Renal Ca2+ and Water Handling in Response to Calcium Sensing Receptor Signaling: Physiopathological Aspects and Role of CaSR-Regulated microRNAs


Abstract
:
1. Introduction
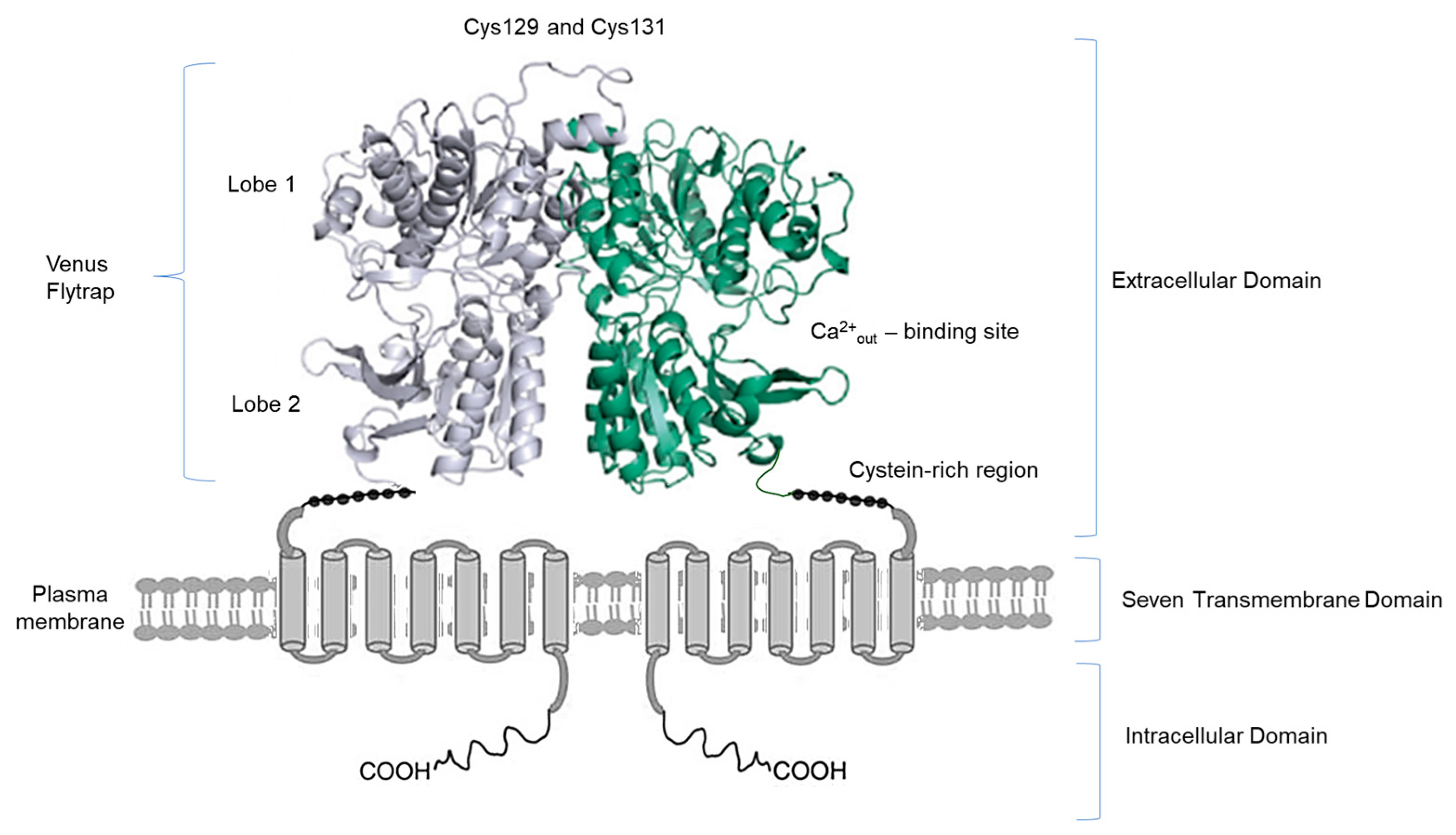
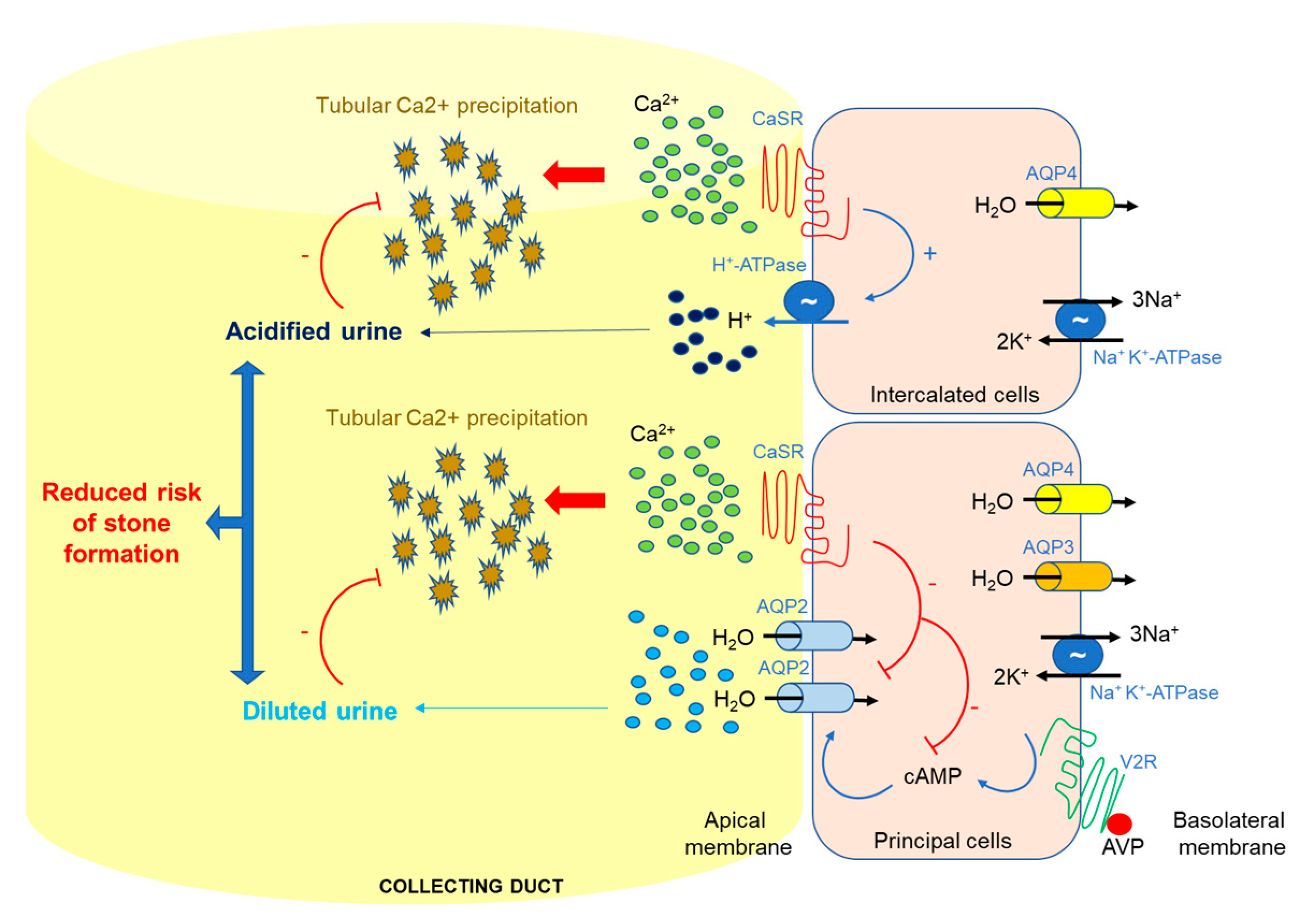
2. Ca2+ Handling and CaSR in the Kidney
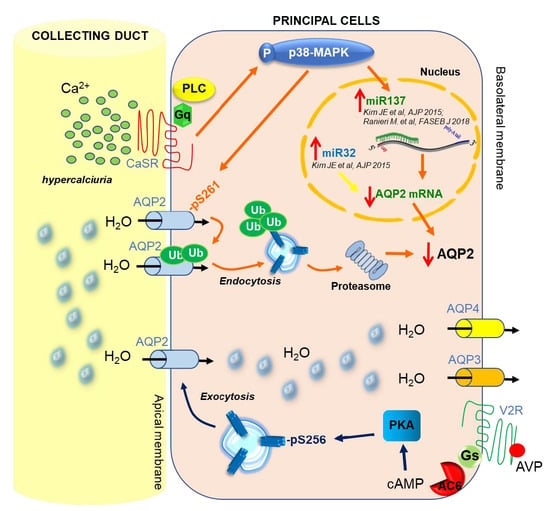
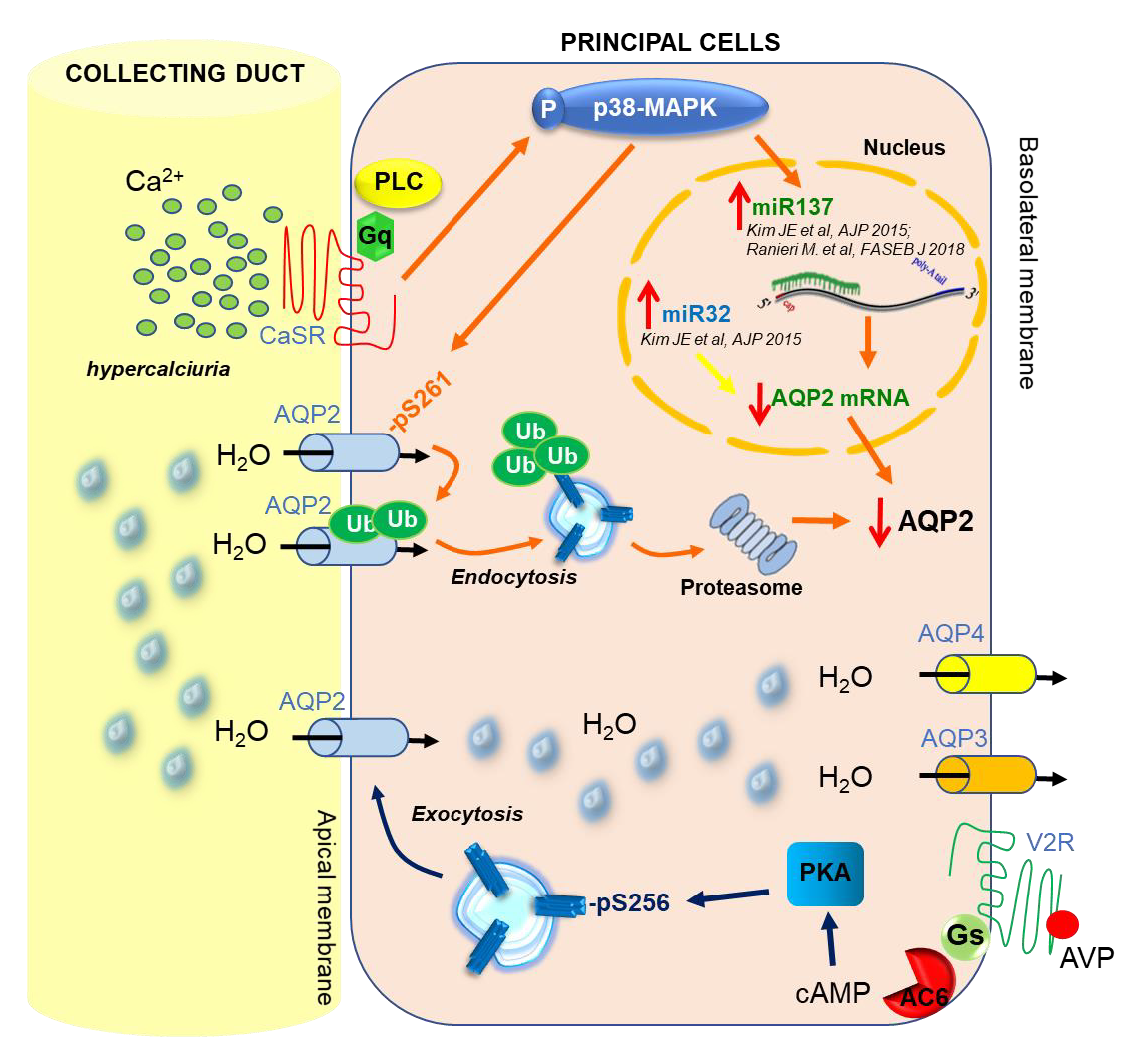
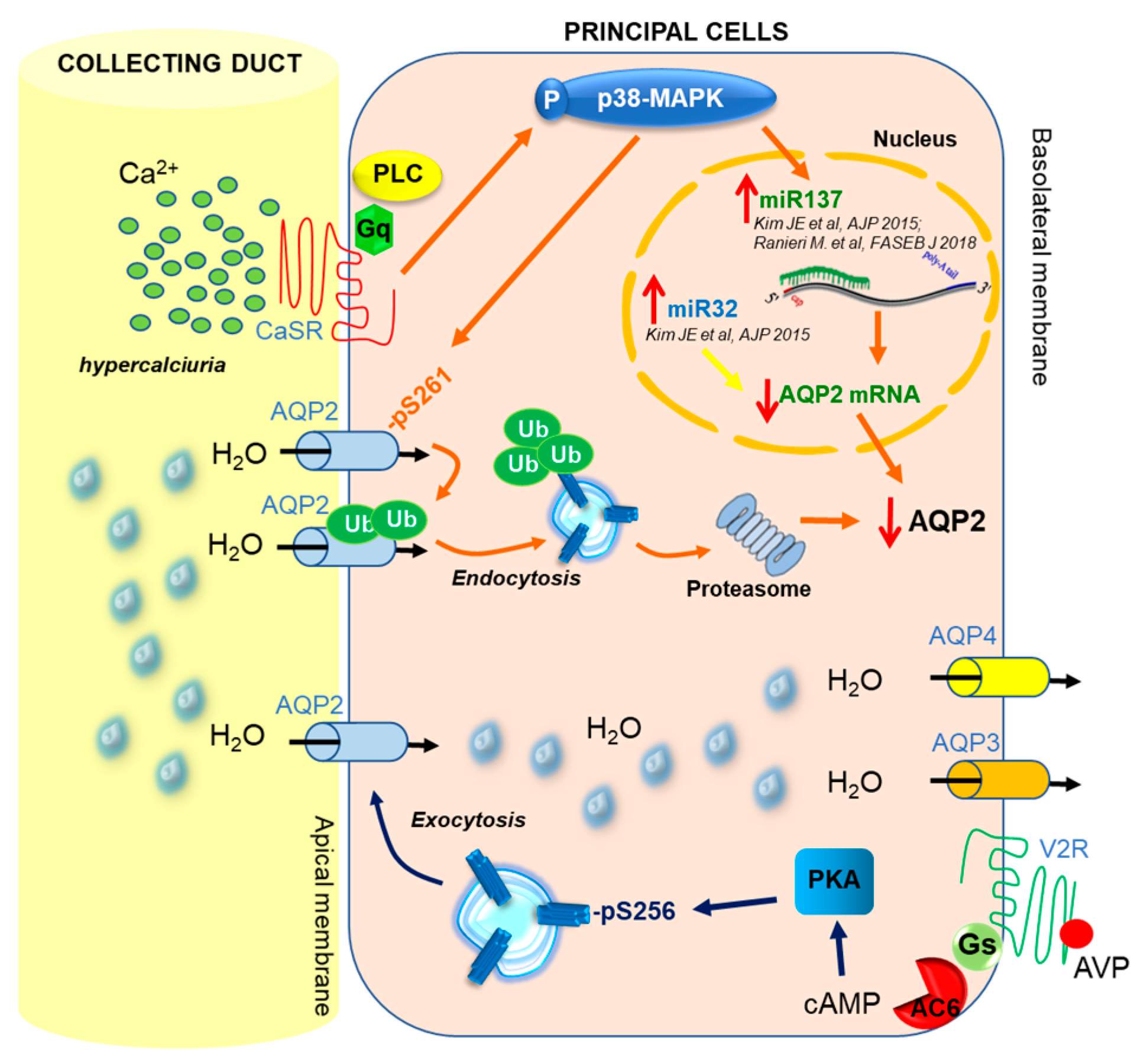
3. CaSR and AQP2 Interplay
4. CaSR-Regulated miRNAs
5. Regulation of miRNA Expression and Therapeutic Perspectives
6. Conclusions
Funding
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Petersen, C.C.; Kasai, H. Calcium and hormone action. Annu. Rev. Physiol. 1994, 56, 297–319. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Collins, T.J.; Peppiatt, C.M.; Prothero, L.S.; MacKenzie, L.; De Smet, P.; Travers, M.; Tovey, S.C.; Seo, J.T.; Berridge, M.J.; et al. Calcium signalling-an overview. Semin. Cell Dev. Biol. 2001, 12, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mellstrom, B.; Naranjo, J.R. Mechanisms of Ca2+-dependent transcription. Curr. Opin. Neurobiol. 2001, 11, 312–319. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium-a life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Civitelli, R.; Ziambaras, K. Calcium and phosphate homeostasis: Concerted interplay of new regulators. J. Endocrinol. Investig. 2011, 34, 3–7. [Google Scholar]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef]
- Tfelt-Hansen, J.; Brown, E.M. The calcium-sensing receptor in normal physiology and pathophysiology: A review. Crit. Rev. Clin. Lab. Sci. 2005, 42, 35–70. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, T.; Zou, J.; Miller, C.L.; Gorkhali, R.; Yang, J.Y.; Schilmiller, A.; Wang, S.; Huang, K.; Brown, E.M.; et al. Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci. Adv. 2016, 2, e1600241. [Google Scholar] [CrossRef]
- Riccardi, D.; Kemp, P.J. The calcium-sensing receptor beyond extracellular calcium homeostasis: Conception, development, adult physiology, and disease. Annu. Rev. Physiol. 2012, 74, 271–297. [Google Scholar] [CrossRef]
- Brown, E.M.; Pollak, M.; Riccardi, D.; Hebert, S.C. Cloning and Characterization of an Extracellular Ca2+-Sensing Receptor from Parathyroid and Kidney-New Insights into the Physiology and Pathophysiology of Calcium-Metabolism. Nephrol Dial. Transpl 1994, 9, 1703–1706. [Google Scholar]
- Riccardi, D.; Valenti, G. Localization and function of the renal calcium-sensing receptor. Nat. Rev. Nephrol. 2016, 12, 414–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.E.; Tamir, H.; Kifor, O.; Simin, R.T.; Rogers, K.V.; Mithal, A.; Gagel, R.F.; Brown, E.M. Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology 1995, 136, 5202–5211. [Google Scholar] [CrossRef]
- Riccardi, D.; Park, J.; Lee, W.S.; Gamba, G.; Brown, E.M.; Hebert, S.C. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. P. Natl. Acad. Sci. USA 1995, 92, 131–135. [Google Scholar] [CrossRef]
