Generation of Mouse Parthenogenetic Epiblast Stem Cells and Their Imprinting Patterns

 , and
, and 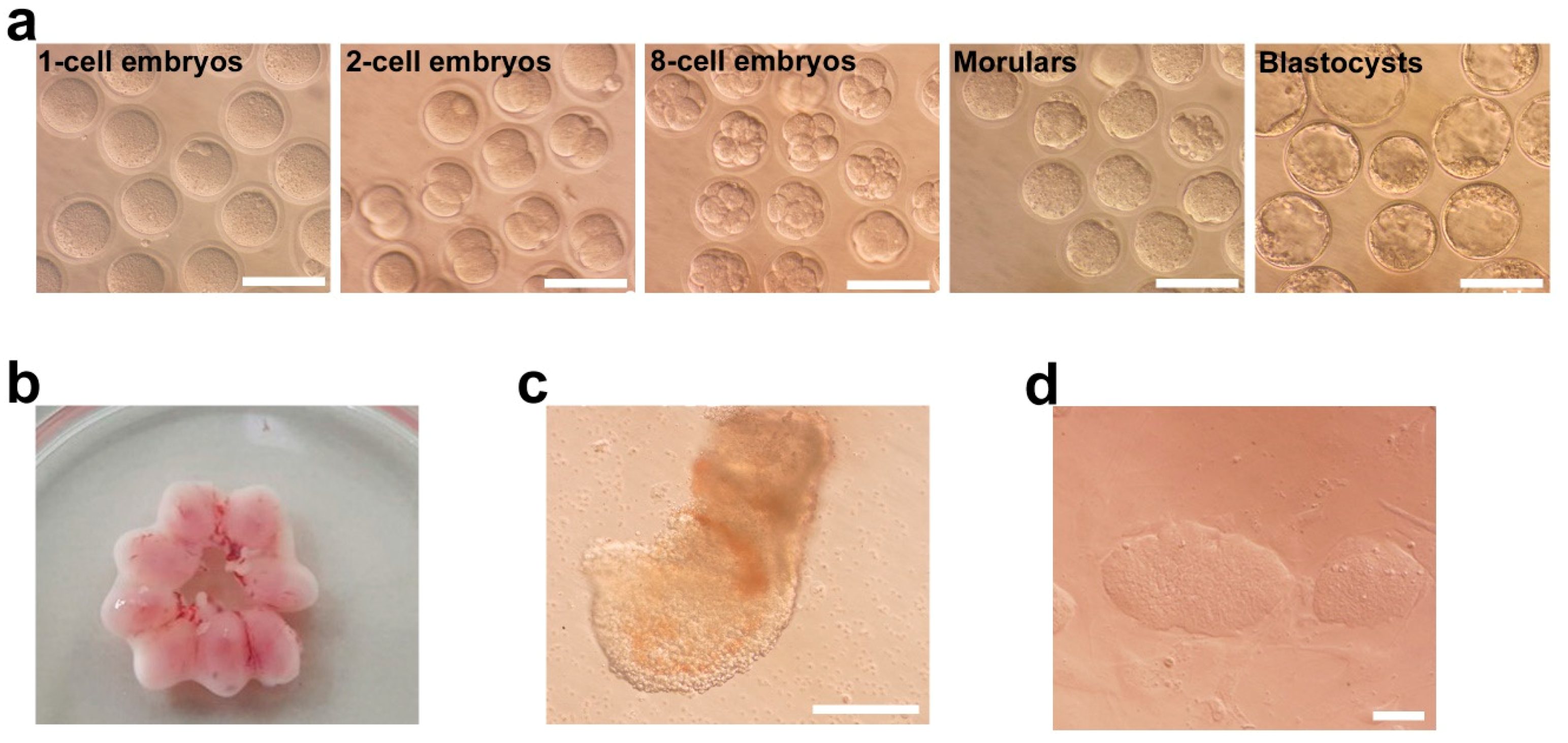{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Derivation of Parthenogenetic EpiSCs
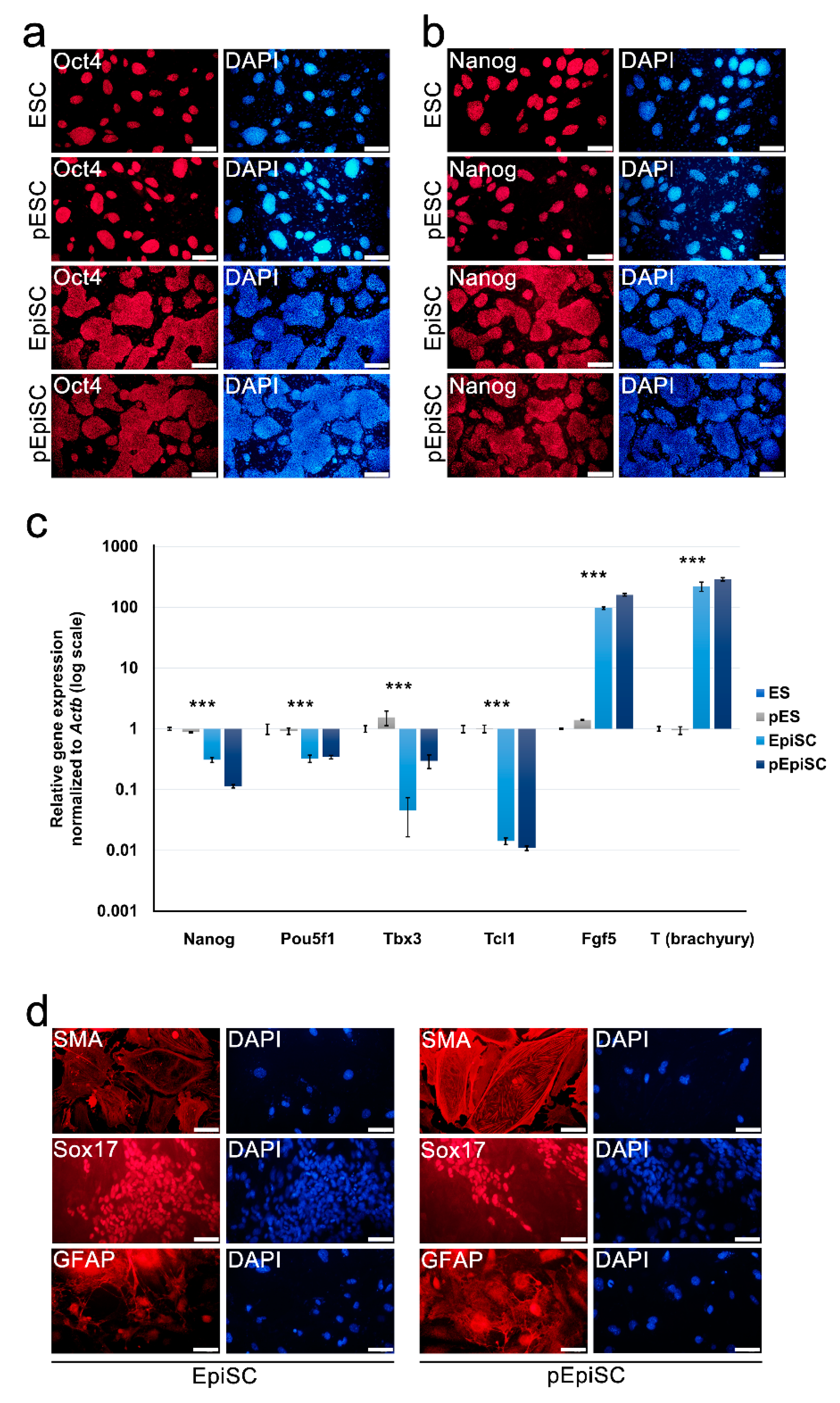
2.2. pEpiSCs Maintained Primed Pluripotency and Differentiation Potential
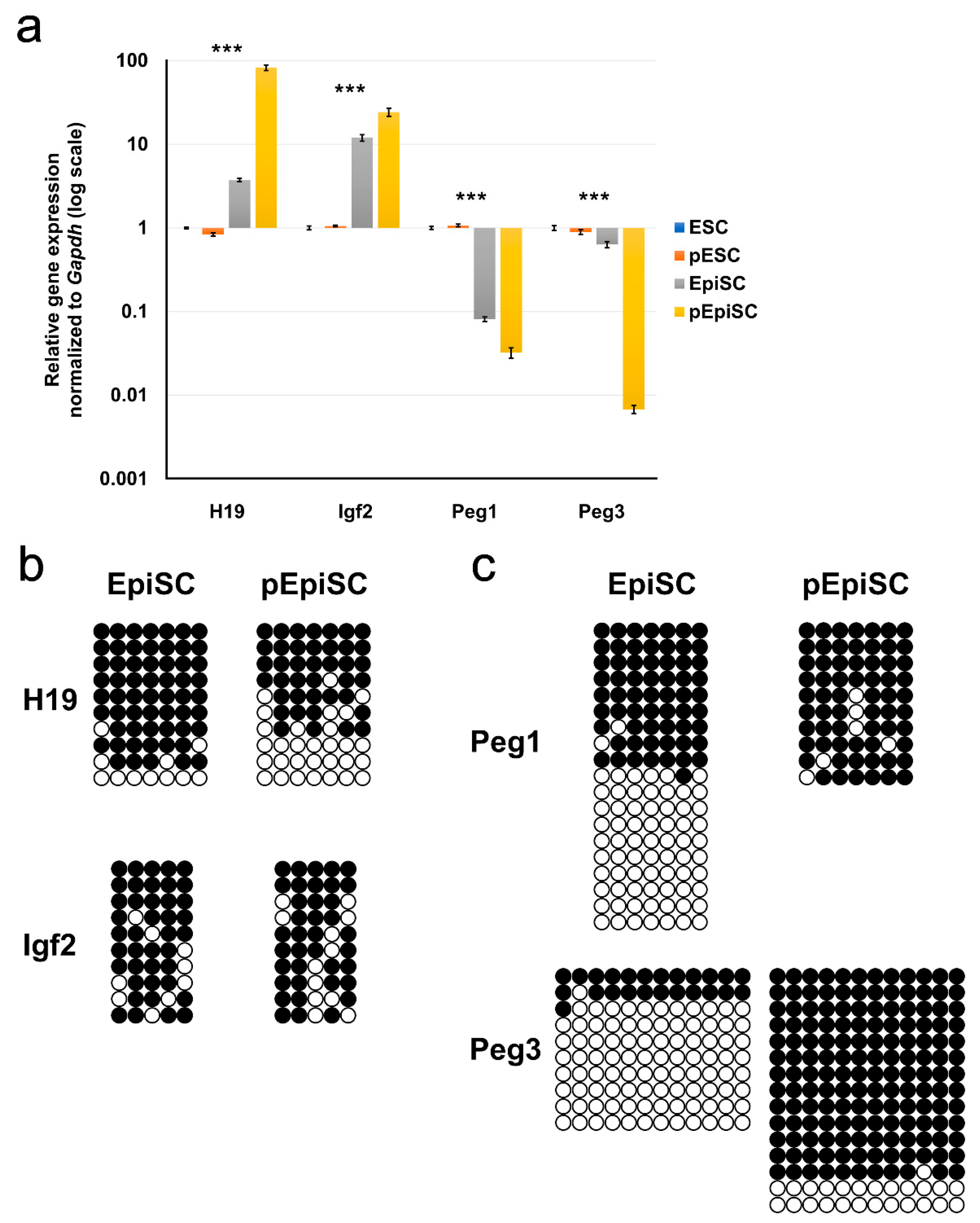
2.3. Expression and Methylation Patterns of Imprinted Genes in pEpiSCs
3. Discussion
4. Materials and Methods
4.1. Generation of Parthenogenetic Epiblast Stem Cells
4.2. Cell Culture
4.3. Immunocytochemistry
4.4. RNA Isolation and Real-Time PCR Analysis
4.5. Bisulfite Genome Sequencing
4.6. Statistical Analysis
4.7. Ethical Statement for Animal Use
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C. Development of gynogenetic eggs in the mouse: Implications for parthenogenetic embryos. Science 1983, 222, 1034–1036. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565. [Google Scholar] [CrossRef] [PubMed]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the Prader–Willi/Angelman region. Am. J. Med Genet. Part. A 2008, 146, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol. 2007, 211, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Dean, W.; Brown, D.; Reik, W.; Feil, R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol. Reprod. 2001, 64, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef]
