Myoferlin Regulates Wnt/β-Catenin Signaling-Mediated Skeletal Muscle Development by Stabilizing Dishevelled-2 Against Autophagy


Abstract
:1. Introduction
2. Results
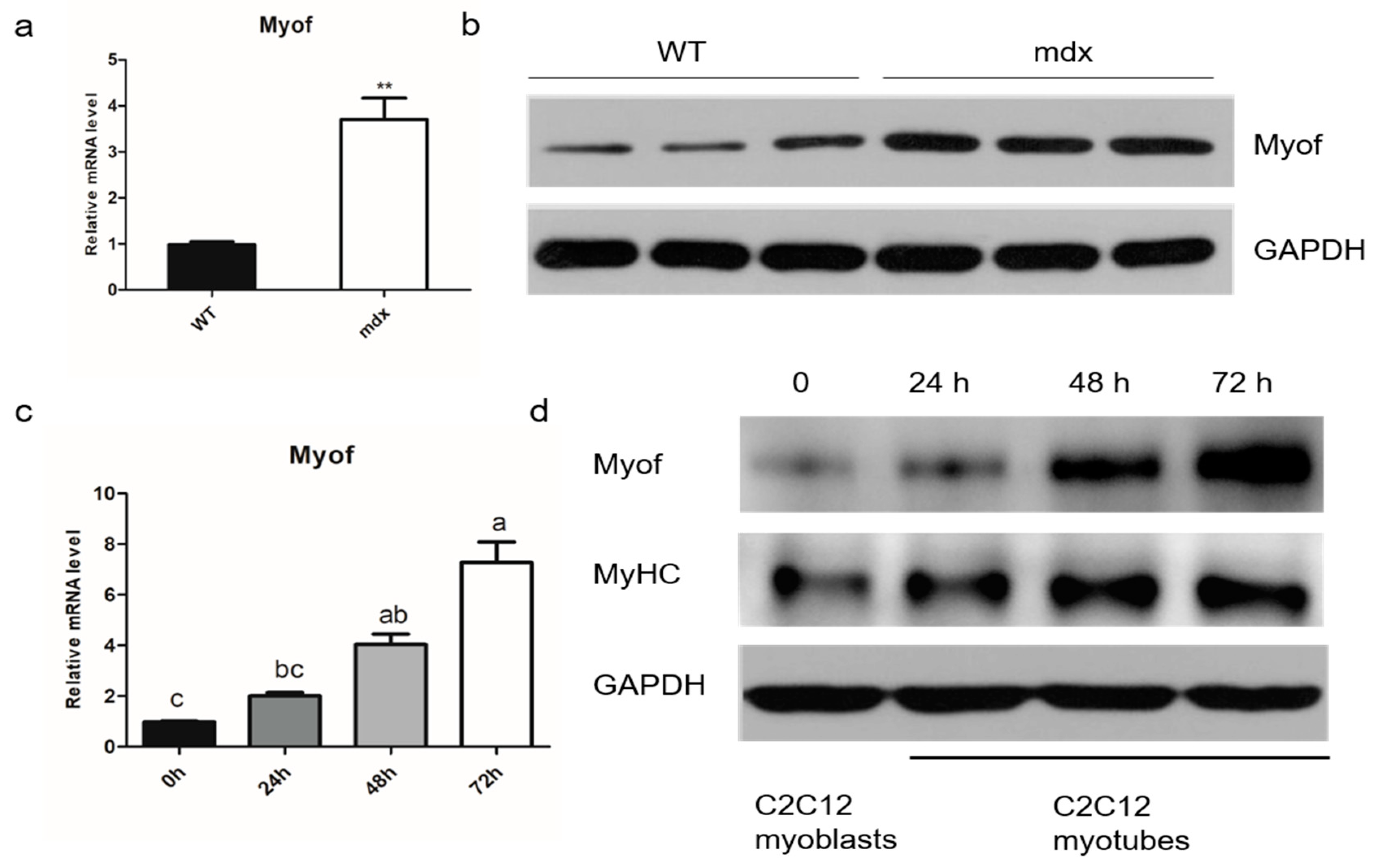
2.1. MyoF is Highly Expressed in Differentiated Myogenic Cells
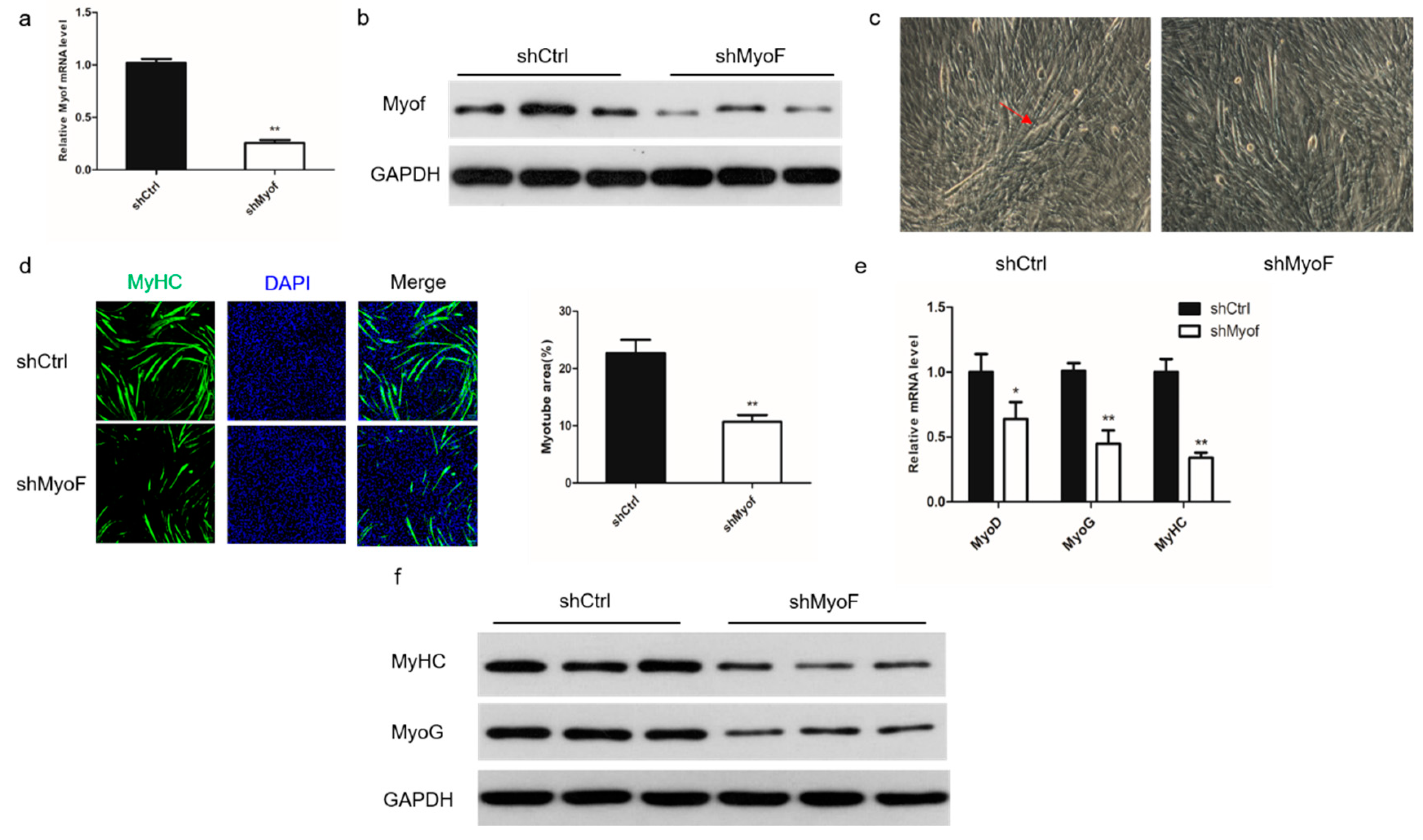
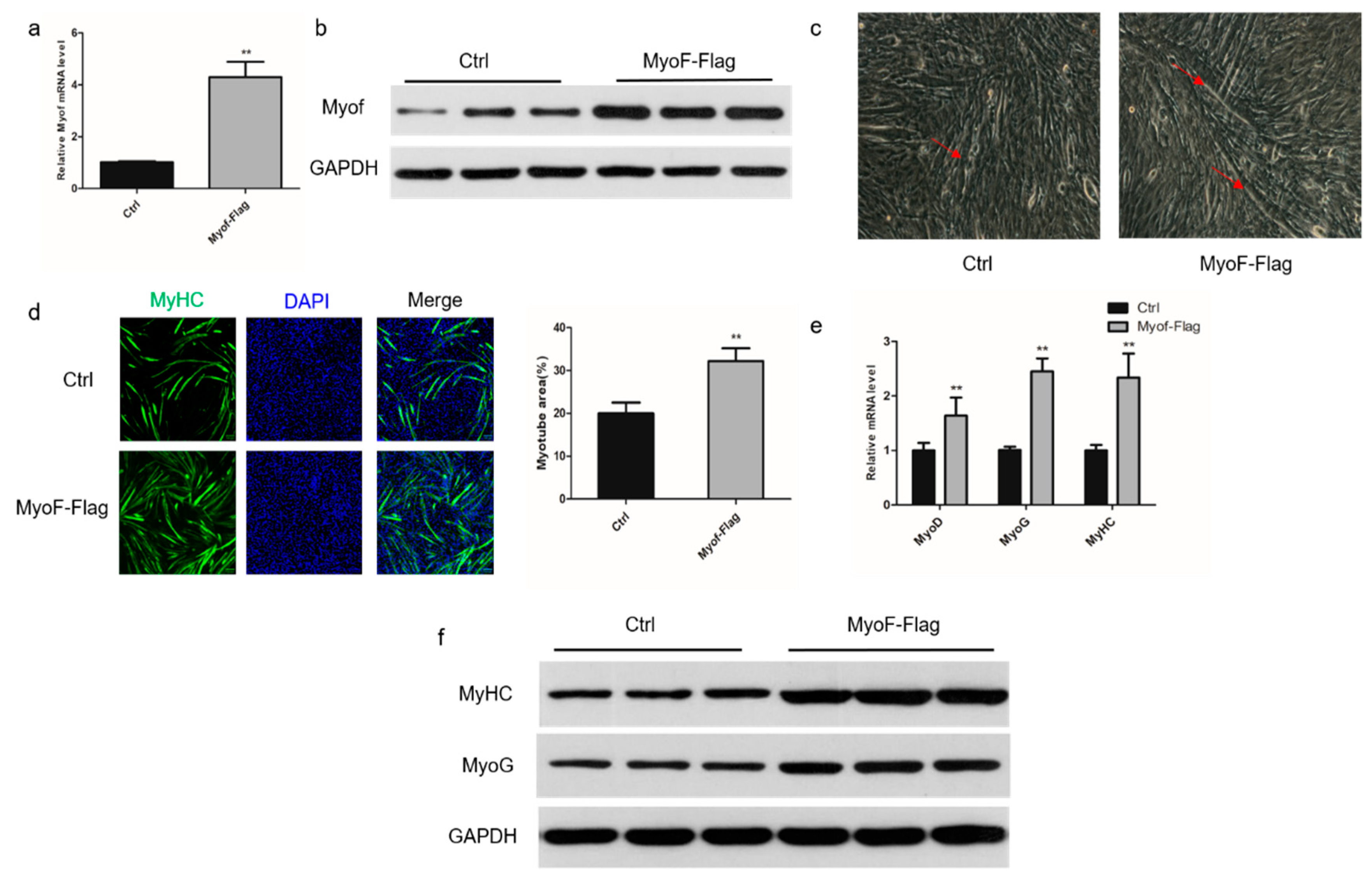
2.2. Role of MyoF in Skeletal Muscle Differentiation
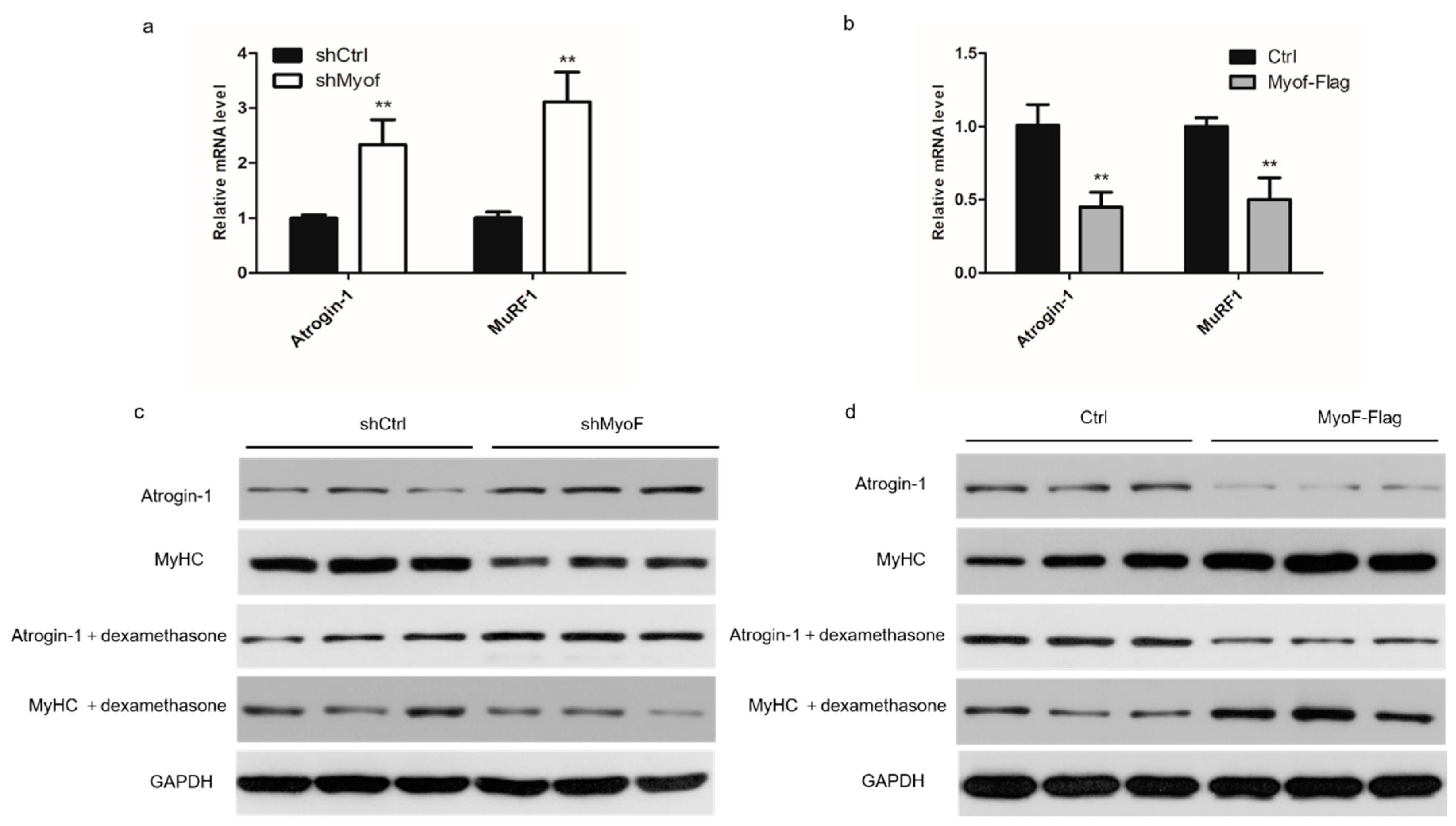
2.3. MyoF Rescues Skeletal Muscle Atrophy
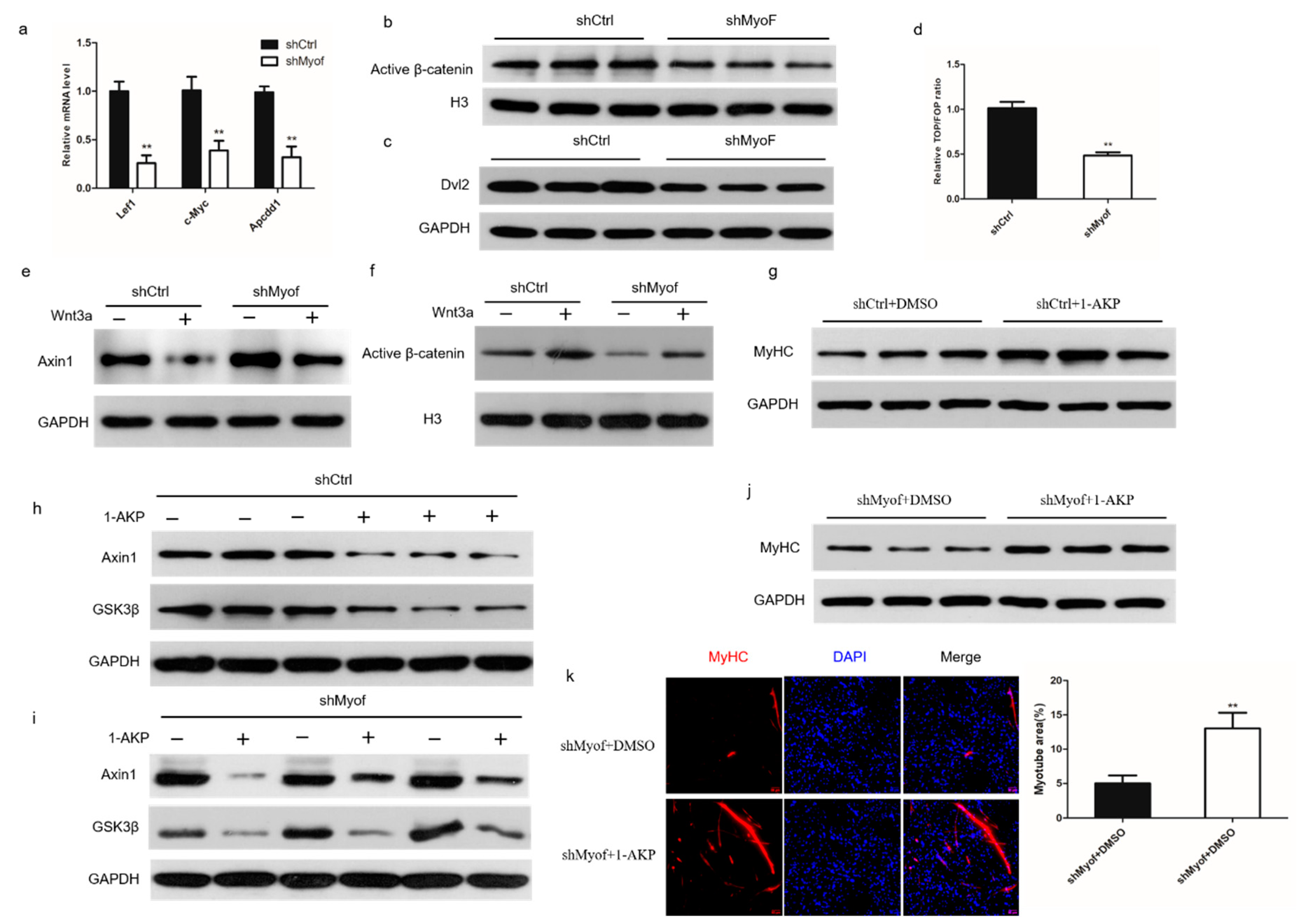
2.4. MyoF Functions by Dvl-2-Mediated Canonical Wnt Signaling
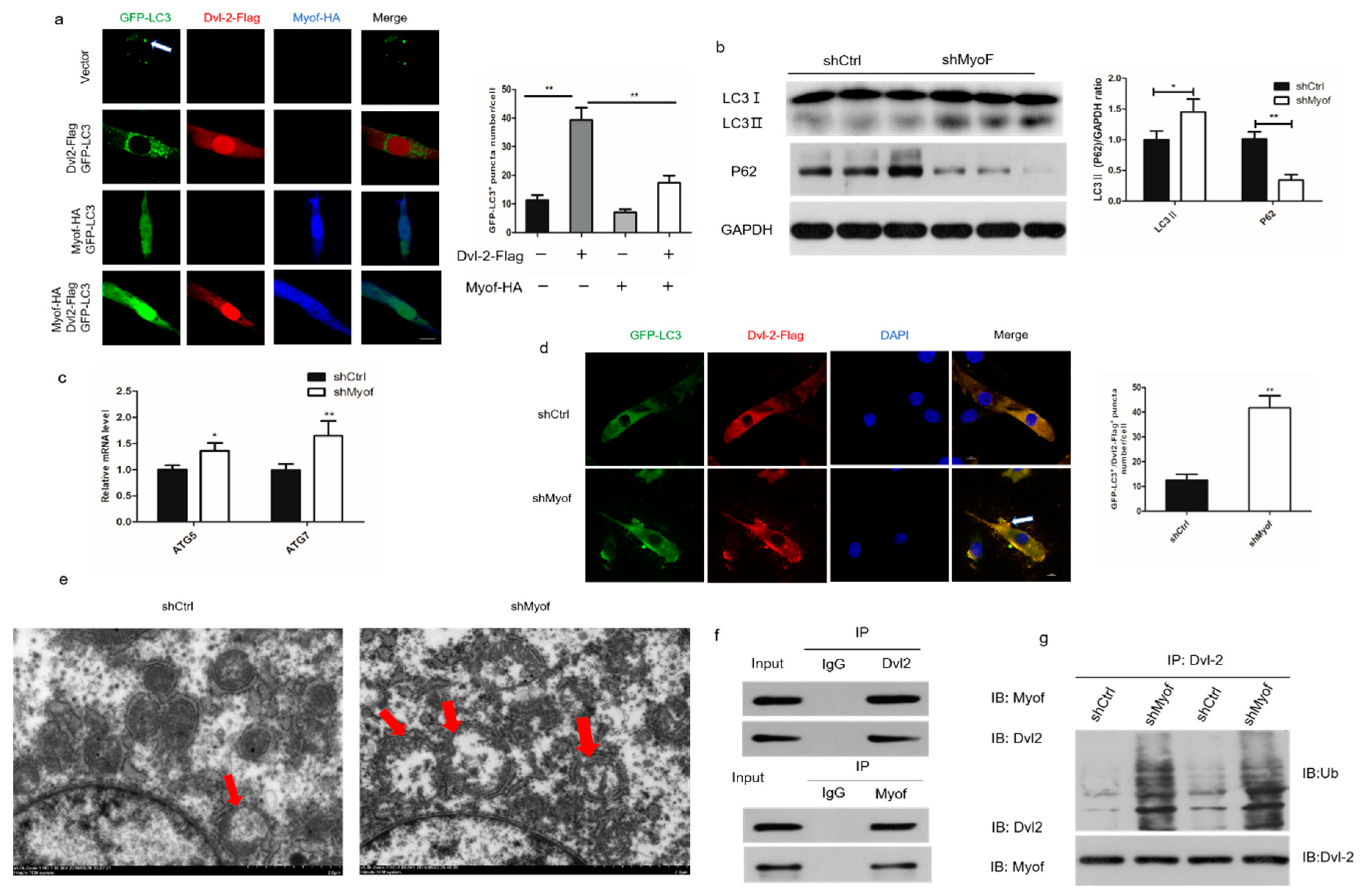
2.5. MyoF Stabilizes Dvl-2 by Preventing Autophagy
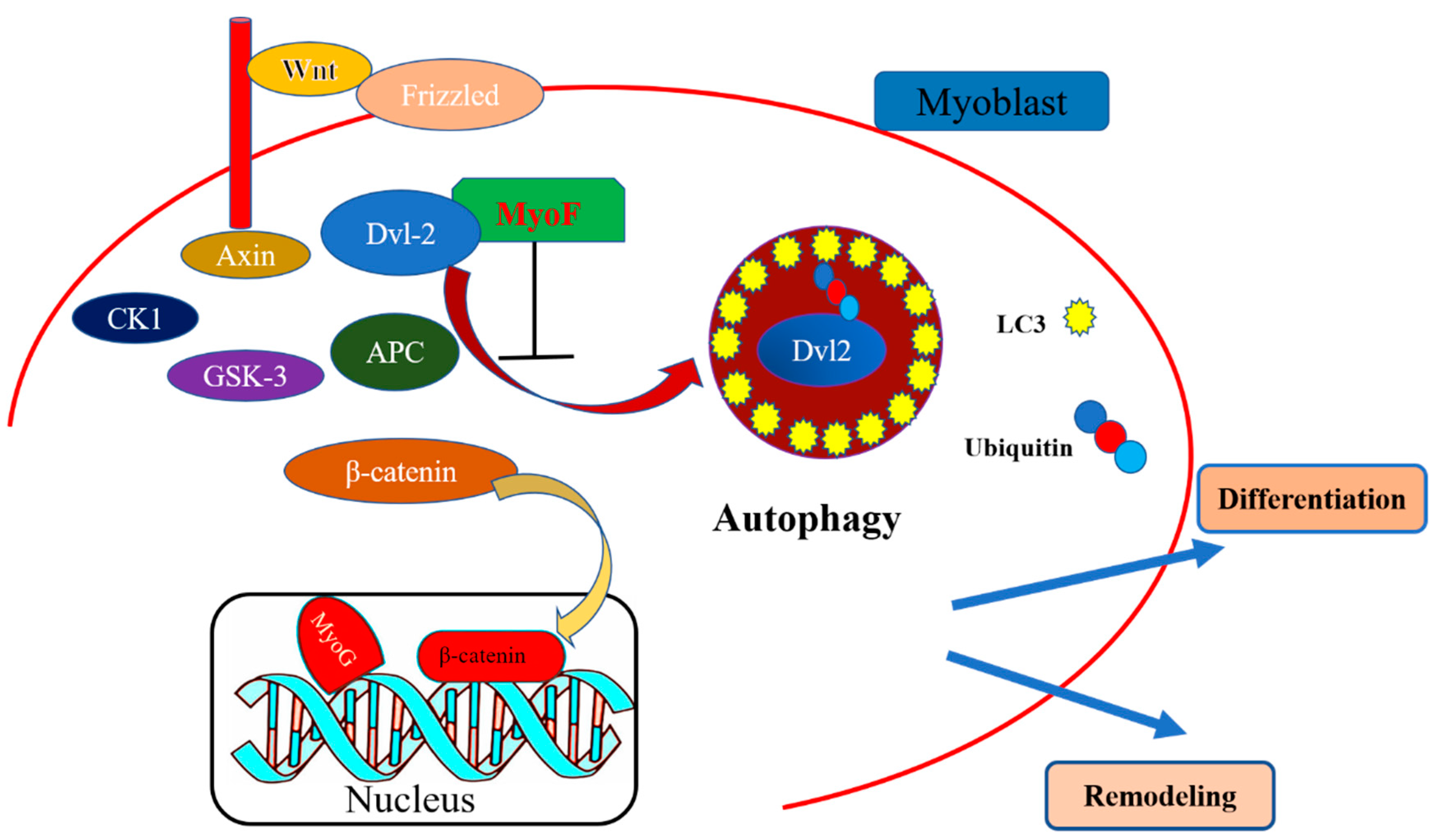
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. MyoF Knockdown and Overexpression
4.3. RNA Extraction and Real-Time PCR
4.4. Cell Treatment Protocols and Antibodies
4.5. Western Blot and Immunoprecipitation (IP) Analysis
4.6. Immunofluorescence and Confocal Microscopy
4.7. Transmission Electron Microscopy
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. cell. 