The Synthetic Dipeptide Pidotimod Shows a Chemokine-Like Activity through CXC Chemokine Receptor 3 (CXCR3)

 ,
,
Abstract
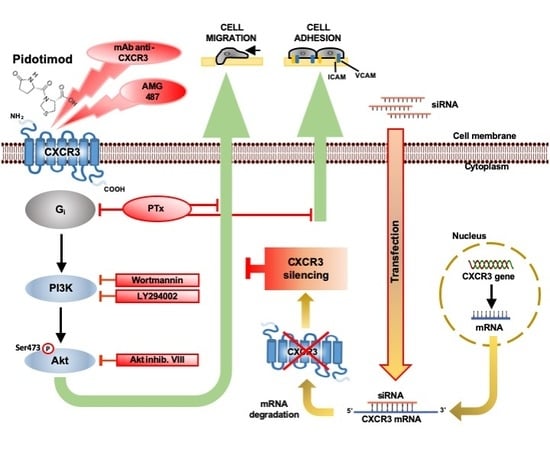
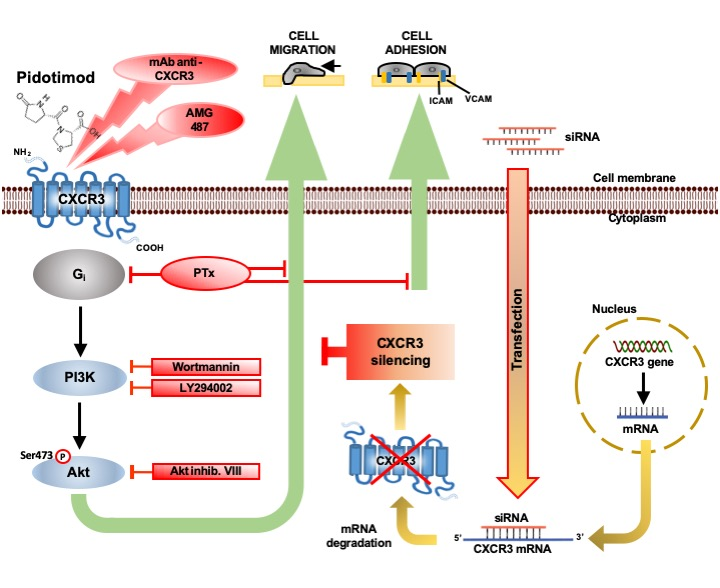
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
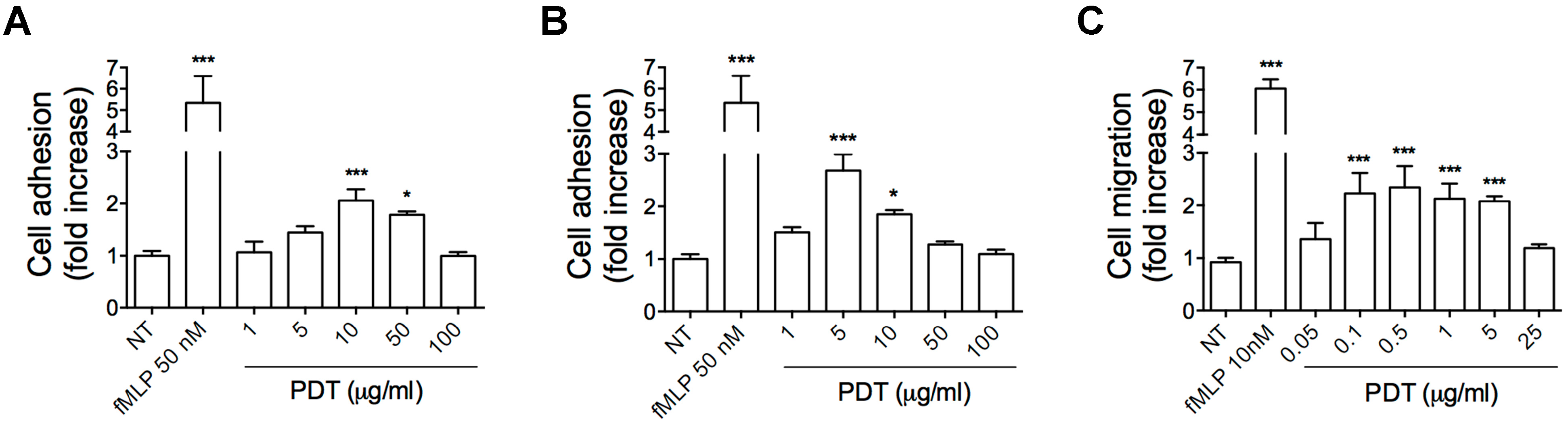
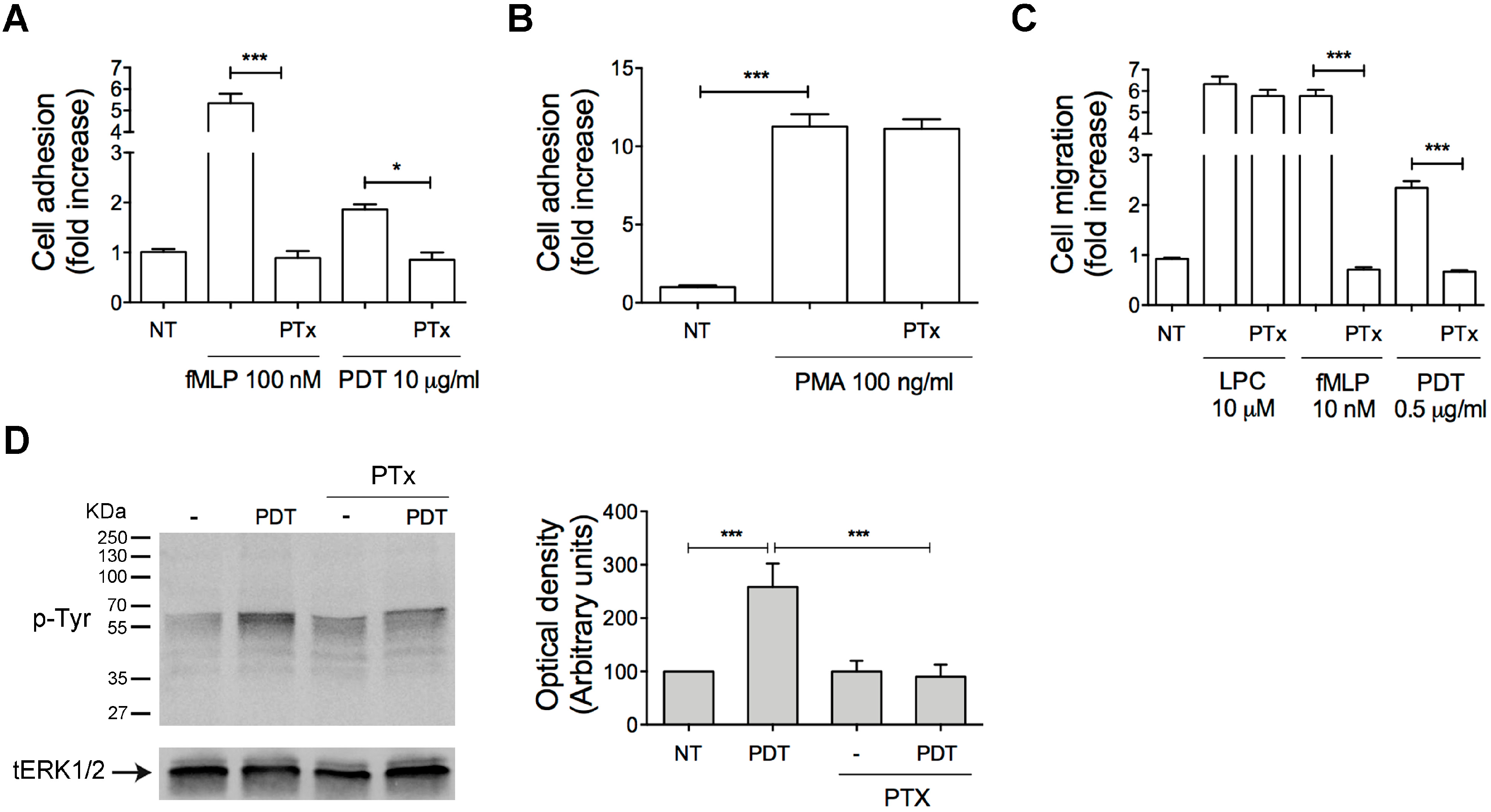
2.1. PDT Induces Monocyte Adhesion and Migration
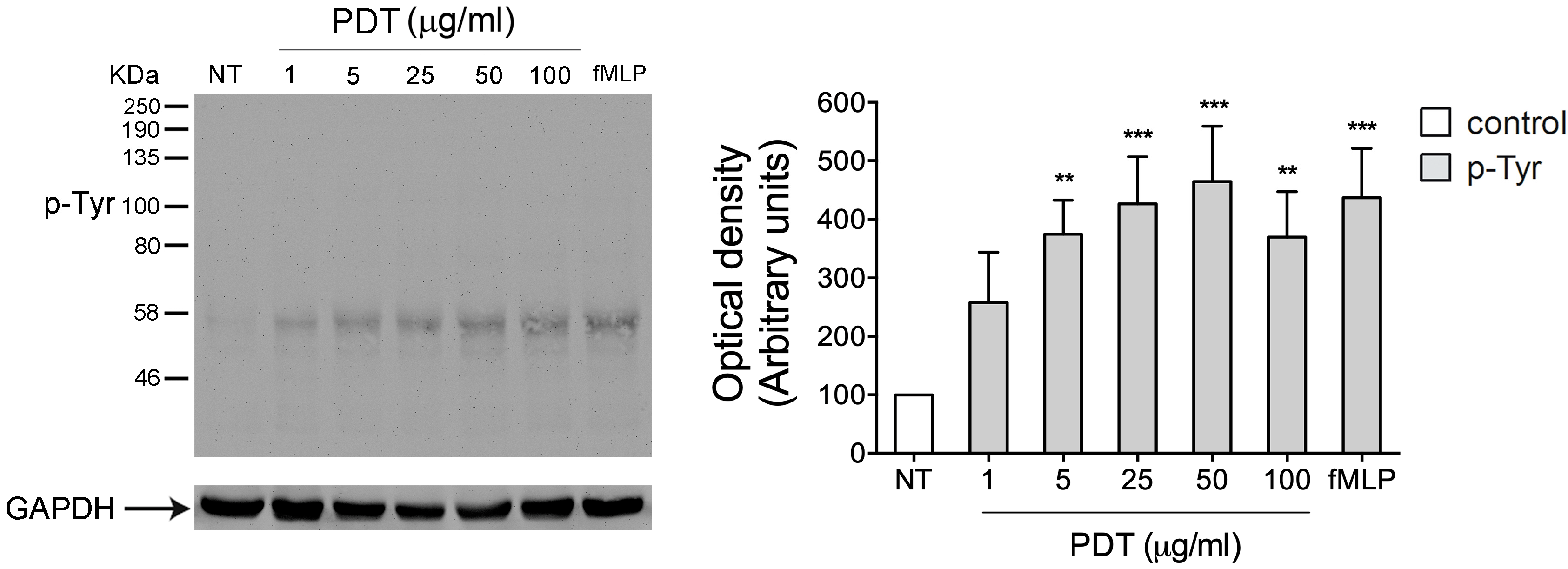
2.2. PTx Treatment Inhibits PDT-Induced Chemokine Activity and Tyrosine Phosphorylation-based Protein Signaling in Monocytes
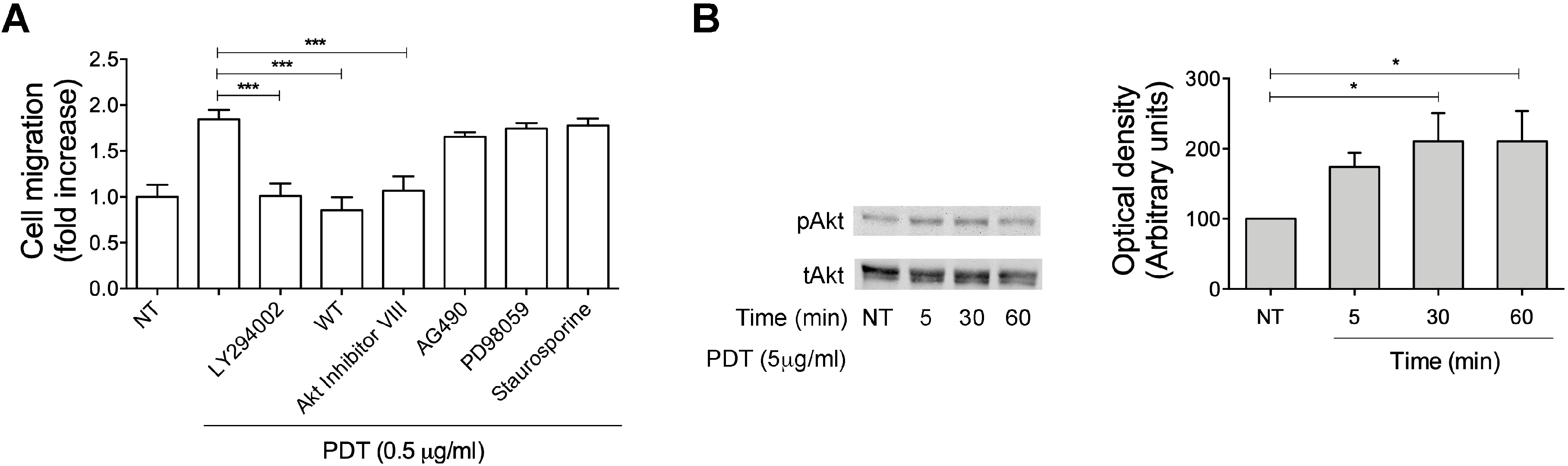
2.3. Monocyte Migration Triggered by PDT Requires the Activation of PI3K/Akt Signaling Pathway
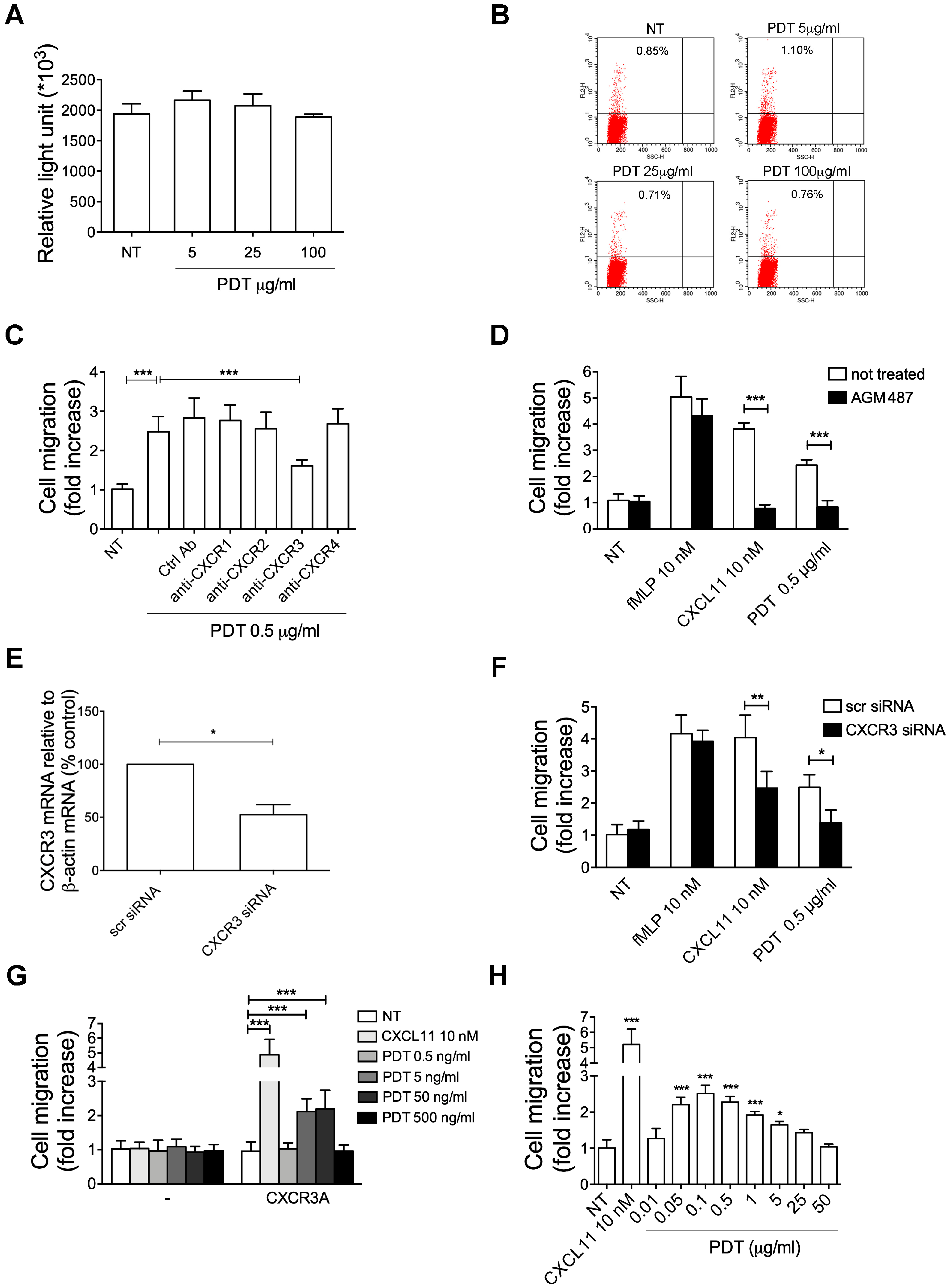
2.4. PDT-Triggered Monocyte Migration Is Mediated by CXCR3
2.5. The PDT-Induced Migratory Activity Is Sustained by the CXCR3A Isoform
2.6. Pidotimod Induces Migration of IL-2-Activated T Lymphocytes
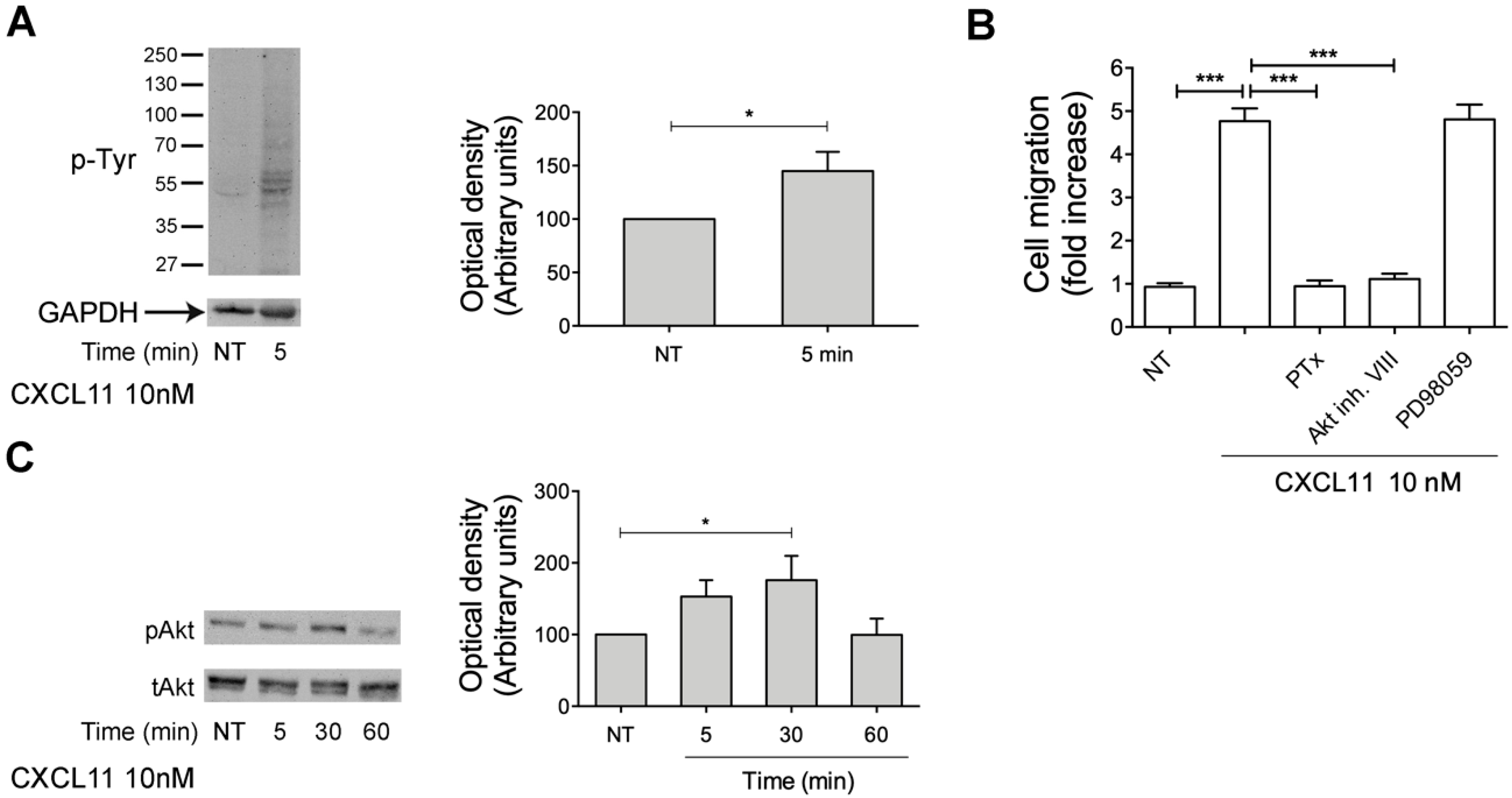
2.7. CXCL11, as PDT, Triggers Monocyte Migration through Akt Kinase Activation
3. Discussion
4. Materials and Methods
4.1. Pidotimod Preparation
4.2. Cell Culture
4.3. Isolation of Human Primary Cells
4.4. Chemotaxis Assays
4.5. Static Adhesion Assay
4.6. Western Blot Analysis
4.7. siRNA Technique
4.8. Real-Time PCR for Gene Expression Analysis
4.9. Cell Viability Assay
4.10. Flow Cytometry Analysis
4.11. Transfections
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffin, M.R.; Walker, F.J.; Iwane, M.K.; Weinberg, G.A.; Staat, M.A.; Erdman, D.D. Epidemiology of respiratory infections in young children: Insights from the new vaccine surveillance network. Pediatr. Infect. Dis. J. 2004, 23, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Schaad, U.B. OM-85 BV, an immunostimulant in pediatric recurrent respiratory tract infections: A systematic review. World J. Pediatr. 2010, 6, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Rozy, A.; Chorostowska-Wynimko, J. Bacterial immunostimulants–mechanism of action and clinical application in respiratory diseases. Pneumonol. Alergol. Pol. 2008, 76, 353–359. [Google Scholar] [PubMed]