- Smith, A.G. Embryo-derived stem cells: Of mice and men. Annu. Rev. Cell Dev. Biol. 2001, 17, 435–462. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Nichols, J.; Smith, A. Naive and primed pluripotent states. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; de Sousa Lopes, S.M.C.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191. [Google Scholar] [CrossRef]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Kaufman-Francis, K.; Studdert, J.B.; Steiner, K.A.; Power, M.D.; Loebel, D.A.; Jones, V.; Hor, A.; de Alencastro, G.; Logan, G.J.; et al. The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell 2014, 14, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; Tong, L.M.; Balestra, M.E.; Javier, R.; Andrews-Zwilling, Y.; Li, G.; Walker, D.; Zhang, W.R.; Kreitzer, A.C.; Huang, Y. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 2012, 11, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Galonska, C.; Ziller, M.J.; Karnik, R.; Meissner, A. Ground state conditions induce rapid reorganization of core pluripotency factor binding before global epigenetic reprogramming. Cell Stem Cell 2015, 17, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Joo, J.Y.; Hong, Y.J.; Kim, J.S.; Song, H.; Lee, J.W.; Wu, G.; Schöler, H.R.; Do, J.T. Distinct enhancer activity of Oct4 in naive and primed mouse pluripotency. Stem Cell Rep. 2016, 7, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.; Yoon, S.; Do, J. Mitochondrial dynamics in stem cells and differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef]