- Chang, W.; Tu, C.; Chen, T.H.; Komuves, L.; Oda, Y.; Pratt, S.A.; Miller, S.; Shoback, D. Expression and signal transduction of calcium-sensing receptors in cartilage and bone. Endocrinology 1999, 140, 5883–5893. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Cheng, I.; Rogers, K.; Riccardi, D.; Hall, A.; Diaz, R.; Hebert, S.C.; Soybel, D.I.; Brown, E.M. Identification and localization of extracellular Ca2+-sensing receptor in rat intestine. Am. J. Physiol. 1998, 274, G122–G130. [Google Scholar] [CrossRef]
- Ward, D.T.; Mughal, M.Z.; Ranieri, M.; Dvorak-Ewell, M.M.; Valenti, G.; Riccardi, D. Molecular and clinical analysis of a neonatal severe hyperparathyroidism case caused by a stop mutation in the calcium-sensing receptor extracellular domain representing in effect a human ‘knockout’. Eur. J. Endocrinol. 2013, 169, K1–K7. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, S.; Quinn, S.J.; Brown, E.M.; Bai, M. The extracellular calcium-sensing receptor dimerizes through multiple types of intermolecular interactions. J. Biol. Chem. 2001, 276, 5316–5322. [Google Scholar] [CrossRef]
- Ranieri, M.; Tamma, G.; Di Mise, A.; Vezzoli, G.; Soldati, L.; Svelto, M.; Valenti, G. Excessive signal transduction of gain-of-function variants of the calcium-sensing receptor (CaSR) are associated with increased ER to cytosol calcium gradient. PLoS ONE 2013, 8, e79113. [Google Scholar] [CrossRef]
- Vezzoli, G.; Terranegra, A.; Arcidiacono, T.; Biasion, R.; Coviello, D.; Syren, M.L.; Paloschi, V.; Giannini, S.; Mignogna, G.; Rubinacci, A.; et al. R990G polymorphism of calcium-sensing receptor does produce a gain-of-function and predispose to primary hypercalciuria. Kidney Int. 2007, 71, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Terranegra, A.; Ferraretto, A.; Dogliotti, E.; Scarpellini, M.; Corbetta, S.; Barbieri, A.M.; Spada, A.; Arcidiacono, T.; Rainone, F.; Aloia, A.; et al. Calcimimetic R-568 effects on activity of R990G polymorphism of calcium-sensing receptor. J. Mol. Endocrinol. 2010, 45, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Vezzoli, G.; Tanini, A.; Ferrucci, L.; Soldati, L.; Bianchin, C.; Franceschelli, F.; Malentacchi, C.; Porfirio, B.; Adamo, D.; Terranegra, A.; et al. Influence of calcium-sensing receptor gene on urinary calcium excretion in stone-forming patients. J. Am. Soc. Nephrol. Jasn 2002, 13, 2517–2523. [Google Scholar] [CrossRef] [PubMed]
- Di Mise, A.; Tamma, G.; Ranieri, M.; Centrone, M.; van den Heuvel, L.; Mekahli, D.; Levtchenko, E.N.; Valenti, G. Activation of Calcium-Sensing Receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci. Rep. 2018, 8, 5704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mise, A.; Ranieri, M.; Centrone, M.; Venneri, M.; Tamma, G.; Valenti, D.; Valenti, G. Activation of the Calcium-Sensing Receptor Corrects the Impaired Mitochondrial Energy Status Observed in Renal Polycystin-1 Knockdown Cells Modeling Autosomal Dominant Polycystic Kidney Disease. Front. Mol. Biosci. 2018, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smajilovic, S.; Yano, S.; Jabbari, R.; Tfelt-Hansen, J. The calcium-sensing receptor and calcimimetics in blood pressure modulation. Br. J. Pharmacol. 2011, 164, 884–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loot, A.E.; Pierson, I.; Syzonenko, T.; Elgheznawy, A.; Randriamboavonjy, V.; Zivkovic, A.; Stark, H.; Fleming, I. Ca2+-sensing receptor cleavage by calpain partially accounts for altered vascular reactivity in mice fed a high-fat diet. J. Cardiovasc. Pharmacol. 