- Ounis, I. Determination of the antiinfectious activity of RU 41740 (Biostim) as an example of an immunomodulator. Adv. Exp. Med. Biol. 1992, 319, 165–174. [Google Scholar] [PubMed]
- Fiocchi, A.; Terracciano, L.; Martelli, A.; Bernardo, L.; Calcinai, E.; Marcassa, S. Ribosome-component immune modulation of respiratory tract infections in children. Allergy Asthma Proc. 2009, 30, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, P.; LeeS, E. Immunomodulators Chronic Rhinosinusitis. World J. Otorhinolaryngol. Head Neck Surg. 2018, 4, 186–192. [Google Scholar] [CrossRef]
- Masihi, K.N. Fighting infection using immunomodulatory agents. Expert Opin. Biol. Ther. 2001, 1, 641–653. [Google Scholar] [CrossRef]
- Burgio, G.R.; Marseglia, G.L.; Severi, F.; de Benedetti, F.; Masarone, M.; Ottolenghi, A.; Pagliano, L.; Serra, U.; Nespoli, L. Immunoactivation by pidotimod in children with recurrent respiratory infections. Arzneimittelforschung 1994, 44, 1525–1529. [Google Scholar]
- Esposito, S.; Musio, A. Immunostimulants and prevention of recurrent respiratory tract infections. J. Biol. Regul. Homeost. Agents 2013, 27, 627–636. [Google Scholar]
- Motta, G.; De Campora, E.; de Vita, C.; Esposito, S.; Galletti, C.; Incutti, V.; Mallardi, V.; Motta, S.; Pucci, V.; Salonna, F. Immunoactivity of Pidotimod against episodes of recurrent tonsillitis in childhood. Arzneimittelforschung 1994, 44, 1521–1524. [Google Scholar]
- Careddu, P.; Mei, V.; Venturoli, V.; Corsini, A. Pidotimod in the treatment of recurrent respiratory infections in paediatric patients. Arzneimittelforschung 1994, 44, 1485–1489. [Google Scholar] [PubMed]
- Caramia, G.; Clemente, E.; Solli, R.; Mei, V.; Cera, R.; Carnelli, V.; Venturoli, V.; Corsini, A. Efficacy and safety of Pidotimod in the treatment of recurrent respiratory infections in children. Arzneimittelforschung 1994, 44, 1480–1484. [Google Scholar] [PubMed]