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy – unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Z.; Gu, Z.; Qin, Z. Molecular mechanism and regulation of autophagy. Acta Pharmacol. Sin. 2005, 26, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Hinds, S.; Bian, W.; Dennis, R.G.; Bursac, N. The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 2011, 32, 3575–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physioll Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Bates, P.C.; Millward, D.J. Myofibrillar protein turnover. Synthesis rates of myofibrillar and sarcoplasmic protein fractions in different muscles and the changes observed during postnatal development and in response to feeding and starvation. Biochem. J. 1983, 214, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Autophagy in skeletal muscle. Febs Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Hong-Guang, X.; Lihong, Z.; Gang, C.; Tao, Z.; Junli, L.; Mingzhi, J.; Xiuquan, M.; Dawei, M.; Junying, Y. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010, 6, 61–66. [Google Scholar] [Green Version]
- Marco, S.; Luisa, C.; Paolo, G.; Paolo, B. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [Green Version]
- Robertson, T.A.; Papadimitriou, J.M.; Grounds, M.D. Fusion of myogenic cells to the newly sealed region of damaged myofibres in skeletal muscle regeneration. Neuropath. App. Neuro. 2010, 19, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Maltzahn, J.V.; Chang, N.C.; Bentzinger, C.F.; Rudnicki, M.A. Wnt signaling in myogenesis. Trends in Cell Biol. 2012, 22, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef]