2013, 61, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Schepelmann, M.; Yarova, P.L.; Lopez-Fernandez, I.; Davies, T.S.; Brennan, S.C.; Edwards, P.J.; Aggarwal, A.; Graca, J.; Rietdorf, K.; Matchkov, V.; et al. The vascular Ca2+-sensing receptor regulates blood vessel tone and blood pressure. Am. J. Physiol.. Cell Physiol. 2016, 310, C193–C204. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fernandez, I.; Schepelmann, M.; Brennan, S.C.; Yarova, P.L.; Riccardi, D. The calcium-sensing receptor: One of a kind. Exp. Physiol. 2015, 100, 1392–1399. [Google Scholar] [CrossRef]
- Berra Romani, R.; Raqeeb, A.; Laforenza, U.; Scaffino, M.F.; Moccia, F.; Avelino-Cruz, J.E.; Oldani, A.; Coltrini, D.; Milesi, V.; Taglietti, V.; et al. Cardiac microvascular endothelial cells express a functional Ca+ -sensing receptor. J. Vasc. Res. 2009, 46, 73–82. [Google Scholar] [CrossRef]
- Molostvov, G.; Hiemstra, T.F.; Fletcher, S.; Bland, R.; Zehnder, D. Arterial Expression of the Calcium-Sensing Receptor Is Maintained by Physiological Pulsation and Protects against Calcification. PLoS ONE 2015, 10, e0138833. [Google Scholar] [CrossRef] [PubMed]
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D., Jr.; Thompson, M.A.; Lowe, A.P.P.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.C.; et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 2015, 7, 284ra260. [Google Scholar] [CrossRef] [PubMed]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Pra, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Abeta42 prompted by exogenous fibrillary or soluble Abeta25-35 in human cortical astrocytes and neurons-therapeutic relevance to Alzheimer’s disease. Biochim. Et Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [CrossRef]
- Dal Pra, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Abeta peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromolecular Med. 2014, 16, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Dal Pra, I.; Chiarini, A.; Armato, U. Antagonizing amyloid-beta/calcium-sensing receptor signaling in human astrocytes and neurons: A key to halt Alzheimer’s disease progression? Neural Regen. Res. 2015, 10, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.C.; Thiem, U.; Roth, S.; Aggarwal, A.; Fetahu, I.; Tennakoon, S.; Gomes, A.R.; Brandi, M.L.; Bruggeman, F.; Mentaverri, R.; et al. Calcium sensing receptor signalling in physiology and cancer. Biochim. Et Biophys. Acta 2013, 1833, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Tennakoon, S.; Aggarwal, A.; Kallay, E. The calcium-sensing receptor and the hallmarks of cancer. Biochim. Et Biophys. Acta 2016, 1863, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Promkan, M.; Liu, G.; Varani, J.; Chakrabarty, S. Role of calcium sensing receptor (CaSR) in tumorigenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 455–463. [Google Scholar] [CrossRef]
- Toka, H.R.; Pollak, M.R.; Houillier, P. Calcium Sensing in the Renal Tubule. Physiology 2015, 30, 317–326. [Google Scholar] [CrossRef]
- Kuo, R.L.; Lingeman, J.E.; Evan, A.P.; Paterson, R.F.; Parks, J.H.; Bledsoe, S.B.; Munch, L.C.; Coe, F.L. Urine calcium and volume predict coverage of renal papilla by Randall’s plaque. Kidney Int. 2003, 64, 2150–2154. [Google Scholar] [CrossRef]
