Optimisation and Benchmarking of Targeted Amplicon Sequencing for Mycobiome Analysis of Respiratory Specimens

 ,
,
Abstract
:1. Introduction
2. Results
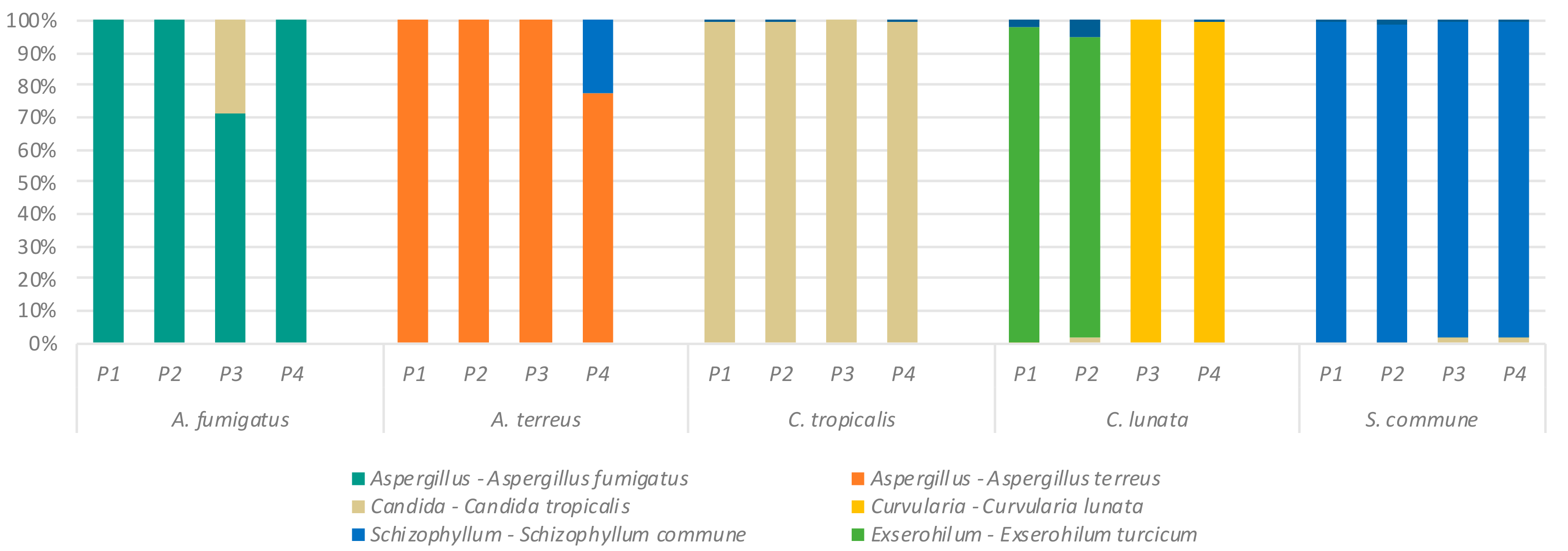
2.1. Detection of Specific Fungal Taxa by ITS Sequencing
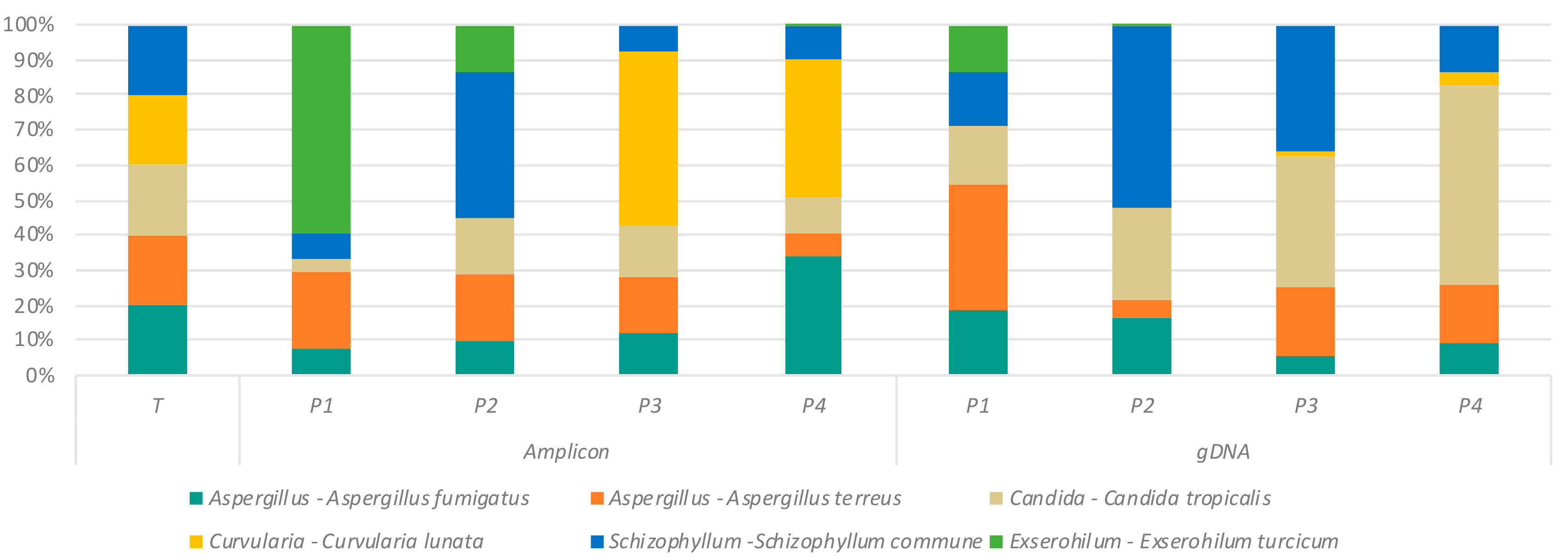
2.2. Effect of Primer Selection on Recovery of Fungal Mock Community Structure
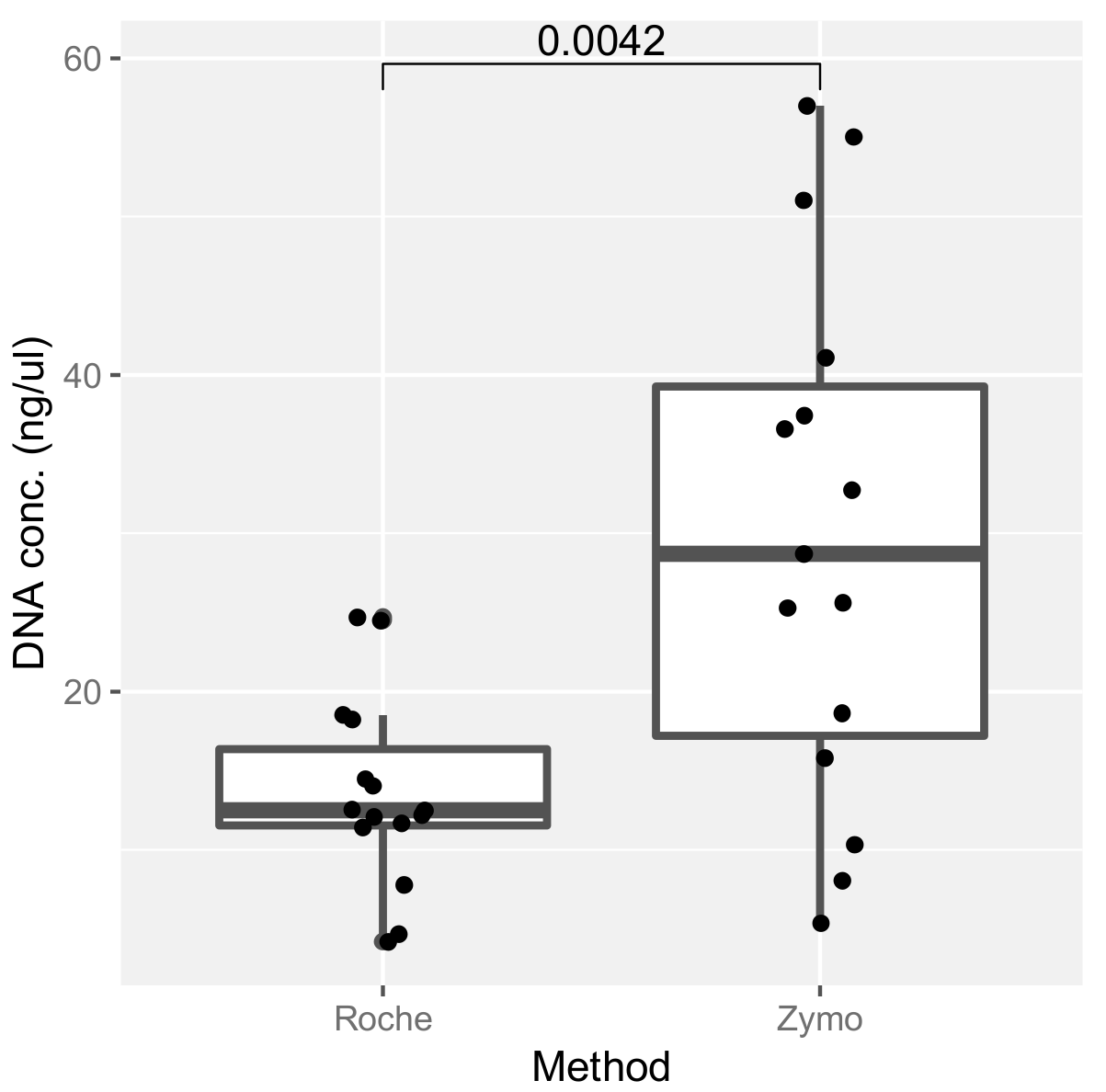
2.3. Assessment of DNA Extraction Methods and Their Effect on ITS Amplification
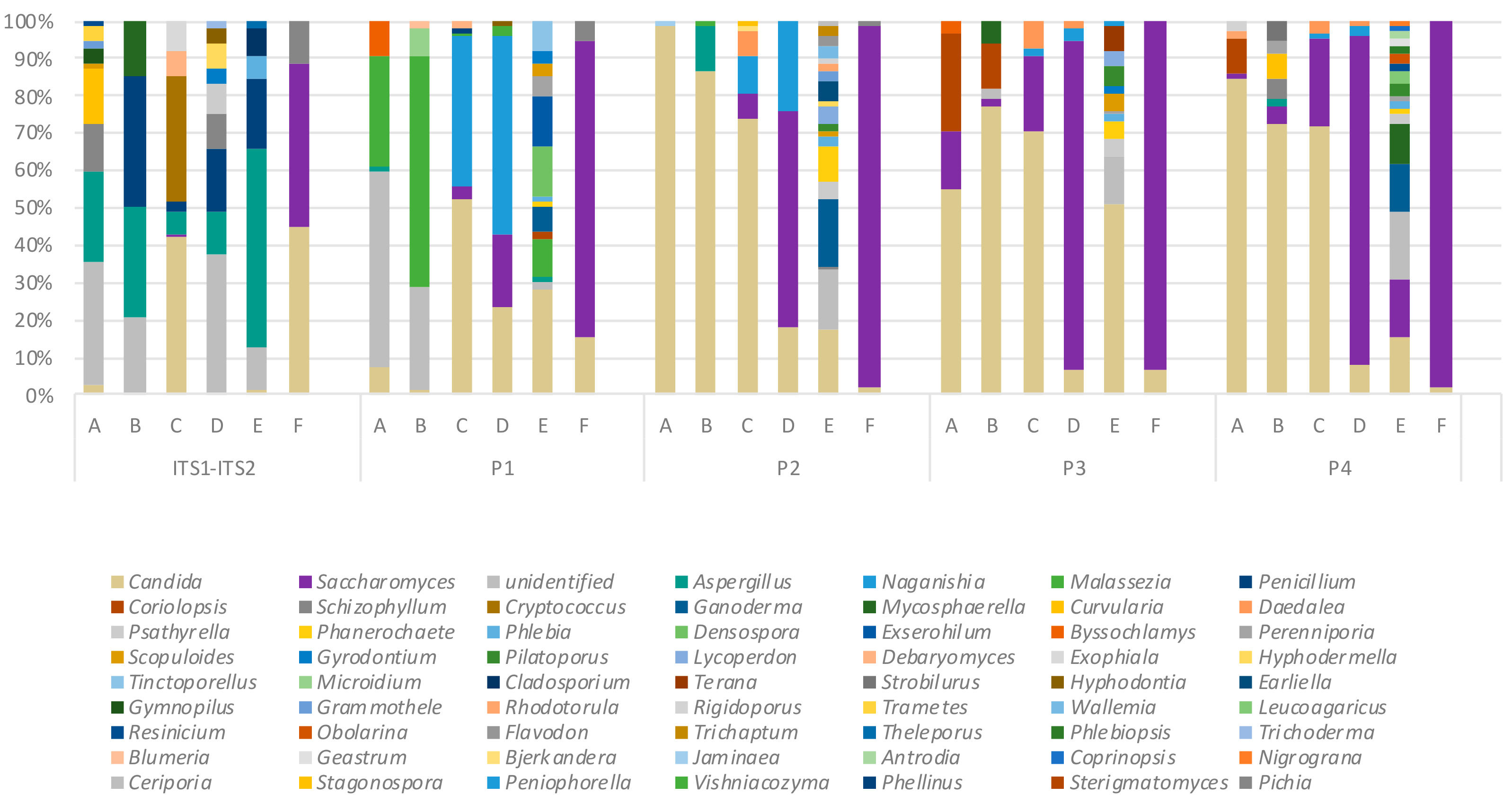
2.4. Variation in Mycobiome Profiles Derived by Primer Sets P1–P4 Compared to Dual ITS1-ITS2 Targeted Amplicon Shotgun Sequencing
2.5. Key Variables in Determining ITS Mycobiome Profiles of Clinical Respiratory Samples
3. Discussion
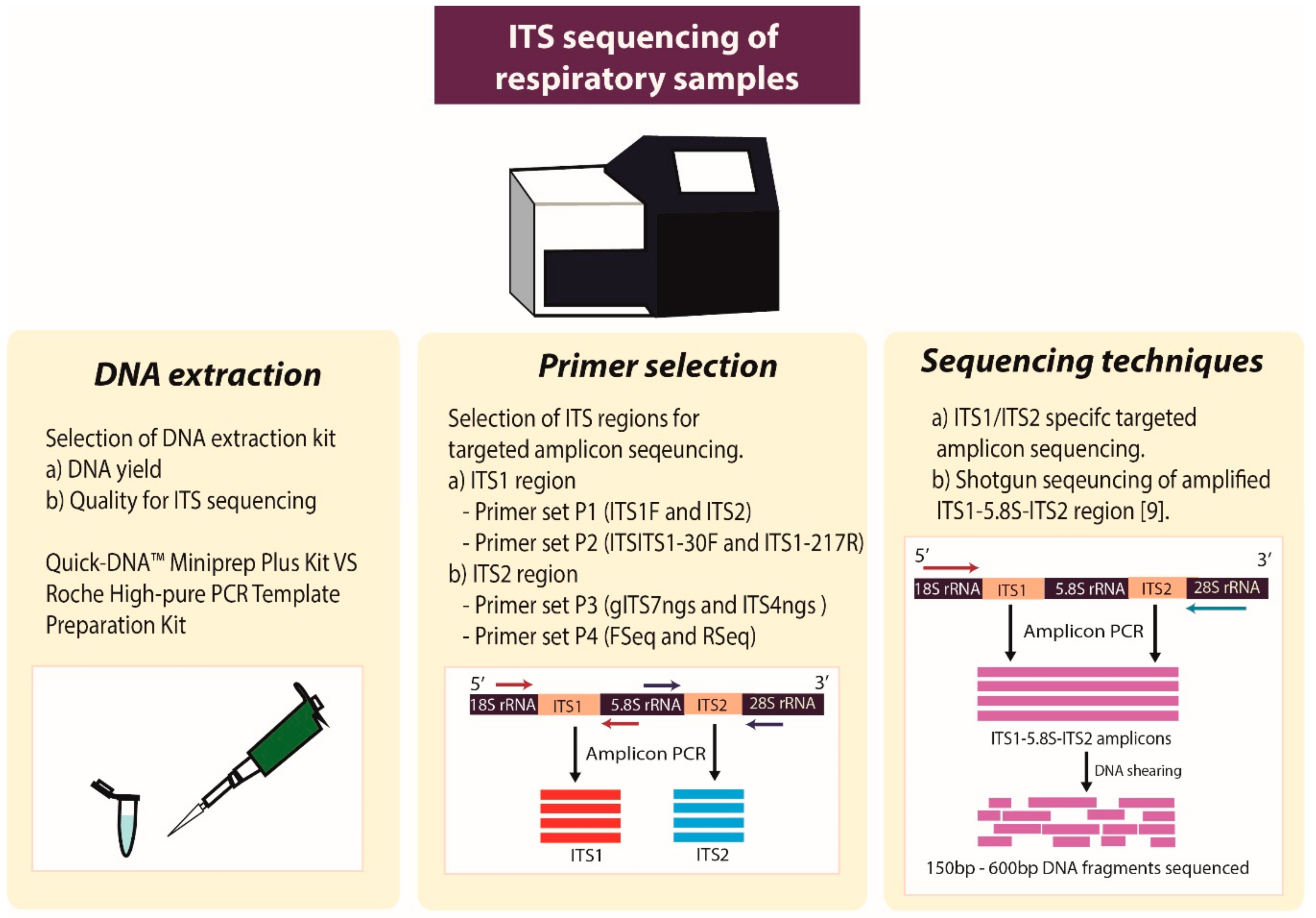
4. Materials and Methods
4.1. Clinical Samples and Cultured Fungal Isolates
4.2. DNA Extraction and Quantification
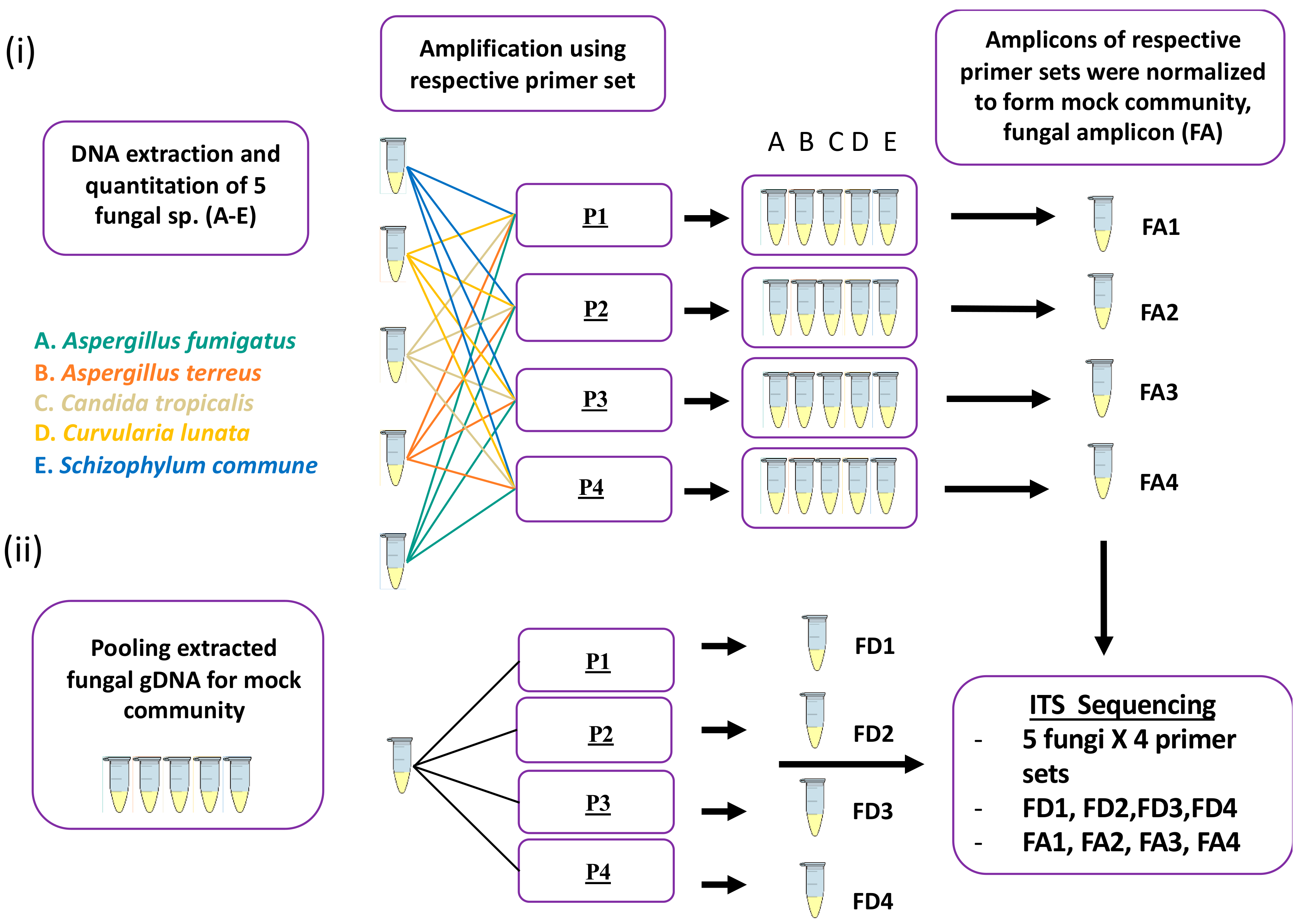
4.3. Mock Community Preparation
4.4. PCR Amplification
4.5. Sequencing: Library Preparation and Subsequent Sequencing
4.6. Data Analysis
4.7. Ethics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ITS | internally transcribed spacer |
| COPD | chronic obstructive pulmonary disease |
References
- Fairlamb, A.H.; Gow, N.A.; Matthews, K.R.; Waters, A.P. Stop neglecting fungi. Nat. Microbol. 2017, 2, 17120. [Google Scholar]
