Molecular Links between Central Obesity and Breast Cancer

 ,
,
Abstract
:1. Introduction
2. Tumor Microenvironment vs. Adipose Tissue Microenvironment and Their Role in Tumor-Promoting Low-Grade Inflammation
3. The Hormones, Their Signaling Pathways, Central Adiposity, and Breast Cancer
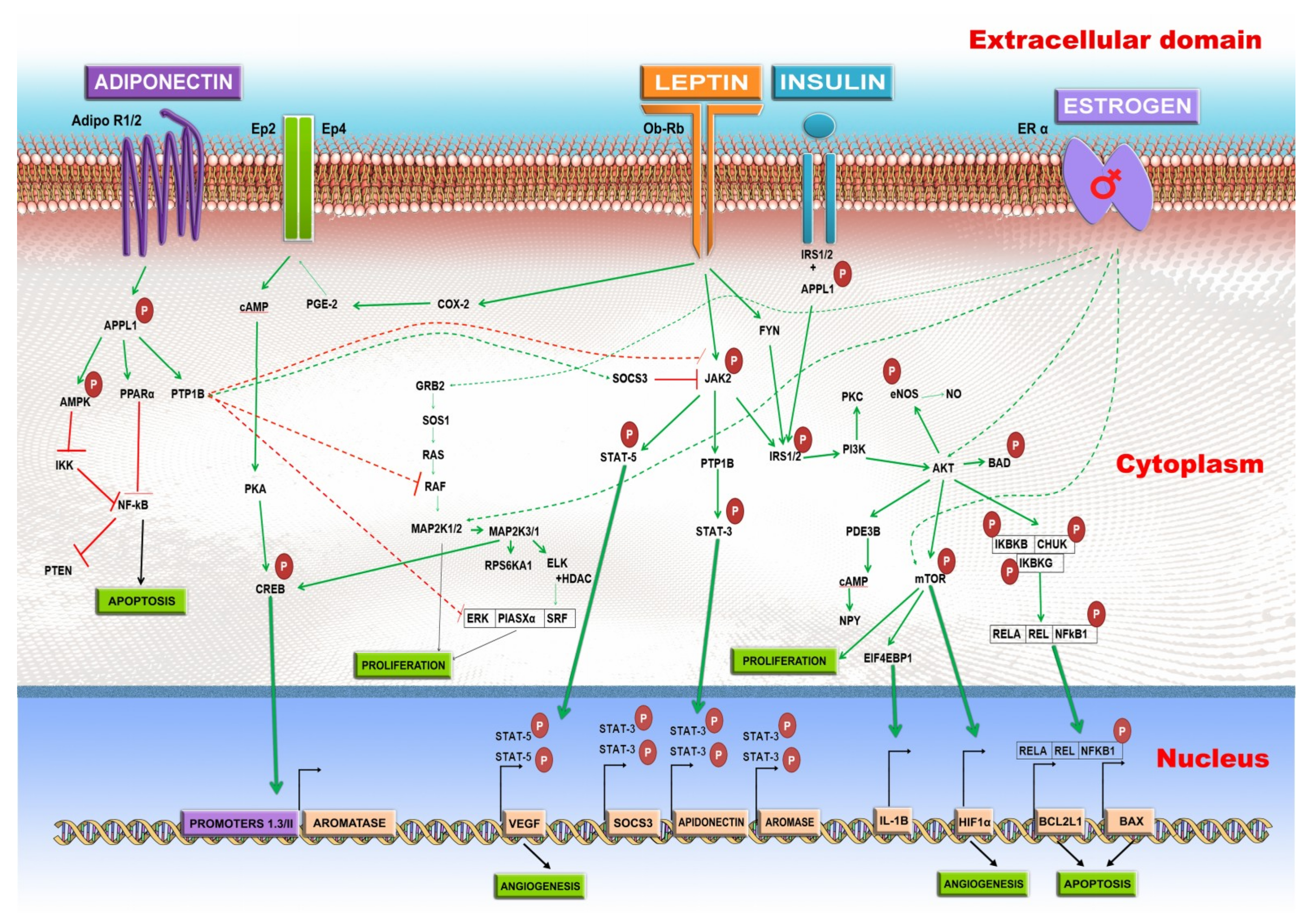
3.1. Leptin
3.2. Adiponectin
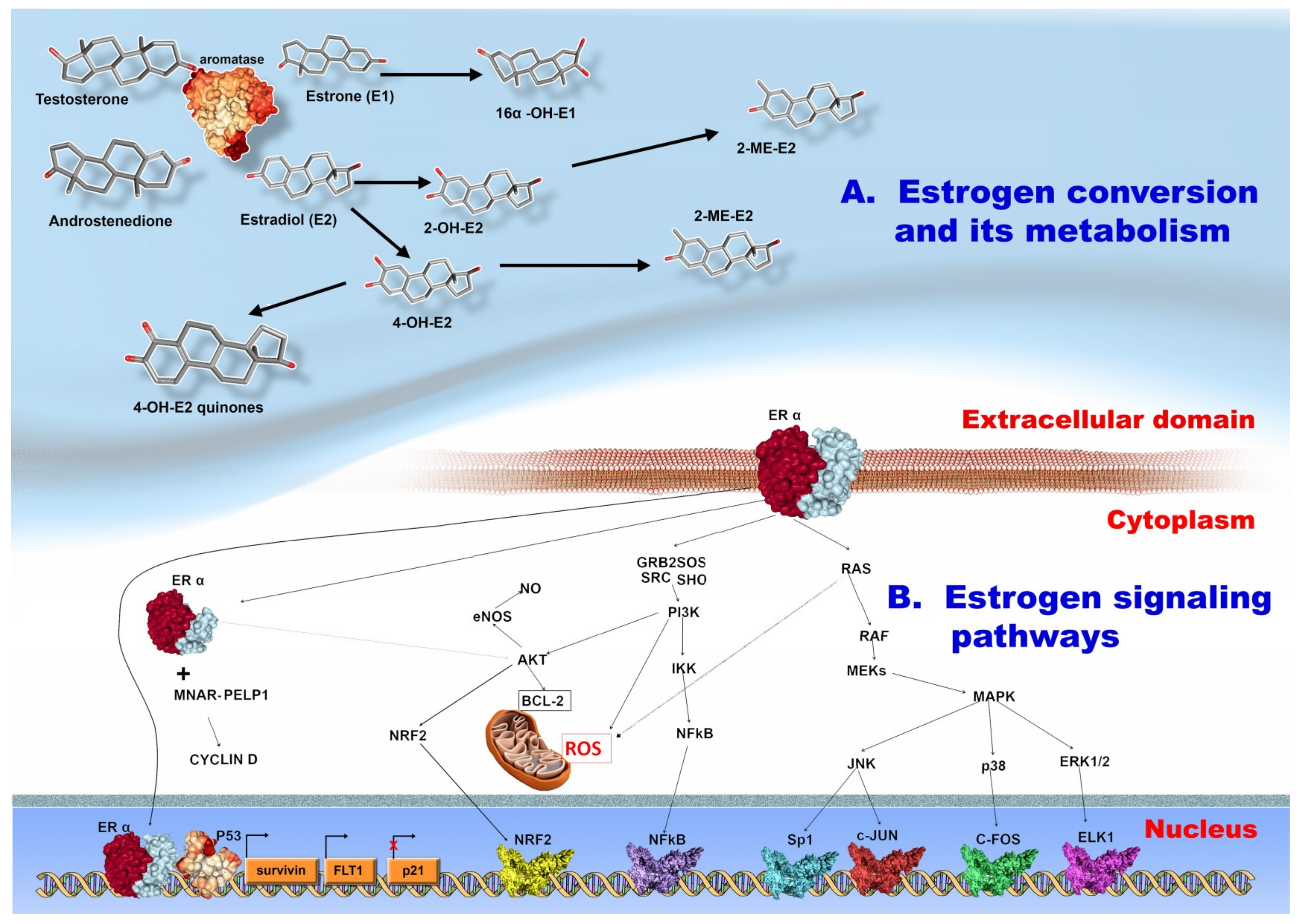
3.3. Estrogen
4. Exosomes, MicroRNAs, and Their Possible Role in the Interplay between Central Adiposity, and BC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BC | Breast Cancer |
| TNBC | Triple Negative Breast Cancer |
| ER | Estrogen Receptor |
| PR | Progesteron Receptor |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| WAT | White Adipose Tissue |
| BMI | Body Mass Index |
| PR+ | Progesterone Receptor Positive |
| VAT | Visceral Adipose Tissue |
| TAMS | Tumor Associated Macrophages |
| Tregs | Regulatory T Cells |
| Th2 | Type 2 T Helper Cells |
| Bregs | Regulatory B Cells |
| CAF | Cancer Associated Fibroblasts |
| M1 | Type 1 Macrophages |
| M2 | Type 2 Macrophages |
| NK | Natural Killer Cells |
| INF γ | Interferon Gamma |
| TNFα | Tumor Necrosis Factor Alpha |
| IL | Interleukin |
| ROS | Reactive Oxygen Species |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| NRF 2 | Nuclear Factor 2 |
| AP1 | Activator Protein 1 |
| HIF 1α | Hypoxia-Inducible Factor 1 Alpha |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| MMPs | Matrix Metalloproteinases |
| AKT | Protein Kinase B |
| NFκβ | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| TLR | Toll-Like Receptor |
| PAMPs | Pathogen-Associated Molecular Patterns |
| CRP | C Reactive Protein |
| LPS | Lipopolysaccharide |
| MCP 1 | Monocyte Chemoattractant Protein 1 |
| ICAM1 | Intercellular Adhesion Molecule 1 (Cd54) |
| MIP | Macrophage Inflammatory Proteins |
| VEGF | Vascular Endothelial Growth Factor |
| ATC | Subcutaneous Stem Cells |
| NLRP3 | Cryopyrin |
| KO | Knock-Out |
| WT | Wild Type |
| COX 2 | Cyclooxygenase 2 |
| PGE2 | Prostaglandin E2 |
| cAMP | Cyclic Amp |
| JNK | Jun N-Terminal Kinase |
| E2 | Estradiol |
| PI3K | Phosphoinositide 3-Kinase |
| BRCA1 | Breast Cancer Type 1 Susceptibility Protein |
| KEAP 1 | Kelch-Like ECH-Associated Protein 1 |
| ARE | Antioxidant Response Element |
| PP | Physagulide P |
| EGFR | Epidermal Growth Factor Receptor |
| MAPK | Mitogen Activated Protein Kinase |
| FGF | Fibroblast Growth Factor |
| EPHA 2 | Ephrin Type-A Receptor 2 |
| ATM | Adipose Tissue Macrophage |
References
- Shah, R.; Rosso, K.; Nathanson, S.D. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Jurj, A.; Braicu, C.; Pop, L.-A.; Tomuleasa, C.; Gherman, C.D.; Berindan-Neagoe, I. The new era of nanotechnology, an alternative to change cancer treatment. Drug Des. Dev. Ther. 2017, 11, 2871–2890. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Braicu, C.; Irimie, A.; Craciun, L.; Berindan-Neagoe, I. Nanopharmacology in translational hematology and oncology. Int. J. Nanomed. 2014, 9, 3465–3479. [Google Scholar] [CrossRef]
- Braicu, C.; Catana, C.; Calin, G.A.; Berindan-Neagoe, I. NCRNA combined therapy as future treatment option for cancer. Curr. Pharm. Des. 2014, 20, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Chiorean, R.; Braicu, C.; Berindan-Neagoe, I. Another review on triple negative breast cancer. Are we on the right way towards the exit from the labyrinth? Breast 2013, 22, 1026–1033. [Google Scholar] [CrossRef]
- Elks, C.M.; Francis, J. Central adiposity, systemic inflammation, and the metabolic syndrome. Curr. Hypertens. Rep. 2010, 12, 99–104. [Google Scholar] [CrossRef]
- Chen, M.J.; Wu, W.Y.; Yen, A.M.; Fann, J.C.; Chen, S.L.; Chiu, S.Y.; Chen, H.H.; Chiou, S.T. Body mass index and breast cancer: Analysis of a nation-wide population-based prospective cohort study on 1 393 985 Taiwanese women. Int. J. Obes. 2016, 40, 524–530. [Google Scholar] [CrossRef]
- Suzuki, R.; Orsini, N.; Saji, S.; Key, T.J.; Wolk, A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status—A meta-analysis. Int. J. Cancer 2009, 124, 698–712. [Google Scholar] [CrossRef]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef]
- Xia, X.; Chen, W.; Li, J.; Chen, X.; Rui, R.; Liu, C.; Sun, Y.; Liu, L.; Gong, J.; Yuan, P. Body mass index and risk of breast cancer: A nonlinear dose-response meta-analysis of prospective studies. Sci. Rep. 2014, 4, 7480. [Google Scholar] [CrossRef] [PubMed]