- Shavit, L.; Jaeger, P.; Unwin, R.J. What is nephrocalcinosis? Kidney Int. 2015, 88, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coe, F.L.; Worcester, E.M.; Evan, A.P. Idiopathic hypercalciuria and formation of calcium renal stones. Nat. Rev. Nephrol. 2016, 12, 519–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezzoli, G.; Soldati, L.; Gambaro, G. Update on primary hypercalciuria from a genetic perspective. J. Urol. 2008, 179, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Moor, M.B.; Bonny, O. Ways of calcium reabsorption in the kidney. Am. J. Physiol. Ren. Physiol. 2016, 310, F1337–F1350. [Google Scholar] [CrossRef] [Green Version]
- Sands, J.M.; Naruse, M.; Baum, M.; Jo, I.; Hebert, S.C.; Brown, E.M.; Harris, H.W. Apical extracellular calcium/polyvalent cation-sensing receptor regulates vasopressin-elicited water permeability in rat kidney inner medullary collecting duct. J. Clin. Investig. 1997, 99, 1399–1405. [Google Scholar] [CrossRef]
- Renkema, K.Y.; Velic, A.; Dijkman, H.B.; Verkaart, S.; van der Kemp, A.W.; Nowik, M.; Timmermans, K.; Doucet, A.; Wagner, C.A.; Bindels, R.J.; et al. The calcium-sensing receptor promotes urinary acidification to prevent nephrolithiasis. J. Am. Soc. Nephrol. 2009, 20, 1705–1713. [Google Scholar] [CrossRef]
- Wagner, C.A.; Mohebbi, N. Urinary pH and stone formation. J. Nephrol. 2010, 23, S165–S169. [Google Scholar]
- Vezzoli, G.; Macrina, L.; Magni, G.; Arcidiacono, T. Calcium-sensing receptor: Evidence and hypothesis for its role in nephrolithiasis. Urolithiasis 2019, 47, 23–33. [Google Scholar] [CrossRef]
- Riccardi, D.; Brown, E.M. Physiology and pathophysiology of the calcium-sensing receptor in the kidney. Am. J. Physiol. Ren. Physiol. 2010, 298, F485–F499. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.T.; Riccardi, D. Renal physiology of the extracellular calcium-sensing receptor. Pflug. Arch. Eur. J. Physiol. 2002, 445, 169–176. [Google Scholar] [CrossRef]
- Houillier, P. Calcium-sensing in the kidney. Curr. Opin. Nephrol. Hypertens. 2013, 22, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.C.; Brown, E.M.; Harris, H.W. Role of the Ca2+-sensing receptor in divalent mineral ion homeostasis. J. Exp. Biol. 1997, 200, 295–302. [Google Scholar] [PubMed]
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Bio. 2003, 4, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.H.; Thakker, R.V. The calcium-sensing receptor: Insights into extracellular calcium homeostasis in health and disease. J. Endocrinol. 1997, 154, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Ba, J.; Brown, D.; Friedman, P.A. Calcium-sensing receptor regulation of PTH-inhibitable proximal tubule phosphate transport. Am. J. Physiol. Ren. Physiol. 2003, 285, F1233–F1243. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.M. Physiology and pathophysiology of the extracellular calcium-sensing receptor. Am. J. Med. 1999, 106, 238–253. [Google Scholar] [CrossRef]
- Nedvetsky, P.I.; Tamma, G.; Beulshausen, S.; Valenti, G.; Rosenthal, W.; Klussmann, E. Regulation of aquaporin-2 trafficking. Handb. Exp. Pharmacol. 2009, 190, 133–157. [Google Scholar] [CrossRef]