- Passali, D.; Calearo, C.; Conticello, S. Pidotimod in the management of recurrent pharyngotonsillar infections in childhood. Arzneimittelforschung 1994, 44, 1511–1516. [Google Scholar] [PubMed]
- La Mantia, I.; Grillo, C.; Mattina, T.; Zaccone, P.; Xiang, M.; Di Mauro, M.; Meroni, P.L.; Nicoletti, F. Prophylaxis with the novel immunomodulator Pidotimod reduces the frequency and severity of upper respiratory tract infections in children with Down’s syndrome. J. Chemother. 1999, 11, 126–130. [Google Scholar] [CrossRef]
- Riboldi, P.; Gerosa, M.; Meroni, P.L. Pidotimod: A reappraisal. Int. J. Immunopathol. Pharmacol. 2009, 22, 255–362. [Google Scholar] [CrossRef]
- Auteri, A.; Pasqui, A.L.; Bruni, F.; Saletti, M.; Di Renzo, M.; Bova, G. Effect of Pidotimod, a new immunostimulating agent, on some aspects of immune response. Vitr. Study. Pharmacol. Res. 1992, 26, 196–197. [Google Scholar] [CrossRef]
- Migliorati, G.; Nicoletti, I.; Riccardi, C. Immunomodulating activity of Pidotimod. Arzneimittelforschung 1994, 44, 1421–1424. [Google Scholar]
- Giagulli, C.; Noerder, M.; Avolio, M.; Becker, P.; Fiorentini, S.; Guzman, C.; Caruso, A. Pidotimod promotes functional maturation of dendritic cells and displays adjuvant properties at the nasal mucosa level. Int. Immunopharmacol. 2009, 9, 1366–1373. [Google Scholar] [CrossRef]
- Zuccotti, G.V.; Mameli, C. Pidotimod: The past and the present. J. Pediatrics 2013, 39, 75–78. [Google Scholar] [CrossRef]
- Di Renzo, M.; Pasqui, A.L.; Bruni, F.; Saletti, M.; Bova, G.; Chiaron, C.; Girardello, R.; Ferri, P.; Auteri, A. The in vitro effect of Pidotimod on some immune functions in cancer patients. Immunopharmacol. Immunotoxicol. 1997, 19, 37–51. [Google Scholar] [CrossRef]
- Fogli, M.; Caccuri, F.; Iaria, M.L.; Giagulli, C.; Corbellini, S.; Campilongo, F.; Caruso, A.; Fiorentini, S. The immunomodulatory molecule Pidotimod induces the expression pf the nod-like receptor NLRP12 and attenuates TLR-induced inflammation. J. Biol. Regul. Homeost. Agents 2014, 28, 753–766. [Google Scholar] [PubMed]