- Harvie, M.; Hooper, L.; Howell, A.H. Central obesity and breast cancer risk: A systematic review. Obes. Rev. 2003, 4, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.; Scott, C.G.; Jensen, M.R.; Norman, A.D.; Bertrand, K.A.; Pankratz, V.S.; Brandt, K.R.; Visscher, D.W.; Shepherd, J.A.; Tamimi, R.M.; et al. Body mass index, mammographic density, and breast cancer risk by estrogen receptor subtype. Breast Cancer Res. BCR 2019, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Godinho-Mota, J.C.M.; Martins, K.A.; Vaz-Goncalves, L.; Mota, J.F.; Soares, L.R.; Freitas-Junior, R. Visceral adiposity increases the risk of breast cancer: A case-control study. Nutr. Hosp. 2018, 35, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, L.; Cui, S.; Tian, F.; Fan, Z.; Geng, C.; Cao, X.; Yang, Z.; Wang, X.; Liang, H.; et al. Distinct Effects of Body Mass Index and Waist/Hip Ratio on Risk of Breast Cancer by Joint Estrogen and Progestogen Receptor Status: Results from a Case-Control Study in Northern and Eastern China and Implications for Chemoprevention. Oncologist 2017, 22, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.W.; Young, E.; Patterson, S.G.; Makey, K.L.; Wells, J.; Huang, M.; Tucker, K.B.; Miele, L. Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biol. Ther. 2011, 11, 910–917. [Google Scholar] [CrossRef]
- Liu, C.R.; Li, Q.; Hou, C.; Li, H.; Shuai, P.; Zhao, M.; Zhong, X.R.; Xu, Z.P.; Li, J.Y. Changes in Body Mass Index, Leptin, and Leptin Receptor Polymorphisms and Breast Cancer Risk. DNA Cell Biol. 2018, 37, 182–188. [Google Scholar] [CrossRef]
- Jung, S.Y.; Papp, J.C.; Sobel, E.M.; Yu, H.; Zhang, Z.F. Breast cancer risk and insulin resistance: Post genome-wide gene-environment interaction study using a random survival forest. Cancer Res. 2019, 79, 2784–2794. [Google Scholar] [CrossRef]
- Sanderson, M.; Lipworth, L.; Shrubsole, M.J.; Andersen, S.W.; Shu, X.O.; Zheng, W.; Hargreaves, M.K.; Blot, W.J. Diabetes, obesity, and subsequent risk of postmenopausal breast cancer among white and black women in the Southern Community Cohort Study. Cancer Causes Control CCC 2019, 30, 425–433. [Google Scholar] [CrossRef]
- Martinez, J.A.; Chalasani, P.; Thomson, C.A.; Roe, D.; Altbach, M.; Galons, J.-P.; Stopeck, A.; Thompson, P.A.; Villa-Guillen, D.E.; Chow, H.H.S. Phase II study of metformin for reduction of obesity-associated breast cancer risk: A randomized controlled trial protocol. BMC Cancer 2016, 16, 500. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Craciun, L.; Tomuleasa, C.; Berindan-Neagoe, I.; Kacso, G.; Florian, I.S.; Crivii, C. Repositioning metformin in cancer: Genetics, drug targets, and new ways of delivery. Tumor Biol. 2014, 35, 5101–5110. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Hadrup, S.; Donia, M.; Thor Straten, P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013, 6, 123–133. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Feuerer, M.; Beckhove, P.; Ahlert, T.; Umansky, V. T cell memory, anergy and immunotherapy in breast cancer. J. Mammary Gland. Boil. Neoplas. 2002, 7, 201–208. [Google Scholar] [CrossRef]
- Varn, F.S.; Mullins, D.W.; Arias-Pulido, H.; Fiering, S.; Cheng, C. Adaptive immunity programmes in breast cancer. Immunology 2017, 150, 25–34. [Google Scholar] [CrossRef]
- He, Y.; Qian, H.; Liu, Y.; Duan, L.; Li, Y.; Shi, G. The roles of regulatory B cells in cancer. J. Immunol. Res. 2014, 2014, 215471. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus white adipose tissue: A mini-review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, C.L.; Doyle, S.L.; Reynolds, J.V. Visceral adiposity, insulin resistance and cancer risk. Diabetol. Metab. Syndr. 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Donninelli, G.; Del Corno, M.; Pierdominici, M.; Scazzocchio, B.; Vari, R.; Varano, B.; Pacella, I.; Piconese, S.; Barnaba, V.; D’Archivio, M.; et al. Distinct Blood and Visceral Adipose Tissue Regulatory T Cell and Innate Lymphocyte Profiles Characterize Obesity and Colorectal Cancer. Front. Immunol. 2017, 8, 643. [Google Scholar] [CrossRef] [PubMed]
- Piconese, S.; Valzasina, B.; Colombo, M.P. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 2008, 205, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Altintas, M.M.; Azad, A.; Nayer, B.; Contreras, G.; Zaias, J.; Faul, C.; Reiser, J.; Nayer, A. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J. Lipid Res. 2011, 52, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yu, X.; Chen, H.; Sjoberg, S.; Roux, J.; Zhang, L.; Ivoulsou, A.H.; Bensaid, F.; Liu, C.L.; Liu, J.; et al. Leptin Deficiency Shifts Mast Cells toward Anti-Inflammatory Actions and Protects Mice from Obesity and Diabetes by Polarizing M2 Macrophages. Cell Metabolism 2015, 22, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.F. The role of mast cells in wound healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Bahr, I.; Goritz, V.; Doberstein, H.; Hiller, G.G.; Rosenstock, P.; Jahn, J.; Portner, O.; Berreis, T.; Mueller, T.; Spielmann, J.; et al. Diet-Induced Obesity Is Associated with an Impaired NK Cell Function and an Increased Colon Cancer Incidence. J. Nutr. Metab. 2017, 2017, 4297025. [Google Scholar] [CrossRef]