- Procino, G.; Carmosino, M.; Tamma, G.; Gouraud, S.; Laera, A.; Riccardi, D.; Svelto, M.; Valenti, G. Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int. 2004, 66, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Procino, G.; Mastrofrancesco, L.; Mira, A.; Tamma, G.; Carmosino, M.; Emma, F.; Svelto, M.; Valenti, G. Aquaporin 2 and apical calcium-sensing receptor: New players in polyuric disorders associated with hypercalciuria. Semin. Nephrol. 2008, 28, 297–305. [Google Scholar] [CrossRef]
- Procino, G.; Mastrofrancesco, L.; Tamma, G.; Lasorsa, D.R.; Ranieri, M.; Stringini, G.; Emma, F.; Svelto, M.; Valenti, G. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study. PLoS ONE 2012, 7, e33145. [Google Scholar] [CrossRef]
- Centrone, M.; Ranieri, M.; Di Mise, A.; Berlingerio, S.P.; Russo, A.; Deen, P.M.T.; Staub, O.; Valenti, G.; Tamma, G. AQP2 Abundance is Regulated by the E3-Ligase CHIP Via HSP70. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Di Mise, A.; Tamma, G.; Valenti, G. Vasopressin-aquaporin-2 pathway: Recent advances in understanding water balance disorders. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Valenti, G.; Laera, A.; Pace, G.; Aceto, G.; Lospalluti, M.L.; Penza, R.; Selvaggi, F.P.; Chiozza, M.L.; Svelto, M. Urinary aquaporin 2 and calciuria correlate with the severity of enuresis in children. J. Am. Soc. Nephrol. 2000, 11, 1873–1881. [Google Scholar] [PubMed]
- Tamma, G.; Di Mise, A.; Ranieri, M.; Svelto, M.; Pisot, R.; Bilancio, G.; Cavallo, P.; De Santo, N.G.; Cirillo, M.; Valenti, G. A decrease in aquaporin 2 excretion is associated with bed rest induced high calciuria. J. Transl. Med. 2014, 12, 133. [Google Scholar] [CrossRef]
- Ranieri, M.; Tamma, G.; Di Mise, A.; Russo, A.; Centrone, M.; Svelto, M.; Calamita, G.; Valenti, G. Negative feedback from CaSR signaling to aquaporin-2 sensitizes vasopressin to extracellular Ca2+. J. Cell Sci. 2015, 128, 2350–2360. [Google Scholar] [CrossRef]
- Ranieri, M.; Zahedi, K.; Tamma, G.; Centrone, M.; Di Mise, A.; Soleimani, M.; Valenti, G. CaSR signaling down-regulates AQP2 expression via a novel microRNA pathway in pendrin and NaCl cotransporter knockout mice. FASEB J. 2018, 32, 2148–2159. [Google Scholar] [CrossRef]
- Soleimani, M.; Barone, S.; Xu, J.; Shull, G.E.; Siddiqui, F.; Zahedi, K.; Amlal, H. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. P. Natl. Acad. Sci. USA 2012, 109, 13368–13373. [Google Scholar] [CrossRef] [Green Version]
- Khositseth, S.; Charngkaew, K.; Boonkrai, C.; Somparn, P.; Uawithya, P.; Chomanee, N.; Payne, D.M.; Fenton, R.A.; Pisitkun, T. Hypercalcemia induces targeted autophagic degradation of aquaporin-2 at the onset of nephrogenic diabetes insipidus. Kidney Int 2017, 91, 1070–1087. [Google Scholar] [CrossRef] [Green Version]
- Nedvetsky, P.I.; Tabor, V.; Tamma, G.; Beulshausen, S.; Skroblin, P.; Kirschner, A.; Mutig, K.; Boltzen, M.; Petrucci, O.; Vossenkämper, A.; et al. Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J. Am. Soc. Nephrol. 2010, 10, 1645–1656. [Google Scholar] [CrossRef]
