Candidate Domestication-Related Genes Revealed by Expression Quantitative Trait Loci Mapping of Narrow-Leafed Lupin (Lupinus angustifolius L.)

 , ,
, ,
Abstract
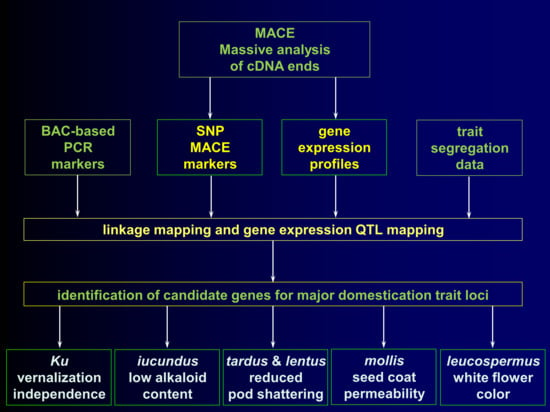
:
1. Introduction
2. Results and Discussion
2.1. Development of New Polymorphic Markers
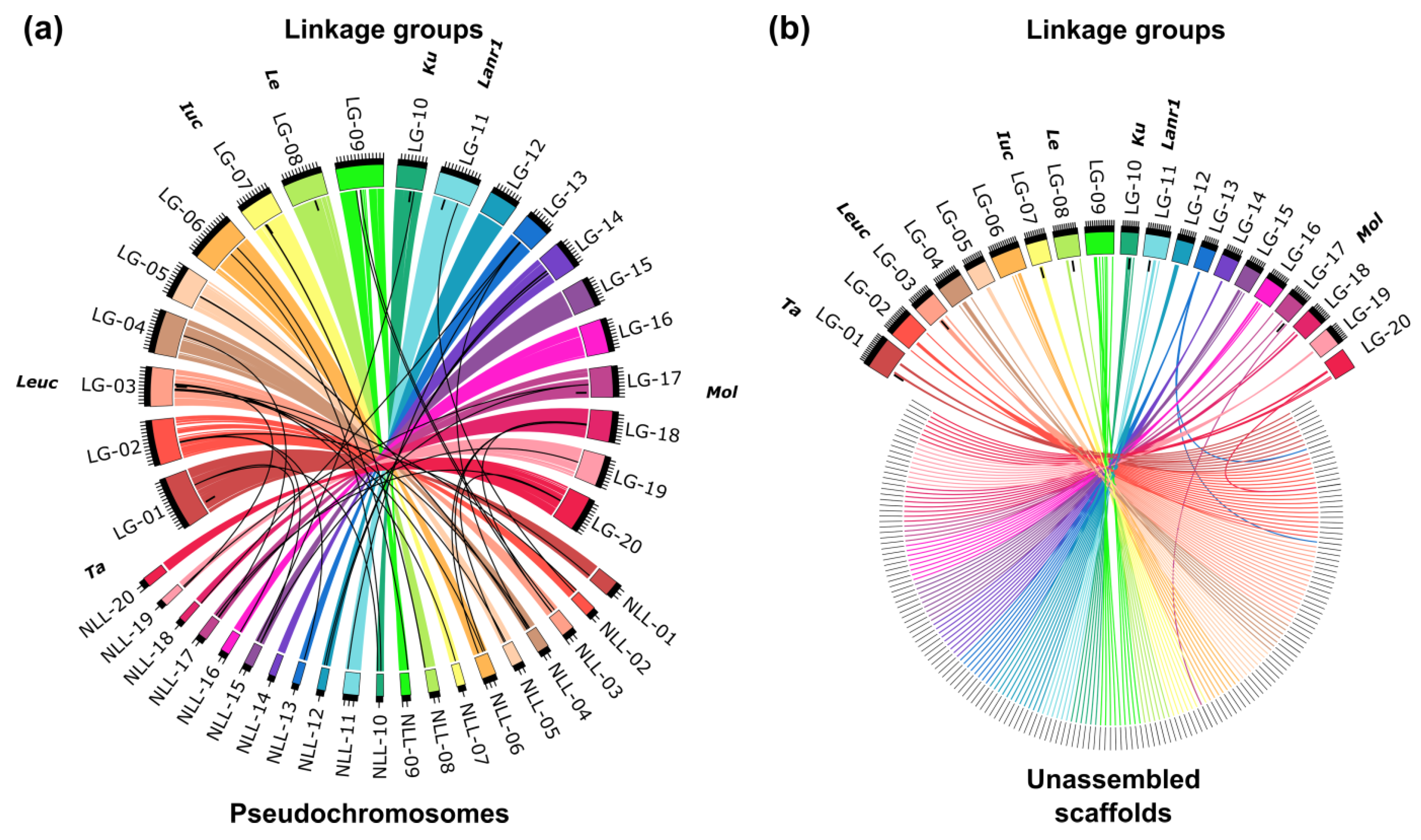
2.2. Construction of a Linkage Map
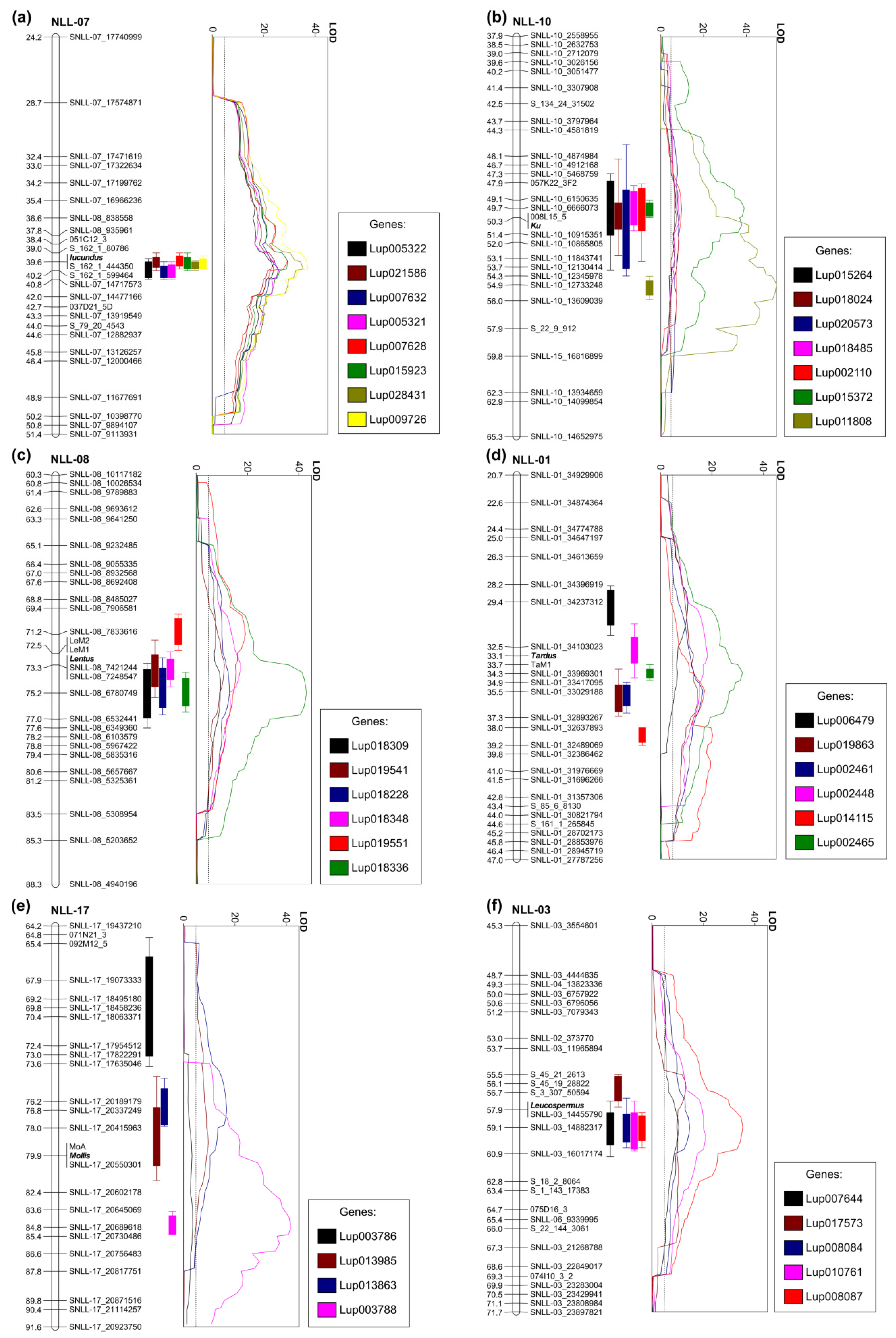
2.3. Gene Expression Profiling, Gene Ontology Enrichment, and Expression Quantitative Trait Loci Mapping
2.4. Genes Identified for Low-Alkaloid Iucundus Locus
2.5. Genes Revealed for Vernalization Independence Ku Locus
2.6. Genes Profiled for Lentus and Tardus Pod Shattering Loci
2.7. Gene Related to the Soft Seededness Mollis Allele
2.8. Genes with eQTL Loci Matching White Flower Color Leucospermus Allele
2.9. Applicability of MACE for Gene-Based Studies
2.10. Recommendations for Improving Narrow-Leafed Lupin As a Crop
3. Materials and Methods
3.1. Plant Material
3.2. Controlled Environment Experiment
3.3. Massive Analysis of cDNA Ends
3.4. Molecular Markers and Linkage Mapping
3.5. Expression Quantitative Trait Loci Mapping
4. Conclusions
- The massive analysis of cDNA ends was revealed to be applicable for molecular marker development and linkage map construction, as well as for gene expression evaluation and expression quantitative trait loci mapping.