- Conroy, M.J.; Fitzgerald, V.; Doyle, S.L.; Channon, S.; Useckaite, Z.; Gilmartin, N.; O’Farrelly, C.; Ravi, N.; Reynolds, J.V.; Lysaght, J. The microenvironment of visceral adipose tissue and liver alter natural killer cell viability and function. J. Leukoc. Biol. 2016, 100, 1435–1442. [Google Scholar] [CrossRef]
- Shoae-Hassani, A.; Behfar, M.; Mortazavi-Tabatabaei, S.A.; Ai, J.; Mohseni, R.; Hamidieh, A.A. Natural Killer Cells from the Subcutaneous Adipose Tissue Underexpress the NKp30 and NKp44 in Obese Persons and Are Less Active against Major Histocompatibility Complex Class I Non-Expressing Neoplastic Cells. Front. Immunol. 2017, 8, 1486. [Google Scholar] [CrossRef] [PubMed]
- Moulin, C.M.; Rizzo, L.V.; Halpern, A. Effect of surgery-induced weight loss on immune function. Expert Rev. Gastroenterol. Hepatol. 2008, 2, 617–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, K.; Gaens, K.; Bijnen, M.; Verboven, K.; Jocken, J.; Wetzels, S.; Wijnands, E.; Hansen, D.; van Greevenbroek, M.; Duijvestijn, A.; et al. Circulating classical monocytes are associated with CD11c(+) macrophages in human visceral adipose tissue. Sci. Rep. 2017, 7, 42665. [Google Scholar] [CrossRef] [PubMed]
- Tallerico, R.; Conti, L.; Lanzardo, S.; Sottile, R.; Garofalo, C.; Wagner, A.K.; Johansson, M.H.; Cristiani, C.M.; Karre, K.; Carbone, E.; et al. NK cells control breast cancer and related cancer stem cell hematological spread. Oncoimmunology 2017, 6, e1284718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczak, A.M.; Jalilvand, A.; Liu, J.; Wright, V.P.; Suzo, A.; Needleman, B.; Noria, S.; Lafuse, W.; Hsueh, W.A.; Bradley, D. Human Visceral Adipose Tissue Macrophages Are Not Adequately Defined by Standard Methods of Characterization. J. Diabetes Res. 2019, 2019, 8124563. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Huda, S.S.; Jordan, F.; Bray, J.; Love, G.; Payne, R.; Sattar, N.; Freeman, D.J. Visceral adipose tissue activated macrophage content and inflammatory adipokine secretion is higher in pre-eclampsia than in healthy pregnancys. Clin. Sci. 2017, 131, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Kralova Lesna, I.; Kralova, A.; Cejkova, S.; Fronek, J.; Petras, M.; Sekerkova, A.; Thieme, F.; Janousek, L.; Poledne, R. Characterisation and comparison of adipose tissue macrophages from human subcutaneous, visceral and perivascular adipose tissue. J. Trans. Med. 2016, 14, 208. [Google Scholar] [CrossRef]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2. [Google Scholar] [CrossRef]
- Stoll, B.A. Upper abdominal obesity, insulin resistance and breast cancer risk. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; Joosten, L.A.; van Velzen, J.F.; Hijmans, A.; Pol, J.A.; van der Vliet, J.A.; Netea, M.G.; Tack, C.J.; et al. The inflammasome and caspase-1 activation: A new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 2011, 152, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Bar Hen, A.; Bedossa, P.; et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johar, R.; Sharma, R.; Kaur, A.; Mukherjee, T.K. Role of Reactive Oxygen Species in Estrogen Dependant Breast Cancer Complication. Anticancer Agents Med. Chem. 2015, 16, 190–199. [Google Scholar] [CrossRef]
- Madeddu, C.; Gramignano, G.; Floris, C.; Murenu, G.; Sollai, G.; Maccio, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J. Cell. Mol. Med. 2014, 18, 2519–2529. [Google Scholar] [CrossRef]
- Teslow, E.A.; Mitrea, C.; Bao, B.; Mohammad, R.M.; Polin, L.A.; Dyson, G.; Purrington, K.S.; Bollig-Fischer, A. Obesity-induced MBD2_v2 expression promotes tumor-initiating triple-negative breast cancer stem cells. Mol. Oncol. 2019, 13, 894–908. [Google Scholar] [CrossRef]
- Bassols, J.; Ortega, F.J.; Moreno-Navarrete, J.M.; Peral, B.; Ricart, W.; Fernandez-Real, J.M. Study of the proinflammatory role of human differentiated omental adipocytes. J. Cell. Biochem. 2009, 107, 1107–1117. [Google Scholar] [CrossRef]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation and breast cancer metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef]
- Ritter, A.; Friemel, A.; Fornoff, F.; Adjan, M.; Solbach, C.; Yuan, J.; Louwen, F. Characterization of adipose-derived stem cells from subcutaneous and visceral adipose tissues and their function in breast cancer cells. Oncotarget 2015, 6, 34475–34493. [Google Scholar] [CrossRef] [Green Version]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Han, J.; Nam, S.J.; Lee, J.E.; Kim, S. Elevated IL-1β expression induces invasiveness of triple negative breast cancer cells and is suppressed by zerumbone. Chem. Biol. Interact. 2016, 258, 126–133. [Google Scholar] [CrossRef]