- Trepiccione, F.; Pisitkun, T.; Hoffert, J.D.; Poulsen, S.B.; Capasso, G.; Nielsen, S.; Knepper, M.A.; Fenton, R.A.; Christensen, B.M. Early targets of lithium in rat kidney inner medullary collecting duct include p38 and ERK1/2. Kidney Int. 2014, 86, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, M.; Hasler, U.; Leroy, V.; de Seigneux, S.; Dimitrov, M.; Mordasini, D.; Rousselot, M.; Martin, P.-Y.; Féraille, E. Calcium-sensing receptor attenuates AVP-induced aquaporin-2 expression via a calmodulin-dependent mechanism. J. Am. Soc. Nephrol. 2008, 1, 109–116. [Google Scholar] [CrossRef]
- Sands, J.M.; Flores, F.X.; Kato, a.; Baum, M.a.; Brown, E.M.; Ward, D.T.; Hebert, S.C.; Harris, H.W. Vasopressin-elicited water and urea permeabilities are altered in IMCD in hypercalcemic rats. Am. J. Physiol. 1998, 274, F978–F985. [Google Scholar] [CrossRef] [PubMed]
- Puliyanda, D.P.; Ward, D.T.; Baum, M.A.; Hammond, T.G.; Harris, H.W., Jr. Calpain-mediated AQP2 proteolysis in inner medullary collecting duct. Biochem. Biophys. Res. Commun. 2003, 303, 52–58. [Google Scholar] [CrossRef]
- Hwang, H.W.; Wentzel, E.A.; Mendell, J.T. A hexanucleotide element directs microRNA nuclear import. Science 2007, 315, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Simpson, K.; Wonnacott, A.; Fraser, D.J.; Bowen, T. MicroRNAs in Diabetic Nephropathy: From Biomarkers to Therapy. Curr. Diabetes Rep. 2016, 16, 35. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Hou, J. Lecture: New light on the role of claudins in the kidney. Organogenesis 2012, 8, 1–9. [Google Scholar] [CrossRef]
- Hou, J. Claudins and mineral metabolism. Curr. Opin. Nephrol. Hypertens. 2016, 25, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Renigunta, V.; Himmerkus, N.; Zhang, J.; Renigunta, A.; Bleich, M.; Hou, J. Claudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathway. Embo J. 2012, 31, 1999–2012. [Google Scholar] [CrossRef]
- Gong, Y.; Hou, J. Claudin-14 underlies Ca++-sensing receptor-mediated Ca++ metabolism via NFAT-microRNA-based mechanisms. J. Am. Soc. Nephrol. 2014, 25, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Himmerkus, N.; Plain, A.; Bleich, M.; Hou, J. Epigenetic regulation of microRNAs controlling CLDN14 expression as a mechanism for renal calcium handling. J. Am. Soc. Nephrol. 2015, 26, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.L. Role of claudins in renal calcium handling. Nefrologia 2015, 35, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Tennakoon, S.; Lines, K.E.; Groschel, C.; Aggarwal, A.; Mesteri, I.; Baumgartner-Parzer, S.; Mader, R.M.; Thakker, R.V.; Kallay, E. miR-135b- and miR-146b-dependent silencing of calcium-sensing receptor expression in colorectal tumors. Int. J. Cancer 2016, 138, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Chakrabarty, S. Induction of CaSR expression circumvents the molecular features of malignant CaSR null colon cancer cells. Int. J. Cancer 2013, 133, 2307–2314. [Google Scholar] [CrossRef]
- Singh, N.; Liu, G.; Chakrabarty, S. Isolation and characterization of calcium sensing receptor null cells: A highly malignant and drug resistant phenotype of colon cancer. Int. J. Cancer 2013, 132, 1996–2005. [Google Scholar] [CrossRef]