- Mertens, I.; Vandingenen, A.; Meeusen, T.; de Loof, A.; Schoofs, L. Postgenomic characterization of G-protein-coupled receptors. Pharmacogenomics 2004, 5, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Littman, D.R. Signal transduction by lymphocyte antigen receptors. Cell 1994, 76, 263–274. [Google Scholar] [CrossRef]
- Seet, B.T.; Dikic, L.; Zhou, M.M.; Pawson, T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006, 7, 473–483. [Google Scholar] [CrossRef]
- Laudanna, C.; Kim, J.Y.; Constantin, G.; Butcher, E. Rapid leukocyte integrin activation by chemokines. Immunol. Rev. 2002, 186, 37–46. [Google Scholar] [CrossRef]
- Campbell, J.J.; Qin, S.; Bacon, K.B.; Mackay, C.R.; Butcher, E.C. Biology of chemokine and classical chemoattractant receptors: Differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J. Cell Biol. 1996, 134, 255–266. [Google Scholar] [CrossRef]
- Giagulli, C.; Magiera, A.; Bugatti, A.; Caccuri, F.; Marsico, S.; Rusnati, M.; Vermi, W.; Fiorentini, S.; Caruso, A. HIV-1 matrix protein p17 binds to the IL-8 receptor CXCR1 and shows IL-8–like chemokine activity on monocytes through Rho/ROCK activation. Blood 2012, 119, 2274–2283. [Google Scholar] [CrossRef]
- Sozzani, S.; Luini, W.; Molino, M.; Jílek, P.; Bottazzi, B.; Cerletti, C.; Matsushima, K.; Mantovani, A. The signal transduction pathway involved in the migration induced by a monocyte chemotactic cytokine. J. Immunol. 1991, 147, 2215–2221. [Google Scholar]
- Fontana, L.; Giagulli, C.; Minuz, P.; Lechi, A.; Laudanna, C. 8-Iso-PGF2 alpha induces beta 2-integrin-mediated rapid adhesion of human polymorphonuclear neutrophils: A link between oxidative stress and ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 55–60. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Lysophosphatidylcholine- and MCP-1-induced chemotaxis of monocytes requires potassium channel activity. Pflug. Arch. 2009, 459, 71–77. [Google Scholar] [CrossRef]