- Lin, S.; Gan, Z.; Han, K.; Yao, Y.; Min, D. Interleukin-6 as a prognostic marker for breast cancer: A meta-analysis. Tumori 2015, 101, 535–541. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, N.J. Interleukin-6 in Breast Tumor Microenvironment, Breast Cancer—Focusing Tumor Microenvironment, Stem Cells and Metastasis; InTech: London, UK, 2011. [Google Scholar]
- Harada, A.; Sekido, N.; Akahoshi, T.; Wada, T.; Mukaida, N.; Matsushima, K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J. Leukoc. Biol. 1994, 56, 559–564. [Google Scholar] [CrossRef]
- Todorovic-Rakovic, N.; Milovanovic, J. Interleukin-8 in breast cancer progression. J. Interf. Cytokine Res. 2013, 33, 563–570. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Kokubu, F.; Fujita, J.; Huang, S.K.; Hizawa, N. Role of interleukin-17F in asthma. Inflamm. Allergy Drug Targets 2009, 8, 383–389. [Google Scholar] [CrossRef]
- Welte, T.; Zhang, X.H. Interleukin-17 Could Promote Breast Cancer Progression at Several Stages of the Disease. Mediat. Inflamm. 2015, 2015, 804347. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Yu, H.T.; Chan, S.H.; Lee, C.L.; Wang, H.S.; Soong, Y.K. Eutopic endometrial interleukin-18 system mRNA and protein expression at the level of endometrial-myometrial interface in adenomyosis patients. Fertil. Steril. 2010, 94, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Shi, W.; Deshmukh, R.R.; Long, J.; Cheng, X.; Ji, W.; Zeng, G.; Chen, X.; Zhang, Y.; Dou, Q.P. Tumor necrosis factor-alpha sensitizes breast cancer cells to natural products with proteasome-inhibitory activity leading to apoptosis. PLoS ONE 2014, 9, e113783. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, Y.; Yuan, F.; Huang, J.; Xu, W.; Mao, B.; Yuan, Z.; Bi, W. TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis 2017, 6, e383. [Google Scholar] [CrossRef]
- Sadashiv; Tiwari, S.; Paul, B.N.; Kumar, S.; Chandra, A.; Dhananjai, S.; Negi, M.P. Resistin gene expression in visceral adipose tissue of postmenopausal women and its association with insulin resistance. Womens Health 2012, 8, 521–528. [Google Scholar] [CrossRef]
- Curat, C.A.; Wegner, V.; Sengenes, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumie, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef]
- Yip, I.; Go, V.L.; Hershman, J.M.; Wang, H.J.; Elashoff, R.; DeShields, S.; Liu, Y.; Heber, D. Insulin-leptin-visceral fat relation during weight loss. Pancreas 2001, 23, 197–203. [Google Scholar] [CrossRef]
- Niu, J.; Jiang, L.; Guo, W.; Shao, L.; Liu, Y.; Wang, L. The Association between Leptin Level and Breast Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e67349. [Google Scholar] [CrossRef] [PubMed]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced ER-alpha-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer 2018, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.K.; Choi, D.Y.; Kim, S.H.; Hong, J.T.; Kwon, T.K.; Jeong, J.H.; Park, P.H. Estrogen receptor signaling mediates leptin-induced growth of breast cancer cells via autophagy induction. Oncotarget 2017, 8, 109417–109435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocino-Rodriguez, M.D.; Santillan-Benitez, J.G.; Dozal-Dominguez, D.S.; Hernandez-Navarro, M.D.; Flores-Merino, M.V.; Sandoval-Cabrera, A.; Garcia Vazquez, F.J. Expression of AdipoR1 and AdipoR2 Receptors as Leptin-Breast Cancer Regulation Mechanisms. Dis. Mark. 2017, 2017, 4862016. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, R.R.; Lanier, V.; Newman, G. Leptin’s Pro-Angiogenic Signature in Breast Cancer. Cancers 2013, 5, 1140–1162. [Google Scholar] [CrossRef]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. BCR 2009, 11, R36. [Google Scholar] [CrossRef]
- Pena-Cano, M.I.; Saucedo, R.; Morales-Avila, E.; Valencia, J.; Zavala-Moha, J.A.; Lopez, A. Deregulated microRNAs and Adiponectin in Postmenopausal Women with Breast Cancer. Gynecol. Obstet. Investig. 2019, 84, 369–377. [Google Scholar] [CrossRef]
- Yu, Z.; Tang, S.; Ma, H.; Duan, H.; Zeng, Y. Association of serum adiponectin with breast cancer: A meta-analysis of 27 case-control studies. Medicine 2019, 98, e14359. [Google Scholar] [CrossRef]
- Macías-Gómez, N.M.; Hernández-Terrones, M.C.; Ramírez-Guerrero, A.A.; Leal-Ugarte, E.; Gutiérrez-Angulo, M.; Peregrina-Sandoval, J. ADIPOQ rs2241766 SNP as protective marker against DIBC development in Mexican population. PLoS ONE 2019, 14, e0214080. [Google Scholar] [CrossRef]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Gyorffy, B.; Gabrielson, E.; Saxena, N.K.; et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.; Naimo, G.D.; Gelsomino, L.; Malivindi, R.; Bruno, L.; Pellegrino, M.; Tarallo, R.; Memoli, D.; Weisz, A.; Panno, M.L.; et al. Uncoupling effects of estrogen receptor alpha on LKB1/AMPK interaction upon adiponectin exposure in breast cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 4343–4355. [Google Scholar] [CrossRef]
- Fruhbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Vansaun, M.N. Molecular pathways: Adiponectin and leptin signaling in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1926–1932. [Google Scholar] [CrossRef]
- Nanjappa, V.; Raju, R.; Muthusamy, B.; Sharma, J.; Thomas, J.K.; Nidhina, P.A.H.; Harsha, H.C.; Pandey, A.; Anilkumar, G.; Prasad, T.S.K. A comprehensive curated reaction map of leptin signaling pathway. J. Proteom. Bioinform. 2011, 4, 184–189. [Google Scholar] [CrossRef]
- Van Kruijsdijk, R.C.; van der Wall, E.; Visseren, F.L. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 2009, 18, 2569–2578. [Google Scholar] [CrossRef]
- Nalabolu, M.R.; Palasamudram, K.; Jamil, K. Adiponectin and leptin molecular actions and clinical significance in breast cancer. Int. J. Hematol. Oncol. Stem Cell Res. 2014, 8, 31–40. [Google Scholar]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Jin, S.W.; Kim, Y.A.; Khanal, T.; Lee, G.H.; Kim, S.J.; Rhee, S.D.; Chung, Y.C.; Hwang, Y.J.; Jeong, T.C.; et al. Leptin induces CREB-dependent aromatase activation through COX-2 expression in breast cancer cells. Food Chem. Toxicol. 2017, 106, 232–241. [Google Scholar] [CrossRef]
- Liao, W.; Yu, C.; Wen, J.; Jia, W.; Li, G.; Ke, Y.; Zhao, S.; Campell, W. Adiponectin induces interleukin-6 production and activates STAT3 in adult mouse cardiac fibroblasts. Biol. Cell 2009, 101, 263–272. [Google Scholar] [CrossRef]
- Magoffin, D.A.; Weitsman, S.R.; Aagarwal, S.K.; Jakimiuk, A.J. Leptin regulation of aromatase activity in adipose stromal cells from regularly cycling women. Ginekol. Pol. 1999, 70, 1–7. [Google Scholar] [PubMed]
- Catalano, S.; Marsico, S.; Giordano, C.; Mauro, L.; Rizza, P.; Panno, M.L.; Ando, S. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J. Biol. Chem. 2003, 278, 28668–28676. [Google Scholar] [CrossRef]
- Tekmal, R.R.; Kirma, N.; Gill, K.; Fowler, K. Aromatase overexpression and breast hyperplasia, an in vivo model—Continued overexpression of aromatase is sufficient to maintain hyperplasia without circulating estrogens, and aromatase inhibitors abrogate these preneoplastic changes in mammary glands. Endocr. Relat. Cancer 1999, 6, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.P.; Grossmann, M.E. Minireview: Obesity and breast cancer: The estrogen connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Hetemaki, N.; Savolainen-Peltonen, H.; Tikkanen, M.J.; Wang, F.; Paatela, H.; Hamalainen, E.; Turpeinen, U.; Haanpaa, M.; Vihma, V.; Mikkola, T.S. Estrogen Metabolism in Abdominal Subcutaneous and Visceral Adipose Tissue in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2017, 102, 4588–4595. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, A.I.; Catalan, V.; Gomez-Ambrosi, J.; Rodriguez, A.; Fruhbeck, G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front. Endocrinol. 2014, 5, 65. [Google Scholar] [CrossRef]
- Liang, J.; Shang, Y. Estrogen and cancer. Annu. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef]
- Gorrini, C.; Gang, B.P.; Bassi, C.; Wakeham, A.; Baniasadi, S.P.; Hao, Z.; Li, W.Y.; Cescon, D.W.; Li, Y.T.; Molyneux, S.; et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 4472–4477. [Google Scholar] [CrossRef]
- Jeffery, E.; Wing, A.; Holtrup, B.; Sebo, Z.; Kaplan, J.L.; Saavedra-Pena, R.; Church, C.D.; Colman, L.; Berry, R.; Rodeheffer, M.S. The Adipose Tissue Microenvironment Regulates Depot-Specific Adipogenesis in Obesity. Cell Metab. 2016, 24, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Kaaks, R.; Rinaldi, S.; Key, T.J.; Berrino, F.; Peeters, P.H.; Biessy, C.; Dossus, L.; Lukanova, A.; Bingham, S.; Khaw, K.T.; et al. Postmenopausal serum androgens, oestrogens and breast cancer risk: The European prospective investigation into cancer and nutrition. Endocr. Relat. Cancer 2005, 12, 1071–1082. [Google Scholar] [CrossRef]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.; Dorgan, J.F.; Longcope, C.; Stanczyk, F.Z.; Stephenson, H.E., Jr.; Falk, R.T.; Miller, R.; et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl. Cancer Instig. 2003, 95, 1218–1226. [Google Scholar]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. New Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, B.J.; Schairer, C.; Gail, M.H.; Boyd-Morin, J.; Xu, X.; Sue, L.Y.; Buys, S.S.; Isaacs, C.; Keefer, L.K.; Veenstra, T.D.; et al. Estrogen metabolism and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2012, 104, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Frasor, J.; Komm, B.; Chang, K.C.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen-regulated gene networks in human breast cancer cells: Involvement of E2F1 in the regulation of cell proliferation. Mol. Endocrinol. 2007, 21, 2112–2123. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Seeger, H.; Wallwiener, D.; Huober, J.; Mueck, A.O. The ratio of the estradiol metabolites 2-hydroxyestrone (2-OHE1) and 16alpha-hydroxyestrone (16-OHE1) may predict breast cancer risk in postmenopausal but not in premenopausal women: Two case-control studies. Arch. Gynecol. Obstet. 2015, 291, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Mani, C.; Somasagara, R.R.; Clark, D.W.; Ananthapur, V.; Vinaya, K.; Palle, K. Detection and evaluation of estrogen DNA-adducts and their carcinogenic effects in cultured human cells using biotinylated estradiol. Mol. Carcinogen. 2017, 56, 1010–1020. [Google Scholar] [CrossRef]