- Davis, D.B.; Delmonte, A.J.; Ly, C.T.; Mcnally, E.M. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum. Mol. Genet. 2000, 9, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, J.L.; Bashir, R. In silico functional and structural characterisation of ferlin proteins by mapping disease-causing mutations and evolutionary information onto three-dimensional models of their C2 domains. J. Neurol. Sci. 2007, 260, 114–123. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Lapidos, K.A.; Konstantina, H.; Samantha, L.; Rodney, D.; Peter, P.; Svensson, E.C.; Mcnally, E.M. Myoferlin regulation by NFAT in muscle injury, regeneration and repair. J. Cell Sci. 2009, 123, 2413–2422. [Google Scholar] [CrossRef]
- Masahiko, I.; Yoshihiro, W.; Hiroko, K.; Seiji, S.; Takahiro, J.; Hiroaki, O.; Ichizo, N.; Ikuya, N. Expression of myoferlin in skeletal muscles of patients with dysferlinopathy. Tohoku J. Exp Med. 2006, 209, 109–116. [Google Scholar]
- Doherty, K.R.; Andrew, C.; Dawn Belt, D.; Delmonte, A.J.; Avery, P.; Earley, J.U.; Michele, H.; Mcnally, E.M. Normal myoblast fusion requires myoferlin. Development 2005, 132, 5565–5575. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Posey, A.D.; Heretis, K.; Swaggart, K.A.; Earley, J.U.; Pytel, P.; Mcnally, E.M. Myoferlin is required for insulin-like growth factor response and muscle growth. Faseb. J. 2010, 24, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Schaale, K.; Neumann, J.; Schneider, D.; Ehlers, S.; Reiling, N. Wnt signaling in macrophages: Augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur. J. Cell Biol. 2011, 90, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Lauenroth, K.; Leost, M.; Meijer, L.; Lemcke, T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3β. Bioorg. Med. Chem. Lett. 2004, 14, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.Q.; Mcnally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef]
- Posey, A.D.; Demonbreun, A.; Mcnally, E.M. Chapter eight–Ferlin Proteins in Myoblast Fusion and Muscle Growth. Curr. Top. Dev. Bio. 2011, 96, 203–230. [Google Scholar]
- Doherty, K.R.; Demonbreun, A.R.; Wallace, G.Q.; Andrew, C.; Posey, A.D.; Konstantina, H.; Peter, P.; Mcnally, E.M. The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion. J. Biol. Chem. 2008, 283, 20252–20260. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Rossi, A.E.; Alvarez, M.G.; Swanson, K.E.; Deveaux, H.K.; Earley, J.U.; Hadhazy, M.; Vohra, R.; Walter, G.A.; Pytel, P. Dysferlin and Myoferlin Regulate Transverse Tubule Formation and Glycerol Sensitivity. Am. J. Pathol. 2014, 184, 248–259. [Google Scholar] [CrossRef]
- Jagoe, R.T.; Goldberg, A.L. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr. Opin. Clin. Nutr. Metab. Care. 2001, 4, 183–190. [Google Scholar] [CrossRef]
- Jackman, R.W.; Kandarian, S.C. The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2004, 287, C834–843. [Google Scholar] [CrossRef]
- Galli, L.M.; Karl, W.; Roel, N.; Zipora, Y.R.; Tsutomu, N.; Wilfred, D.; Burrus, L.W. A proliferative role for Wnt-3a in chick somites. Dev. Biolo. 2004, 269, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Anthony, O.; Corina, S.; Graham, L.; Steve, A.; Petr, V.; Francesco, M.; Diana, L.W.; Ketan, P. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J. Cell Sci. 2008, 121, 2939–2950. [Google Scholar] [Green Version]
- Lacour, F.; Vezin, E.; Bentzinger, F.C.; Sincennes, M.-C.; Giordani, L.; Ferry, A.; Mitchell, R.D.; Patel, K.; Rudnicki, M.A.; Chaboissier, M.-C. R-spondin1 regulates muscle progenitor cell differentiation and fusion through dual regulation of canonical and non-canonical Wnt signaling pathways. Cell Rep. 2017, 18, 2320–2330. [Google Scholar] [CrossRef]