- Humpherys, D.; Eggan, K.; Akutsu, H.; Hochedlinger, K.; Rideout, W.M.; Biniszkiewicz, D.; Yanagimachi, R.; Jaenisch, R. Epigenetic instability in ES cells and cloned mice. Science 2001, 293, 95–97. [Google Scholar] [CrossRef]
- Heo, J.; Lim, J.; Lee, S.; Jeong, J.; Kang, H.; Kim, Y.; Kang, J.W.; Yu, H.Y.; Jeong, E.M.; Kim, K. Sirt1 regulates DNA methylation and differentiation potential of embryonic stem cells by antagonizing Dnmt3l. Cell Rep. 2017, 18, 1930–1945. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Stel, J.M.; Shioda, K.; Qu, N.; Odajima, J.; Mitsunaga, S.; Zhang, X.; Nagano, M.; Hochedlinger, K.; Isselbacher, K.J.; et al. Erasure of DNA methylation, genomic imprints, and epimutations in a primordial germ-cell model derived from mouse pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9545–9550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenisch, R.; Young, R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008, 132, 567–582. [Google Scholar] [CrossRef]
- Do, J.T.; Schöler, H.R. Regulatory circuits underlying pluripotency and reprogramming. Trends Pharmacol. Sci. 2009, 30, 296–302. [Google Scholar] [CrossRef]
- Jang, H.S.; Hong, Y.J.; Choi, H.W.; Song, H.; Byun, S.J.; Uhm, S.J.; Seo, H.G.; Do, J.T. Changes in parthenogenetic imprinting patterns during reprogramming by cell fusion. PLoS ONE 2016, 11, e0156491. [Google Scholar] [CrossRef]
- Surani, M.A.; Kothary, R.; Allen, N.D.; Singh, P.B.; Fundele, R.; Ferguson-Smith, A.C.; Barton, S.C. Genome imprinting and development in the mouse. Development 1990, 108, 89–98. [Google Scholar]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415. [Google Scholar] [CrossRef]