- Welf, E.S.; Haugh, J.M. Signaling pathways that control cell migration: Models and analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Cantrell, D. Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann. N. Y. Acad. Sci. 2010, 1183, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt7PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J. Natl. Cancer Inst. 2015, 11, 107. [Google Scholar]
- Xue, G.; Hemmings, B.A. PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 2013, 105, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, D.; Schulz, A.; Lede, V.; Schulze, A.; Thor, D.; Brüser, A.; Schöneberg, T. P2Y Receptors in Immune Response and Inflammation. Adv. Immunol. 2017, 136, 85–121. [Google Scholar] [PubMed]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef]
- Struckhoff, A.P.; Vitko, J.R.; Rana, M.K.; Davis, C.T.; Foderingham, K.E.; Liu, C.H.; Vanhoy-Rhodes, L.; Elliot, S.; Zhu, Y.; Burow, M.; et al. Dynamic regulation of ROCK in tumor cells controls CXCR4-driven adhesion events. J. Cell Sci. 2010, 123, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Traves, S.L.; Tudhope, S.J.; Fenwick, P.S.; Belchamber, K.B.; Russell, R.E.; Barne, P.J.; Donnelly, L.E. Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. Eur. Respir. J. 2016, 47, 1093–1102. [Google Scholar] [CrossRef]
- Billottet, C.; Quemener, C.; Bikfalvi, A. CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim. Biophys. Acta 2013, 1836, 287–295. [Google Scholar] [CrossRef]
- Lasagni, L.; Francalanci, M.; Annunziato, F.; Lazzeri, E.; Giannini, S.; Cosmi, L.; Sagrinati, C.; Mazzinghi, B.; Orlando, C.; Maggi, E.; et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J. Exp. Med. 2003, 197, 1537–1549. [Google Scholar] [CrossRef]
- Sallusto, F.; Mackay, C.R. Chemoattractants and their receptors in homeostasis and inflammation. Curr. Opin. Immunol. 2004, 16, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Bonacchi, A.; Romagnani, P.; Romanelli, R.G.; Efsen, E.; Annunziato, F.; Lasagni, L.; Francalanci, M.; Serio, M.; Laffi, G.; Pinzani, M.; et al. Signal transduction by the chemokine receptor CXCR3: Activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J. Biol. Chem. 2001, 276, 9945–9954. [Google Scholar] [CrossRef] [PubMed]