- Bradlow, H.L.; Telang, N.T.; Sepkovic, D.W.; Osborne, M.P. 2-hydroxyestrone: The ‘good’ estrogen. J. Endocrinol. 1996, 150 (Suppl. S3), S259–S265. [Google Scholar]
- Obi, N.; Vrieling, A.; Heinz, J.; Chang-Claude, J. Estrogen metabolite ratio: Is the 2-hydroxyestrone to 16alpha-hydroxyestrone ratio predictive for breast cancer? Int. J. Womens Health 2011, 3, 37–51. [Google Scholar] [CrossRef]
- Yue, W.; Yager, J.D.; Wang, J.P.; Jupe, E.R.; Santen, R.J. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids 2013, 78, 161–170. [Google Scholar] [CrossRef]
- Berindan-Neagoe, I.; Calin, G.A. Molecular pathways: MicroRNAs, cancer cells, and microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6247–6253. [Google Scholar] [CrossRef]
- Ferrante, S.C.; Nadler, E.P.; Pillai, D.K.; Hubal, M.J.; Wang, Z.; Wang, J.M.; Gordish-Dressman, H.; Koeck, E.; Sevilla, S.; Wiles, A.A.; et al. Adipocyte-derived exosomal miRNAs: A novel mechanism for obesity-related disease. Pediatric Res. 2015, 77, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, R.; Rivenbark, A.G.; Coleman, W.B. Loss of post-transcriptional regulation of DNMT3b by microRNAs: A possible molecular mechanism for the hypermethylation defect observed in a subset of breast cancer cell lines. Int. J. Oncol. 2012, 41, 721–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Cimino, D.; De Pitta, C.; Orso, F.; Zampini, M.; Casara, S.; Penna, E.; Quaglino, E.; Forni, M.; Damasco, C.; Pinatel, E.; et al. miR148b is a major coordinator of breast cancer progression in a relapse-associated microRNA signature by targeting ITGA5, ROCK1, PIK3CA, NRAS, and CSF1. FASEB J. Off. Publ. Fed. of Am. Soc. Exp. Biol. 2013, 27, 1223–1235. [Google Scholar] [CrossRef]
- Orso, F.; Quirico, L.; Virga, F.; Penna, E.; Dettori, D.; Cimino, D.; Coppo, R.; Grassi, E.; Elia, A.R.; Brusa, D.; et al. miR-214 and miR-148b Targeting Inhibits Dissemination of Melanoma and Breast Cancer. Cancer Res. 2016, 76, 5151–5162. [Google Scholar] [CrossRef]
- Lai, Y.; Chen, Y.; Lin, Y.; Ye, L. Down-regulation of LncRNA CCAT1 enhances radiosensitivity via regulating miR-148b in breast cancer. Cell. Biol. Int. 2018, 42, 227–236. [Google Scholar] [CrossRef]
- Cuk, K.; Zucknick, M.; Madhavan, D.; Schott, S.; Golatta, M.; Heil, J.; Marme, F.; Turchinovich, A.; Sinn, P.; Sohn, C.; et al. Plasma microRNA panel for minimally invasive detection of breast cancer. PLoS ONE 2013, 8, e76729. [Google Scholar] [CrossRef]
- Cuk, K.; Zucknick, M.; Heil, J.; Madhavan, D.; Schott, S.; Turchinovich, A.; Arlt, D.; Rath, M.; Sohn, C.; Benner, A.; et al. Circulating microRNAs in plasma as early detection markers for breast cancer. Int. J. Cancer 2013, 132, 1602–1612. [Google Scholar] [CrossRef]
- Shen, J.; Hu, Q.; Schrauder, M.; Yan, L.; Wang, D.; Medico, L.; Guo, Y.; Yao, S.; Zhu, Q.; Liu, B.; et al. Circulating miR-148b and miR-133a as biomarkers for breast cancer detection. Oncotarget 2014, 5, 5284–5294. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Jia, S.; Zhang, W.H.; Zhang, Y.; Kang, Y.; Li, P.S. Differential distribution of microRNAs in breast cancer grouped by clinicopathological subtypes. Asian Pac. J. Cancer Prev. 2013, 14, 3197–3203. [Google Scholar] [CrossRef]
- Do Canto, L.M.; Marian, C.; Willey, S.; Sidawy, M.; Da Cunha, P.A.; Rone, J.D.; Li, X.; Gusev, Y.; Haddad, B.R. MicroRNA analysis of breast ductal fluid in breast cancer patients. Int. J. Oncol. 2016, 48, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.J.; Muluhngwi, P.; Alizadeh-Rad, N.; Green, M.A.; Rouchka, E.C.; Waigel, S.J.; Klinge, C.M. Genome-wide miRNA response to anacardic acid in breast cancer cells. PLoS ONE 2017, 12, e0184471. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Adamczyk-Grochala, J.; Deregowska, A.; Wnuk, M. Sulforaphane-Induced Cell Cycle Arrest and Senescence are accompanied by DNA Hypomethylation and Changes in microRNA Profile in Breast Cancer Cells. Theranostics 2017, 7, 3461–3477. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, L.; Stebbing, J.; Braga, V.M.; Frampton, A.E.; Jacob, J.; Buluwela, L.; Jiao, L.R.; Periyasamy, M.; Madsen, C.D.; Caley, M.P.; et al. miR-23b regulates cytoskeletal remodeling, motility and metastasis by directly targeting multiple transcripts. Nucl. Acids Res. 2013, 41, 5400–5412. [Google Scholar] [CrossRef]