- Ferrari, L.; Bragato, C.; Brioschi, L.; Spreafico, M.; Esposito, S.; Pezzotta, A.; Pizzetti, F.; Moreno-Fortuny, A.; Bellipanni, G.; Giordano, A. HDAC8 regulates canonical Wnt pathway to promote differentiation in skeletal muscles. J. Cell Physiol. 2018, 234, 6067–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, M.J.; Sara, E.; Paterson, H.; Naylor, S.; Cook, D.; Jayatilake, H.; Fryer, L.G.; Hutchinson, L.; Fry, M.J.; Dale, T.C. Interaction of Axin and Dvl-2 proteins regulates Dvl-2-stimulated TCF-dependent transcription. EMBO J. 1999, 18, 2823–2835. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.; Haegebarth, A.; Kuper, I. Loss of the Tumor Suppressor CYLD Enhances Wnt/β-Catenin Signaling through K63-Linked Ubiquitination of Dvl. Mol. Cell. 2010, 37, 607–619. [Google Scholar] [CrossRef]
- Geng, Y.; Ju, Y.; Ren, F.; Qiu, Y.; Tomita, Y.; Tomoeda, M.; Kishida, M.; Wang, Y.; Jin, L.; Su, F. Insulin Receptor Substrate 1/2 (IRS1/2) Regulates Wnt/β-Catenin Signaling through Blocking Autophagic Degradation of Dishevelled2. J. Biol. Chem. 2014, 289, 11230–11241. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Weipeng, C.; Lan, B.; Wei, Z.; Guoming, X.; Tiantian, C.; Wei, F.; Jian, Z.; Wei, W.; Xu, Z. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 2011, 12, 781–790. [Google Scholar]