- Korniejewska, A.; McKnight, A.J.; Johnson, Z.; Watson, M.L.; Ward, S.G. Expression and agonist responsiveness of CXCR3 variants in human T lymphocytes. Immunology 2011, 132, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.J.; Canals, M.; Wijtmans, M.; de Munnik, S.; Nguyen, P.; Verzijl, D.; de Esch, I.J.; Vischer, H.F.; Smit, M.J.; Leurs, R. Pharmacological characterization of a small-molecule agonist for the chemokine receptor CXCR3. Br. J. Pharmacol. 2012, 166, 898–911. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signaling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Kenakin, T. Collateral efficacy in drug discovery: Taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol. Sci. 2007, 28, 407–415. [Google Scholar] [CrossRef]
- Baker, J.G.; Hill, S.J. Multiple GPCR conformations and signaling pathways: Implications for antagonist affinity estimates. Trends Pharmacol. Sci. 2007, 28, 374–381. [Google Scholar] [CrossRef]
- Blanpain, C.; Doranz, B.J.; Bondue, A.; Govaerts, C.; De Leener, A.; Vassart, G.; Doms, R.W.; Proudfoot, A.; Parmentier, M. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J. Biol. Chem. 2003, 101, 746–754. [Google Scholar] [CrossRef]
- Kofuku, Y.; Yoshiura, C.; Ueda, T.; Terasawa, H.; Hirai, T.; Tominaga, S.; Hirose, M.; Maeda, Y.; Takahashi, H.; Terashima, Y.; et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J. Biol. Chem. 2009, 284, 35240–35250. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Xanthou, G.; Williams, T.J.; Pease, J.E. Molecular characterization of the chemokine receptor CXCR3: Evidence for the involvement of distinct extracellular domains in a multi-step model of ligand binding and receptor activation. Eur. J. Immunol. 2003, 33, 2927–2936. [Google Scholar] [CrossRef] [PubMed]
- Kondru, R.; Zhang, J.; Ji, C.; Mirzadegan, T.; Rotstein, D.; Sankuratri, S.; Dioszegi, M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 2008, 73, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, D.; Storelli, S.; Scholten, D.J.; Bosch, L.; Reinhart, T.A.; Streblow, D.N.; Tensen, C.P.; Fitzsimons, C.P.; Zaman, G.J.; Pease, J.E.; et al. Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J. Pharmacol. Exp. Ther. 2008, 325, 544–555. [Google Scholar] [CrossRef]