- Chotirmall, S.H.; Martin-Gomez, M.T. Aspergillus Species in Bronchiectasis: Challenges in the Cystic Fibrosis and Non-cystic Fibrosis Airways. Mycopathologia 2018, 183, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, G.B.; Zain, N.M.; Bruce, K.D.; Burr, L.D.; Chen, A.C.; Rivett, D.W.; McGuckin, M.A.; Serisier, D.J. A Novel Microbiota Stratification System Predicts Future Exacerbations in Bronchiectasis. Ann. Am. Thorac. Soc. 2014, 11, 496–503. [Google Scholar] [CrossRef]
- Cox, M.J.; Turek, E.M.; Hennessy, C.; Mirza, G.K.; James, P.L.; Coleman, M.; Jones, A.; Wilson, R.; Bilton, D.; Cookson, W.O.C.; et al. Longitudinal Assessment of Sputum Microbiome by Sequencing of the 16S rRNA Gene in Non-Cystic Fibrosis Bronchiectasis Patients. PLoS ONE 2017, 12, e0170622. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Viscogliosi, E.; Delhaes, L. The Lung Mycobiome: an Emerging Field of the Human Respiratory Microbiome. Front. Microbiol. 2015, 6, 89. [Google Scholar] [CrossRef]
- Fraczek, M.G.; Chishimba, L.; Niven, R.M.; Bromley, M.; Simpson, A.; Smyth, L.; Denning, D.W.; Bowyer, P. Corticosteroid Treatment is Associated with Increased Filamentous Fungal Burden in Allergic Fungal Disease. J. Allergy Clin. Immunol. 2018, 142, 407–414. [Google Scholar] [CrossRef]