- Shilo, V.; Ben-Dov, I.Z.; Nechama, M.; Silver, J.; Naveh-Many, T. Parathyroid-specific deletion of dicer-dependent microRNAs abrogates the response of the parathyroid to acute and chronic hypocalcemia and uremia. FASEB J. 2015, 29, 3964–3976. [Google Scholar] [CrossRef] [Green Version]
- Vaira, V.; Verdelli, C.; Forno, I.; Corbetta, S. MicroRNAs in parathyroid physiopathology. Mol. Cell. Endocrinol. 2017, 456, 9–15. [Google Scholar] [CrossRef]
- Hasler, U.; Leroy, V.; Martin, P.Y.; Feraille, E. Aquaporin-2 abundance in the renal collecting duct: New insights from cultured cell models. Am. J. Physiol Ren. Physiol 2009, 297, F10–F18. [Google Scholar] [CrossRef]
- Moeller, H.B.; Olesen, E.T.B.; Fenton, R.A. Regulation of the water channel aquaporin-2 by posttranslational modification. Am. J. Physiol. Ren. Physiol. 2011, 300, F1062–F1073. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.; da Silva, I.V.; Rodrigues, C.M.P.; Castro, R.E.; Soveral, G. The Emerging Role of microRNAs in Aquaporin Regulation. Front. Chem. 2018, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Chao, G.; Wang, Y.; Zhang, S.; Yang, W.; Ni, Z.; Zheng, X. MicroRNA-29a increased the intestinal membrane permeability of colonic epithelial cells in irritable bowel syndrome rats. Oncotarget 2017, 8, 85828–85837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Shi, Y.; Liu, M.; Sun, J. circHIPK3 regulates cell proliferation and migration by sponging miR-124 and regulating AQP3 expression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Huebert, R.C.; Jagavelu, K.; Hendrickson, H.I.; Vasdev, M.M.; Arab, J.P.; Splinter, P.L.; Trussoni, C.E.; Larusso, N.F.; Shah, V.H. Aquaporin-1 promotes angiogenesis, fibrosis, and portal hypertension through mechanisms dependent on osmotically sensitive microRNAs. Am. J. Pathol. 2011, 179, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Jung, H.J.; Lee, Y.J.; Kwon, T.H. Vasopressin-regulated miRNAs and AQP2-targeting miRNAs in kidney collecting duct cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F749–F764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Shi, H.; Gao, M.; Ma, N.; Sun, R. Long non-coding RNA CASC2 improved acute lung injury by regulating miR-144-3p/AQP1 axis to reduce lung epithelial cell apoptosis. Cell Biosci. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yang, R.; Zhao, S.; Chen, Y.; Hong, S.; Wang, K.; Wang, T.; Cheng, J.; Zhang, T.; Chen, D. Decreased miR-320 expression is associated with breast cancer progression, cell migration, and invasiveness via targeting Aquaporin 1. Acta Biochim. Et Biophys. Sin. 2018, 50, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Sepramaniam, S.; Armugam, A.; Lim, K.Y.; Karolina, D.S.; Swaminathan, P.; Tan, J.R.; Jeyaseelan, K. MicroRNA 320a functions as a novel endogenous modulator of aquaporins 1 and 4 as well as a potential therapeutic target in cerebral ischemia. J. Biol. Chem 2010, 285, 29223–29230. [Google Scholar] [CrossRef]
- Tang, R.; Pei, L.; Bai, T.; Wang, J. Down-regulation of microRNA-126-5p contributes to overexpression of VEGFA in lipopolysaccharide-induced acute lung injury. Biotechnol. Lett. 2016, 38, 1277–1284. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Ma, Y.; Tang, G.; Liu, Y.; Chen, X.; Zhang, Z.; Zeng, L.; Wang, Y.; Ouyang, Y.B.; et al. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J. Cereb. Blood Flow Metab. 2015, 35, 1977–1984. [Google Scholar] [CrossRef]