- Jaiprakash, S.; Shalaka, M.; Diptendu, M.; Nihar Ranjan, J. Malin regulates Wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 2012, 287, 6830–6839. [Google Scholar]
- Benyu, M.; Bofeng, L.; Weipeng, C.; Chan, G.; Zhen, Q.; Yuanheng, N.; Ye-Guang, C. The Wnt Signaling Antagonist Dapper1 Accelerates Dishevelled2 Degradation via Promoting Its Ubiquitination and Aggregate-induced Autophagy. J. Biol. Chem. 2015, 290, 12346–12354. [Google Scholar] [Green Version]
- Han, S.; Cui, C.; Wang, Y.; He, H.; Liu, Z.; Shen, X.; Chen, Y.; Li, D.; Zhu, Q.; Yin, H. Knockdown of CSRP3 inhibits differentiation of chicken satellite cells by promoting TGF-β/Smad3 signaling. Gene. 2019, 707, 36–43. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| MyoF | TGCCACTAACATCCCTAA | ATCACCACCGATTCTACTC |
| MyoD | AGCACTACAGTGGCGACTCA | GGCCGCTGTAATCCATCA |
| MyoG | TACAGCGACCAACAGTACGC | TCTGCATTGTTTCCATCCTG |
| MyHC | CGGCTGCCTAAAGTGGAGAT | AGGCCTGTAGGCGCTCAA |
| Atrogin-1 | GCAAACACTGCCACATTCTCTC | CTTGAGGGGAAAGTGAGACG |
| MuRF1 | ACCTGCTGGTGGAAAACATC | CTTCGTGTTCCTTGCACATC |
| Apcdd1 | CTGAAGCATCTCCACAACGG | GGACCCGACCTTACTTCACA |
| c-Myc | TAGTGCTGCATGAGGAGACA | CTCCACAGACACCACATCAA |