- Chandrasekaran, R.; Mac Aogain, M.; Chalmers, J.D.; Elborn, S.J.; Chotirmall, S.H. Geographic Variation in the Aetiology, Epidemiology and Microbiology of Bronchiectasis. BMC Pulm. Med. 2018, 18, 83. [Google Scholar] [CrossRef]
- Mac Aogain, M.; Chandrasekaran, R.; Lim, A.Y.H.; Low, T.B.; Tan, G.L.; Hassan, T.; Ong, T.H.; Hui Qi Ng, A.; Bertrand, D.; Koh, J.Y.; et al. Immunological Corollary of the Pulmonary Mycobiome in Bronchiectasis: the CAMEB Study. Eur. Respir. J. 2018, 52, 1800766. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Nilsson, R.H.; Tedersoo, L.; Abarenkov, K.; Carlsen, T.; Kjoller, R.; Koljalg, U.; Pennanen, T.; Rosendahl, S.; Stenlid, J.; et al. Fungal Community Analysis by High-throughput Sequencing of Amplified Markers--a User’s Guide. New Phytol. 2013, 199, 288–299. [Google Scholar] [CrossRef]
- McTaggart, L.R.; Copeland, J.K.; Surendra, A.; Wang, P.W.; Husain, S.; Coburn, B.; Guttman, D.S.; Kus, J.V. Mycobiome Sequencing and Analysis Applied to Fungal Community Profiling of the Lower Respiratory Tract During Fungal Pathogenesis. Front. Microbiol. 2019, 10, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huseyin, C.E.; O’Toole, P.W.; Cotter, P.D.; Scanlan, P.D. Forgotten Fungi- the Gut Mycobiome in Human Health and Disease. FEMS Microbiol. Rev. 2017, 41, 479–511. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome Diversity: High-Throughput Sequencing and Identification of Fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Begerow, D.; Nilsson, H.; Unterseher, M.; Maier, W. Current State and SPrspectives of Fungal DNA Barcoding and Rapid Identification Procedures. Appl. Microbiol. Biotechnol. 2010, 87, 99–108. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear Ribosomal Internal Transcribed Spacer (ITS) Region as a Universal DNA Barcode Marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Usyk, M.; Zolnik, C.P.; Patel, H.; Levi, M.H.; Burk, R.D. Novel ITS1 Fungal Primers for Characterization of the Mycobiome. mSphere 2017, 2, e00488–17. [Google Scholar] [CrossRef]
- Bates, S.T.; Ahrendt, S.; Bik, H.M.; Bruns, T.D.; Caporaso, J.G.; Cole, J.; Dwan, M.; Fierer, N.; Gu, D.; Houston, S.; et al. Meeting Report: Fungal ITS Workshop (October 2012). Stand. Genom. Sci. 2013, 8, 118–123. [Google Scholar] [CrossRef]
- Koetschan, C.; Forster, F.; Keller, A.; Schleicher, T.; Ruderisch, B.; Schwarz, R.; Muller, T.; Wolf, M.; Schultz, J. The ITS2 Database III--Sequences and Structures for Phylogeny. Nucleic Acids Res. 2010, 38, D275–279. [Google Scholar] [CrossRef]
- Coleman, A.W. Is there a Molecular Key to the level of “Biological Species” in Eukaryotes? A DNA guide. Mol. Phylogenet. Evol. 2009, 50, 197–203. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA Genes for Phylogenetics; Elsevier Inc.: Amsterdam, The Netherlands, 1990; pp. 315–322. [Google Scholar]