- Xiong, W.; Ran, J.; Jiang, R.; Guo, P.; Shi, X.; Li, H.; Lv, X.; Li, J.; Chen, D. miRNA-320a inhibits glioma cell invasion and migration by directly targeting aquaporin 4. Oncol. Rep. 2018, 39, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cheng, W.; Wang, X.; Yang, Z.; Zhou, X.; Pan, C. Overexpression of MicroRNA-145 Ameliorates Astrocyte Injury by Targeting Aquaporin 4 in Cerebral Ischemic Stroke. Biomed. Res. Int. 2017, 2017, 9530951. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J. Pathol. 2013, 229, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, L.F.; Chanyshev, M.D.; Kolmykov, S.K.; Ushakov, D.S.; Nechkin, S.S. Effect of xenobiotics on microRNA expression in rat liver. Biomed. Khim. 2016, 62, 154. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E. The art of microRNA research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef]
- Remsburg, C.; Konrad, K.; Sampilo, N.F.; Song, J.L. Analysis of microRNA functions. Method. Cell Biol. 2019, 151, 323–334. [Google Scholar]
- Bijkerk, R.; Trimpert, C.; van Solingen, C.; de Bruin, R.G.; Florijn, B.W.; Kooijman, S.; van den Berg, R.; van der Veer, E.P.; Bredewold, E.O.W.; Rensen, P.C.N.; et al. MicroRNA-132 controls water homeostasis through regulating MECP2-mediated vasopressin synthesis. Am. J. Physiol. Ren. Physiol. 2018, 315, F1129–F1138. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Target mRNA | Target Protein | Target Organ | References | |
|---|---|---|---|---|---|
| miR-9 miR-374 |  | CLDN14 | Claudin-14 | Thick Ascending Limb cell, kidney | Hou J, Organogenesis 2012 [79]; Gong Y, Hou J, JASN 2014 [82]; Gong Y et al., JASN 2015 [83]; Hou J, Curr Opin Nephrol Hypert 2016 [80] |
| miR-21 miR-135a miR-135b | | Tumor suppressors | Tumor suppressor proteins | Human colon carcinoma cell lines, colon | Singh N et al., Int J Cancer, 2013 [86,87] |
| miR-145 |  | Oncogenes | Oncoproteins | Human colon carcinoma cell lines, colon | Singh N et al., Int J Cancer, 2013 [86,87] |
| miR-375 miR-429 miR-361 | | PTH | Parathormone | Parathyroid | Shilo V et al., FASEB J 2015 [88] |
| miR-137 | | AQP2 | Aquaporin-2 | Collecting duct, kidney | Ranieri M et al., FASEB J 2018 [66] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranieri, M. Renal Ca2+ and Water Handling in Response to Calcium Sensing Receptor Signaling: Physiopathological Aspects and Role of CaSR-Regulated microRNAs. Int. J. Mol. Sci. 2019, 20, 5341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215341
Ranieri M. Renal Ca2+ and Water Handling in Response to Calcium Sensing Receptor Signaling: Physiopathological Aspects and Role of CaSR-Regulated microRNAs. International Journal of Molecular Sciences. 2019; 20(21):5341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215341
Chicago/Turabian StyleRanieri, Marianna. 2019. "Renal Ca2+ and Water Handling in Response to Calcium Sensing Receptor Signaling: Physiopathological Aspects and Role of CaSR-Regulated microRNAs" International Journal of Molecular Sciences 20, no. 21: 5341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215341