- De Kruijf, P.; van Heteren, J.; Lim, H.; Conti, P.; van der Lee, M.; Bosch, L.; Ho, K.K.; Auld, D.; Ohlmeyer, M.; Smit, M.J.; et al. Nonpeptidergic allosteric antagonists differentially bind to the CXCR2 chemokine receptor. J. Pharmacol. Exp. Ther. 2009, 329, 783–790. [Google Scholar] [CrossRef]
- Garcia-Perez, J.; Rueda, P.; Staropoli, I.; Kellenberger, E.; Alcami, J.; Arenzana-Seisdedos, F.; Lagane, B. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem. 2011, 286, 4978–4990. [Google Scholar] [CrossRef]
- Qin, S.; Rottman, J.B.; Myers, P.; Kassam, N.; Weinblatt, M.; Loetscher, M.; Koch, A.E.; Moser, B.; Mackay, C.R. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest. 1998, 101, 746–754. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef]
- Thomas, S.Y.; Hou, R.; Boyson, J.E.; Means, T.K.; Hess, C.; Olson, D.P.; Strominger, J.L.; Brenner, M.B.; Gumperz, J.E.; Wilson, S.B.; et al. CD1d-restricted NKT cells express a chemokine receptor profile indicative of Th1-type inflammatory homing cells. J. Immunol. 2003, 171, 2571–2580. [Google Scholar] [CrossRef]
- Nanki, T.; Takada, K.; Komano, Y.; Morio, T.; Kanegane, H.; Nakajima, A.; Lipsky, P.E.; Miyasaka, N. Chemokine receptor expression and functional effects of chemokines on B cells: Implication in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R149. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Whaley, D.; Kulasekeran, P.; Hancock, W.W.; Lu, B.; Bodnar, R.; Newsome, J.; Hebda, P.A.; Wells, A. Delayed and deficient dermal maturation in mice lacking the CXCR3 ELR-negative CXC chemokine receptor. Am. J. Pathol. 2007, 171, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Whaley, D.; Y-Chen, A.; Kulasekeran, P.; Hebda, P.A.; Wells, A. ELR-negative CXC chemokine CXCL11 (IP-9/I-TAC) facilitates dermal and epidermal maturation during wound repair. Am. J. Pathol. 