- Ell, B.; Qiu, Q.; Wei, Y.; Mercatali, L.; Ibrahim, T.; Amadori, D.; Kang, Y. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 2014, 289, 21888–21895. [Google Scholar] [CrossRef]
- Adi, N.; Adi, J.; Cesar, L.; Kurlansky, P.; Agatston, A.; Webster, K.A. Role of Micro RNA-205 in Promoting Visceral Adiposity of NZ10 Mice with Polygenic Susceptibility for Type 2 Diabetes. J. Diabetes Metab. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef]
- Tsouko, E.; Wang, J.; Frigo, D.E.; Aydogdu, E.; Williams, C. miR-200a inhibits migration of triple-negative breast cancer cells through direct repression of the EPHA2 oncogene. Carcinogenesis 2015, 36, 1051–1060. [Google Scholar] [CrossRef]
- Yao, J.; Xu, F.; Zhang, D.; Yi, W.; Chen, X.; Chen, G.; Zhou, E. TP73-AS1 promotes breast cancer cell proliferation through miR-200a-mediated TFAM inhibition. J. Cell. Biochem. 2018, 119, 680–690. [Google Scholar] [CrossRef]
- Li, P.; Xu, T.; Zhou, X.; Liao, L.; Pang, G.; Luo, W.; Han, L.; Zhang, J.; Luo, X.; Xie, X.; et al. Downregulation of miRNA-141 in breast cancer cells is associated with cell migration and invasion: Involvement of ANP32E targeting. Cancer Med. 2017, 6, 662–672. [Google Scholar] [CrossRef]
- Abedi, N.; Mohammadi-Yeganeh, S.; Koochaki, A.; Karami, F.; Paryan, M. miR-141 as potential suppressor of beta-catenin in breast cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 9895–9901. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.B.; Ma, G.; Zhao, Y.H.; Xiao, Y.; Zhan, Y.; Jing, C.; Gao, K.; Liu, Z.H.; Yu, S.J. miR-429 inhibits migration and invasion of breast cancer cells in vitro. Int. J. Oncol. 2015, 46, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cospons. Am. Soc. Prev. Oncol. 2012, 21, 1236–1243. [Google Scholar] [CrossRef]
- Zhang, C.M.; Zhao, J.; Deng, H.Y. MiR-155 promotes proliferation of human breast cancer MCF-7 cells through targeting tumor protein 53-induced nuclear protein 1. J. Biomed. Sci. 2013, 20, 79. [Google Scholar] [CrossRef]
- Cojocneanu Petric, R.; Braicu, C.; Raduly, L.; Zanoaga, O.; Dragos, N.; Monroig, P.; Dumitrascu, D.; Berindan-Neagoe, I. Phytochemicals modulate carcinogenic signaling pathways in breast and hormone-related cancers. OncoTargets Ther. 2015, 8, 2053–2066. [Google Scholar] [CrossRef]
- Braicu, C.; Mehterov, N.; Vladimirov, B.; Sarafian, V.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. Nutrigenomics in cancer: Revisiting the effects of natural compounds. Semin. Cancer Biol. 2017, 46, 84–106. [Google Scholar] [CrossRef]
- Zhou, W.; Shi, G.; Zhang, Q.; Wu, Q.; Li, B.; Zhang, Z. MicroRNA-20b promotes cell growth of breast cancer cells partly via targeting phosphatase and tensin homologue (PTEN). Cell Biosci. 2014, 4, 62. [Google Scholar] [CrossRef]
- Gentile, A.M.; Lhamyani, S.; Coin-Araguez, L.; Clemente-Postigo, M.; Oliva Olivera, W.; Romero-Zerbo, S.Y.; Garcia-Serrano, S.; Garcia-Escobar, E.; Zayed, H.; Doblado, E.; et al. miR-20b, miR-296, and Let-7f Expression in Human Adipose Tissue is Related to Obesity and Type 2 Diabetes. Obesity 2019, 27, 245–254. [Google Scholar] [CrossRef]
- Dinami, R.; Buemi, V.; Sestito, R.; Zappone, A.; Ciani, Y.; Mano, M.; Petti, E.; Sacconi, A.; Blandino, G.; Giacca, M.; et al. Epigenetic silencing of miR-296 and miR-512 ensures hTERT dependent apoptosis protection and telomere maintenance in basal-type breast cancer cells. Oncotarget 2017, 8, 95674–95691. [Google Scholar] [CrossRef]
- Tao, W.-Y.; Liang, X.-S.; Liu, Y.; Wang, C.-Y.; Pang, D. Decrease of let-7f in low-dose metronomic Paclitaxel chemotherapy contributed to upregulation of thrombospondin-1 in breast cancer. Int. J. Biol. Sci. 2015, 11, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Robado de Lope, L.; Alcíbar, O.L.; Amor López, A.; Hergueta-Redondo, M.; Peinado, H. Tumour-adipose tissue crosstalk: Fuelling tumour metastasis by extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160485. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Guo, X. The clinical potential of circulating microRNAs in obesity. Nat. Rev. Endocrinol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Groza, M.; Zimta, A.A.; Irimie, A.; Achimas-Cadariu, P.; Cenariu, D.; Stanta, G.; Berindan-Neagoe, I. Recent advancements in the study of breast cancer exosomes as mediators of intratumoral communication. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gernapudi, R.; Yao, Y.; Zhang, Y.; Wolfson, B.; Roy, S.; Duru, N.; Eades, G.; Yang, P.; Zhou, Q. Targeting exosomes from preadipocytes inhibits preadipocyte to cancer stem cell signaling in early-stage breast cancer. Breast Cancer Res. Treat. 2015, 150, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Su, X.; Xu, M.; Xiao, X.; Li, X.; Li, H.; Keating, A.; Zhao, R. Exosomes secreted by mesenchymal stromal/stem cell-derived adipocytes promote breast cancer cell growth via activation of Hippo signaling pathway. Stem Cell Res. Ther. 2019, 10. [Google Scholar] [CrossRef]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Wang, C.; et al. Breast cancer-released exosomes trigger cancer-associated cachexia to promote tumor progression. Adipocyte 2019, 8, 31–45. [Google Scholar] [CrossRef]