- Surani, M.; Barton, S.C.; Norris, M. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548. [Google Scholar] [CrossRef]
- Wu, J.; Okamura, D.; Li, M.; Suzuki, K.; Luo, C.; Ma, L.; He, Y.; Li, Z.; Benner, C.; Tamura, I.; et al. An alternative pluripotent state confers interspecies chimaeric competency. Nature 2015, 521, 316. [Google Scholar] [CrossRef]
- Kim, M.J.; Choi, H.W.; Jang, H.J.; Chung, H.M.; Arauzo-Bravo, M.J.; Schöler, H.R.; Do, J.T. Conversion of genomic imprinting by reprogramming and redifferentiation. J. Cell Sci. 2013, 126, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Kobayashi, S.; Hiratani, I. Epigenetic differences between naïve and primed pluripotent stem cells. Cell. Mol. Life Sci. 2018, 75, 1191–1203. [Google Scholar] [CrossRef]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, H.W.; Araúzo-Bravo, M.J.; Schöler, H.R.; Do, J.T. Reactivation of the inactive X chromosome and post-transcriptional reprogramming of Xist in iPSCs. J. Cell Sci. 2015, 128, 81–87. [Google Scholar] [CrossRef]
- Choi, H.W.; Kim, J.S.; Jang, H.J.; Choi, S.; Kim, J.-H.; Schöler, H.R.; Do, J.T. Reestablishment of the inactive X chromosome to the ground state through cell fusion-induced reprogramming. Cell. Mol. Life Sci. 2012, 69, 4067–4077. [Google Scholar] [CrossRef]
- Guo, G.; von Meyenn, F.; Rostovskaya, M.; Clarke, J.; Dietmann, S.; Baker, D.; Sahakyan, A.; Myers, S.; Bertone, P.; Reik, W.; et al. Epigenetic resetting of human pluripotency. Development 2017, 144, 2748–2763. [Google Scholar] [CrossRef] [Green Version]