- Tedersoo, L.; Lindahl, B. Fungal identification biases in microbiome projects. Environ. Microbiol. Rep. 2016, 8, 774–779. [Google Scholar] [CrossRef]
- Ihrmark, K.; Bodeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandstrom-Durling, M.; Clemmensen, K.E.; et al. New Primers to Amplify the Fungal ITS2 Region--Evaluation by 454-sequencing of Artificial and Natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.L.; Dominguez-Bello, M.G.; Heisel, T.; Al-Ghalith, G.; Knights, D.; Gale, C.A. Development of the Human Mycobiome over the First Month of Life and across Body Sites. mSystems 2018, 3, e00140–17. [Google Scholar] [CrossRef] [PubMed]
- Heisel, T.; Podgorski, H.; Staley, C.M.; Knights, D.; Sadowsky, M.J.; Gale, C.A. Complementary Amplicon-Based Genomic Approaches for the Study of Fungal Communities in Humans. PLoS ONE 2015, 10, e0116705. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Dickson, R.P.; Moore, B.B. The Lung Microbiome, Immunity, and the Pathogenesis of Chronic Lung Disease. J. Immunol. 2016, 196, 4839–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An Open Annotation Tool for Parsing Fungal Community Datasets by Ecological Guild. Fungal. Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Puusepp, R.; Nilsson, R.H.; James, T.Y. Novel Soil-Inhabiting Clades Fill Gaps in the Fungal Tree of Life. Microbiome 2017, 5, 42. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Gellatly, S.L.; Budden, K.F.; Mac Aogain, M.; Shukla, S.D.; Wood, D.L.; Hugenholtz, P.; Pethe, K.; Hansbro, P.M. Microbiomes in Respiratory Health and Disease: An Asia-Pacific Perspective. Respirology. 2017, 22, 240–250. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional Effects of the Microbiota in Chronic Respiratory Disease. Lancet. Respir. Med. 2019. [Google Scholar] [CrossRef]
- Tipton, L.; Ghedin, E.; Morris, A. The Lung Mycobiome in the Next-Generation Sequencing Era. Virulence 2017, 8, 334–341. [Google Scholar] [CrossRef]
- Leaw, S.N.; Chang, H.C.; Sun, H.F.; Barton, R.; Bouchara, J.P.; Chang, T.C. Identification of Medically Important Yeast Species by Sequence Analysis of the Internal Transcribed Spacer Regions. J. Clin. Microbiol. 2006, 44, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.H.; Su, J.H.; Shang, J.J.; Wu, Y.Y.; Li, Y.; Bao, D.P.; Yao, Y.J. Evaluation of the Ribosomal DNA Internal Transcribed Spacer (ITS), Specifically ITS1 and ITS2, for the Analysis of Fungal Diversity by Deep Sequencing. PLoS One 2018, 13, e0206428. [Google Scholar] [CrossRef] [PubMed]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an Environmental DNA Barcode for Fungi: an In Silico Approach Reveals Potential PCR Biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Motooka, D.; Fujimoto, K.; Tanaka, R.; Yaguchi, T.; Gotoh, K.; Maeda, Y.; Furuta, Y.; Kurakawa, T.; Goto, N.; Yasunaga, T.; et al. Fungal ITS1 Deep-Sequencing Strategies to Reconstruct the Composition of a 26-Species Community and Evaluation of the Gut Mycobiota of Healthy Japanese Individuals. Front. Microbiol. 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halwachs, B.; Madhusudhan, N.; Krause, R.; Nilsson, R.H.; Moissl-Eichinger, C.; Högenauer, C.; Thallinger, G.G.; Gorkiewicz, G. Critical Issues in Mycobiota Analysis. Front. Microbiol. 2017, 8, 180. [Google Scholar] [CrossRef] [Green Version]
- Frau, A.; Kenny, J.G.; Lenzi, L.; Campbell, B.J.; Ijaz, U.Z.; Duckworth, C.A.; Burkitt, M.D.; Hall, N.; Anson, J.; Darby, A.C.; et al. DNA Extraction and Amplicon Production Strategies Deeply Influence the Outcome of Gut Mycobiome Studies. Sci. Rep. 2019, 9, 9328. [Google Scholar] [CrossRef]
- Vesty, A.; Biswas, K.; Taylor, M.W.; Gear, K.; Douglas, R.G. Evaluating the Impact of DNA Extraction Method on the Representation of Human Oral Bacterial and Fungal Communities. PLoS ONE 2017, 12, e0169877. [Google Scholar] [CrossRef]
- Illumina. Fungal Metagenomic Sequencing Demonstrated Protocol (1000000064940 v01). Available online: https://support.illumina.com/downloads/fungal-metagenomic-sequencing-demonstrated-protocol-1000000064940.html (accessed on 2 May 2019).