- Meerson, A.; Eliraz, Y.; Yehuda, H.; Knight, B.; Crundwell, M.; Ferguson, D.; Lee, B.P.; Harries, L.W. Obesity impacts the regulation of miR-10b and its targets in primary breast tumors. BMC Cancer 2019, 19, 86. [Google Scholar] [CrossRef]
- Lee, J.; Suk Ryu, H.; Sil Hong, B.; Lee, H.-B.; Lee, M.; Ae Park, I.; Kim, J.; Han, W.; Noh, D.-Y.; Moon, H.-G. Transition into inflammatory cancer-associated adipocytes in breast cancer microenvironment requires microRNA regulatory mechanism. PLoS ONE 2017, 12, e0174126. [Google Scholar] [CrossRef] [PubMed]
- Picon-Ruiz, M.; Pan, C.; Drews-Elger, K.; Jang, K.; Besser, A.H.; Zhao, D.; Morata-Tarifa, C.; Kim, M.; Ince, T.A.; Azzam, D.J.; et al. Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b–Mediated Malignant Progression. Cancer Res. 2016, 76, 491–504. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cytokine | Source of Secretion | Immune Significance | Role in BC | Ref. |
|---|---|---|---|---|
| IL-1β | Myeloid cells, Macrophages, T cells | - stimulates secretion of other pro-inflammatory mediators | - leads to poor prognostic - promotes tumor growth, and metastasis - induces invasiveness in TNBC (triple negative BC) | [54,55,56] |
| IL-6 | T cells, Macrophages | - binds to IL6sR (soluble receptor) and stimulates STAT3 activation in cells that express gp130 - enhances inflammation - fights against infections | - leads to poor prognostic - increased levels in advanced breast tumor stages | [57,58] |
| IL-8 | Macrophages, Epithelial cells, Smooth muscle cells, Endothelial cells | - has increased levels in inflammation - plays a key role in neutrophil degranulation | - increases metastatic potential of ER-, and ER+ BC cells - highly expressed in ER- BC - induces BC progression by stimulating invasion/metastasis | [59,60] |
| IL-17 | Th17 | - leads to inflammatory diseases such as Rheumatoid arthritis. - induces allergic responses - enhances inflammation by recruiting other proinflammatory cytokines | - induces overexpression of VEGF - promotes tumor cells survival - inhibits the antitumor immune response | [61,62] |
| IL-18 | Macrophages | - together with IL-12, induces cell mediated immunity | - inhibits BC cell proliferation - induces inflammatory reactions - enhances BC migration - poor prognostic in TNBC | [56,63,64,65] |
| TNFα | Macrophages, NK cells, Neutrophils, Mast cells, Eosinophils | - relevant in inflammatory setup in BC cells - key proinflammatory cytokine | - enhances inflammatory process -mediates NFκB signaling pathway - regulates the immune cells - induces fever, and apoptosis - promotes BC migration | [66,67,68] |
| Name | Expression in Obese VAT | Expression in BC | Target Gene(s) | Biological Effect in BC | Ref. |
|---|---|---|---|---|---|
| miR-148b | ↓ | ↓ | Dnmt3b, Itga5, Alcam | proliferation, malignant transformation, pro-apoptosis, radio sensitivity, invasion, migration | [122,123,124,125,126] |
| miR-23 | ↑ | ↑ | Par2 | invasion, lung metastasis | [122,133,134,135,136] |
| miR-200 | ↑ | ↑ | E-Cadherin EPHA2 | invasion, migration, proliferation | [137,138,139,140] |
| miR-141 | ↑ | ↓ | [137,141,142] | ||
| miR-429 | ↑ | ↓ | [137,143] | ||
| miR-155 | ↑ | ↑ | Smad1/5, Casp3, Fadd, Rip1, Irak, Pka, Apaf-1, Foxo3a, Bcl-2, Socs1, Smad2, Hif, Pu.1,Mcsf, Tp53inp1 | Invasion, apoptosis, proliferation, angiogenesis, differentiation | [144,145,146] |
| miR-20b | ↑ | ↑ | Pten* | Cell growth and proliferation | [149,150] |
| miR-296 | ↑ | ↓ | hTERT | Shorter telomeres, induce apoptosis | [150,151] |
| let-7f | ↑ | ↑ | THBS1 | No effect on apoptosis or proliferation | [150,152] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimta, A.-A.; Tigu, A.B.; Muntean, M.; Cenariu, D.; Slaby, O.; Berindan-Neagoe, I. Molecular Links between Central Obesity and Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215364
Zimta A-A, Tigu AB, Muntean M, Cenariu D, Slaby O, Berindan-Neagoe I. Molecular Links between Central Obesity and Breast Cancer. International Journal of Molecular Sciences. 2019; 20(21):5364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215364
Chicago/Turabian StyleZimta, Alina-Andreea, Adrian Bogdan Tigu, Maximilian Muntean, Diana Cenariu, Ondrej Slaby, and Ioana Berindan-Neagoe. 2019. "Molecular Links between Central Obesity and Breast Cancer" International Journal of Molecular Sciences 20, no. 21: 5364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215364