- Gu, K.-L.; Zhang, Q.; Yan, Y.; Li, T.-T.; Duan, F.-F.; Hao, J.; Wang, X.-W.; Shi, M.; Wu, D.-R.; Guo, W.-T.; et al. Pluripotency-associated miR-290/302 family of microRNAs promote the dismantling of naive pluripotency. Cell Res. 2016, 26, 350. [Google Scholar] [CrossRef]
- Pastor, W.A.; Chen, D.; Liu, W.; Kim, R.; Sahakyan, A.; Lukianchikov, A.; Plath, K.; Jacobsen, S.E.; Clark, A.T. Naive human pluripotent cells feature a methylation landscape devoid of blastocyst or germline memory. Cell Stem Cell 2016, 18, 323–329. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, B.J.; Jang, H.S.; Song, H.; Park, C.; Hong, K.; Lee, J.W.; Do, J.T. Generation of Mouse Parthenogenetic Epiblast Stem Cells and Their Imprinting Patterns. Int. J. Mol. Sci. 2019, 20, 5428. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215428
Seo BJ, Jang HS, Song H, Park C, Hong K, Lee JW, Do JT. Generation of Mouse Parthenogenetic Epiblast Stem Cells and Their Imprinting Patterns. International Journal of Molecular Sciences. 2019; 20(21):5428. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215428
Chicago/Turabian StyleSeo, Bong Jong, Hyun Sik Jang, Hyuk Song, Chankyu Park, Kwonho Hong, Jeong Woong Lee, and Jeong Tae Do. 2019. "Generation of Mouse Parthenogenetic Epiblast Stem Cells and Their Imprinting Patterns" International Journal of Molecular Sciences 20, no. 21: 5428. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215428