| Lef1 | GACAGATCACCCCACCCATT | ATAGCTGGATGAGGGATGCC |
| ATG5 | AGCAGCTCTGGATGGGACTGC | GCCGCTCCGTCGTGGTCTGA |
| ATG7 | GCTCCTCATCACTTTTTGCCAACA | GGAGCCACCACATCATTGC |
| GAPDH | GTGCCGCCTGGAGAAACCT | AAGTCGCAGGAGACAACC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Cui, C.; He, H.; Shen, X.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q.; Yin, H. Myoferlin Regulates Wnt/β-Catenin Signaling-Mediated Skeletal Muscle Development by Stabilizing Dishevelled-2 Against Autophagy. Int. J. Mol. Sci. 2019, 20, 5130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205130
Han S, Cui C, He H, Shen X, Chen Y, Wang Y, Li D, Zhu Q, Yin H. Myoferlin Regulates Wnt/β-Catenin Signaling-Mediated Skeletal Muscle Development by Stabilizing Dishevelled-2 Against Autophagy. International Journal of Molecular Sciences. 2019; 20(20):5130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205130
Chicago/Turabian StyleHan, Shunshun, Can Cui, Haorong He, Xiaoxu Shen, Yuqi Chen, Yan Wang, Diyan Li, Qing Zhu, and Huadong Yin. 2019. "Myoferlin Regulates Wnt/β-Catenin Signaling-Mediated Skeletal Muscle Development by Stabilizing Dishevelled-2 Against Autophagy" International Journal of Molecular Sciences 20, no. 20: 5130. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205130