutamine metabolism in isolated canine pup liver cells. Biochim. Et Biophys. Acta (BBA)—Gen. Subj. 1983, 760, 230–237. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Warburg, O.; Posener, K.; Negelein, E. Über den Stoffwechsel der Carcinomzelle. Biochemische Zeitschrift 1924, 152, 309–344. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.R.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Algra, A.; Chen, Z.; Diener, H.-C.; Norrving, B.; Mehta, Z. Effects of aspirin on risk and severity of early recurrent stroke after transient ischaemic attack and ischaemic stroke: Time-course analysis of randomised trials. Lancet 2016, 388, 365–375. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.U.; Munn, D.; Currie, M.J.; Robinson, B.A.; Pearson, J.F.; Vissers, M.C.M. Increased Tumor Ascorbate is Associated with Extended Disease-Free Survival and Decreased Hypoxia-Inducible Factor-1 Activation in Human Colorectal Cancer. Front. Oncol. 2014, 4, 10. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.U.; Currie, M.J.; Vissers, M.C.M. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic. Biol. Med. 2014, 69, 308–317. [Google Scholar] [CrossRef]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.; Potente, M. New Q(ues) to keep blood vessels growing. EMBO J. 2017, 36, 2315–2317. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: Its importance in health and disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Sontakke, P.; Koczula, K.M.; Jaques, J.; Wierenga, A.T.J.; Brouwers-Vos, A.Z.; Pruis, M.; Gunther, U.L.; Vellenga, E.; Schuringa, J.J. Hypoxia-Like Signatures Induced by BCR-ABL Potentially Alter the Glutamine Uptake for Maintaining Oxidative Phosphorylation. PLoS ONE 2016, 11, e0153226. [Google Scholar] [CrossRef]
- Stegen, S.; van Gastel, N.; Eelen, G.; Ghesquière, B.; D’Anna, F.; Thienpont, B.; Goveia, J.; Torrekens, S.; van Looveren, R.; Luyten, F.P.; et al. HIF-1α Promotes Glutamine-Mediated Redox Homeostasis and Glycogen-Dependent Bioenergetics to Support Postimplantation Bone Cell Survival. Cell Metab. 2016, 23, 265–279. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Guo, L.; Zhou, B.; Liu, Z.; Xu, Y.; Lu, H.; Xia, M.; Guo, E.; Shan, W.; Chen, G.; Wang, C. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11007–11015. [Google Scholar] [CrossRef]
- Eckert, A.W.; Wickenhauser, C.; Salins, P.C.; Kappler, M.; Bukur, J.; Seliger, B. Clinical relevance of the tumor microenvironment and immune escape of oral squamous cell carcinoma. J. Transl. Med. 2016, 14, 85. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Cui, H.; Qin, Q.; Yang, M.; Zhang, H.; Liu, Z.; Yang, Y.; Chen, X.; Zhu, H.; Wang, D.; Meng, C.; et al. Bortezomib enhances the radiosensitivity of hypoxic cervical cancer cells by inhibiting HIF-1α expression. Int. J. Clin. Exp. Pathol. 2015, 8, 9032–9041. [Google Scholar]
- Kappler, M.; Rot, S.; Taubert, H.; Greither, T.; Bartel, F.; Dellas, K.; Hänsgen, G.; Trott, K.-R.; Bache, M. The effects of knockdown of wild-type survivin, survivin-2B or survivin-3 on the radiosensitization in a soft tissue sarcoma cells in vitro under different oxygen conditions. Cancer Gene Ther. 2007, 14, 994–1001. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics 2018, 28, 2520–2522. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33)[Software]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 14 March 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear Models for Microarray Data; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Team, R.C. R: A language and environment for statistical computing. Computing 2013, 1, 201. [Google Scholar]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kappler, M.; Pabst, U.; Weinholdt, C.; Taubert, H.; Rot, S.; Kaune, T.; Kotrba, J.; Porsch, M.; Güttler, A.; Bache, M.; et al. Causes and Consequences of A Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism. Int. J. Mol. Sci. 2019, 20, 4742. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194742
Kappler M, Pabst U, Weinholdt C, Taubert H, Rot S, Kaune T, Kotrba J, Porsch M, Güttler A, Bache M, et al. Causes and Consequences of A Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism. International Journal of Molecular Sciences. 2019; 20(19):4742. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194742
Chicago/Turabian StyleKappler, Matthias, Ulrike Pabst, Claus Weinholdt, Helge Taubert, Swetlana Rot, Tom Kaune, Johanna Kotrba, Martin Porsch, Antje Güttler, Matthias Bache, and et al. 2019. "Causes and Consequences of A Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism" International Journal of Molecular Sciences 20, no. 19: 4742. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194742