- Anslan, S.; Nilsson, R.H.; Wurzbacher, C.; Baldrian, P.; Leho, T.; Bahram, M. Great Differences in Performance and Outcome of High-throughput Sequencing Data Analysis Platforms for Fungal Metabarcoding. MycoKeys 2018, 39, 29–40. [Google Scholar] [CrossRef]
- Gangneux, J.P.; Guegan, H.; Vandenborght, L.E.; Buffet-Bataillon, S.; Enaud, R.; Delhaes, L.; ECMM-ESCMID NGS Study Group. A European ECMM-ESCMID Survey on Goals and Practices for Mycobiota Characterization using Next Generation Sequencing. Mycoses 2019. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Fungal Species |
|---|---|
| Ascomycota | Aspergillus fumigatus |
| Aspergillus terreus | |
| Candida tropicalis | |
| Curvularia lunata | |
| Basidiomycota | Schizophyllum commune |
| Primer Set | Target | Forward Primer (5′ > 3′) | Reverse Primer (3′ > 5′) | References |
|---|---|---|---|---|
| P1 | ITS1 | ITS1F: CTTGGTCATTTAGAGGAAGTAA | ITS2: GCTGCGTTCTTCATCGATGC | [20] |
| P2 | ITS1 | ITS1-30F: GTCCCTGCCCTTTGTACACA | ITS1-217R: TTTCGCTGCGTTCTTCATCG | [16] |
| P3 | ITS2 | gITS7ngs: GTGARTCATCRARTYTTTG | ITS4ngs: TCCTSCGCTTATTGATATGC | [13,21,22] |
| P4 | ITS2 | Fseq: ATGCCTGTTTGAGCGTC | Rseq: CCTACCTGATTTGAGGTC | [23,24] |
| Method | Primer Set | |||
|---|---|---|---|---|
| P1 | P2 | P3 | P4 | |
| Roche | 53% | 80% | 40% | 93% |
| Zymo | 53% | 60% | 33% | 80% |
| Characteristics | COPD Samples (n = 15) | Bronchiectasis Samples (n = 6) |
|---|---|---|
| Age (years): Mean ± SD | 69 ± 9.2 | 64.6 ± 9.2 |
| Gender (male): n (%) | 14 (93.3) | 3 (50.0) |
| BMI (kg/m2): Mean ± SD | 22.2 ± 5.5 | 22.5 ± 6.1 |
| Smoking history: n (%) | ||
| Nonsmoker | 0 (0) | 3 (50.0) |
| Current | 8 (53.3) | 0 (0) |
| Ex-smoker | 7 (46.7) | 3 (50.0) |
| COPD assessment test (CAT): Mean ± SD | 17.6 ± 7.9 | - |
| Post BD FEV1 (% predicted): Mean ± SD | 58.1 ± 14.5 | 70.4 ± 24.3 |
| Post BD FEV1/FVC (% predicted): Mean ± SD | 51.0 ± 8.4 | - |
| BSI score | - | 7 (5–13) |
| No. of exacerbations in previous year: Median (IQR) | 1 (0–2) | 0 (0–6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, N.A.B.M.; Mac Aogáin, M.; Morales, R.F.; Tiew, P.Y.; Chotirmall, S.H. Optimisation and Benchmarking of Targeted Amplicon Sequencing for Mycobiome Analysis of Respiratory Specimens. Int. J. Mol. Sci. 2019, 20, 4991. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204991
Ali NABM, Mac Aogáin M, Morales RF, Tiew PY, Chotirmall SH. Optimisation and Benchmarking of Targeted Amplicon Sequencing for Mycobiome Analysis of Respiratory Specimens. International Journal of Molecular Sciences. 2019; 20(20):4991. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204991
Chicago/Turabian StyleAli, Nur A’tikah Binte Mohamed, Micheál Mac Aogáin, Raika Francesca Morales, Pei Yee Tiew, and Sanjay H. Chotirmall. 2019. "Optimisation and Benchmarking of Targeted Amplicon Sequencing for Mycobiome Analysis of Respiratory Specimens" International Journal of Molecular Sciences 20, no. 20: 4991. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204991