2008, 173, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Whaley, D.; Hooda, S.; Hebda, P.A.; Bodnar, R.J.; Wells, A. Delayed reepithelialization and basement membrane regeneration after wounding in mice lacking CXCR3. Wound Repair Regen. 2009, 17, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luster, A.D.; Leder, P. IP-10, a -C-X-C- chemokine, elicits a potent thymus-dependent antitumor response in vivo. J. Exp. Med. 1993, 178, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Sgadari, C.; Farber, J.M.; Angiolillo, A.L.; Liao, F.; Teruya-Feldstein, J.; Burd, P.R.; Yao, L.; Gupta, G.; Kanegane, C.; Tosato, G. Mig, the monokine induced by interferon-gamma, promotes tumor necrosis in vivo. Blood 1997, 89, 2635–2643. [Google Scholar] [CrossRef]
- Hensbergen, P.J.; Wijnands, P.G.; Schreurs, M.W.; Scheper, R.J.; Willemze, R.; Tensen, C.P. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J. Immunother. 2005, 28, 343–351. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caccuri, F.; Bugatti, A.; Corbellini, S.; Roversi, S.; Zani, A.; Mazzuca, P.; Marsico, S.; Caruso, A.; Giagulli, C. The Synthetic Dipeptide Pidotimod Shows a Chemokine-Like Activity through CXC Chemokine Receptor 3 (CXCR3). Int. J. Mol. Sci. 2019, 20, 5287. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215287
Caccuri F, Bugatti A, Corbellini S, Roversi S, Zani A, Mazzuca P, Marsico S, Caruso A, Giagulli C. The Synthetic Dipeptide Pidotimod Shows a Chemokine-Like Activity through CXC Chemokine Receptor 3 (CXCR3). International Journal of Molecular Sciences. 2019; 20(21):5287. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215287
Chicago/Turabian StyleCaccuri, Francesca, Antonella Bugatti, Silvia Corbellini, Sara Roversi, Alberto Zani, Pietro Mazzuca, Stefania Marsico, Arnaldo Caruso, and Cinzia Giagulli. 2019. "The Synthetic Dipeptide Pidotimod Shows a Chemokine-Like Activity through CXC Chemokine Receptor 3 (CXCR3)" International Journal of Molecular Sciences 20, no. 21: 5287. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215287