- The analysis of vernalization independence Ku locus shed light on vernalization response via FLOWERING LOCUS T and FD regulon, providing transcriptomic evidence for contribution of several genes acting in C-repeat binding factor (CBF) cold responsiveness and in UDP-glycosyltransferases pathways. This information can be relevant to decipher vernalization pathway in legumes, because legume genomes do not contain a major vernalization-responsive gene FLOWERING LOCUS C (FLC) but other genes from this pathway, including activators and repressors of FLC, are present.
- The study of low-alkaloid iucundus locus highlighted a high number of cis- and trans-regulated alkaloid biosynthesis genes with gene expression orchestrated by a regulatory agent localized at iucundus locus, supporting the concept that the ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7 gene may control low-alkaloid phenotype in narrow-leafed lupin.
- Research on reduced pod shattering lentus locus selected a DUF1218 domain homolog as a candidate gene controlling the orientation of the sclerified endocarp and a DETOXIFICATION14 homolog for purplish hue of young pods.
- An ABCG transporter gene was identified as a hypothetical contributor to sclerenchyma fortification underlying reduced pod shattering tardus locus.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2OG | DMR6-like OXYGENASE 2 |
| AAP | Amino acid permease |
| ABCG5 | G-family ATP-binding ABC transporter 5 |
| AGL8 | AGAMOUS-LIKE 8 |
| AK1 | Aspartate kinase 1 |
| AP1 | APETALA1 |
| ASDH | Aspartate-semialdehyde dehydrogenase |
| BAC | Bacterial artificial chromosome |
| BAN | Anthocyanidin reductase |
| BolA2 | BolA-like family protein 2 |
| CBF | C-repeat binding factor |
| CCR1 | Cinnamoyl-CoA reductase 1 |
| CRLK1 | CALCIUM/CALMODULIN-REGULATED RECEPTOR-LIKE KINASE 1 |
| CXE1 | Carboxylesterase 1 |
| DAPDC1 | Diaminopimelate decarboxylase 1 |
| DapL | LL-diaminopimelate aminotransferase |
| DArT | Diversity Arrays Technology |
| DFR | Dihydroflavonol 4-reductase |
| DHDPS | 4-Hydroxy-tetrahydrodipicolinate synthase |
| DTX14 | MATE efflux family protein DETOXIFICATION14 |
| DUF1218 | Fiber protein Fb34, domain of unknown function 1218 |
| eQTL | Expression quantitative trait locus |
| FKF1 | FLAVIN-BINDING KELCH REPEAT, F-BOX 1 |
| FLC | FLOWERING LOCUS C |
| FSD2 | Fe superoxide dismutase 2 |
| FT | FLOWERING LOCUS T |
| FUL | FRUITFULL |
| GAUT10 | Galacturonosyltransferase 10-like |
| GLT1 | Glutamate synthase 1 |
| GO | GENE Gene ontology |
| GTF2H2 | General transcription factor IIH subunit 2 |
| GWAS | Genome-wide association study |
| ICE1 | INDUCER OF CBF EXPRESSION 1 |
| KAB1 | Voltage-gated potassium channel subunit beta 1 |
| KNAT1 | Homeobox protein knotted-1-like |
| LaAT | HXXXD-type ACYL-TRANSFERASE |
| LanFTc1 | L. angustifolius FLOWERING LOCUS T c1 |
| LDC | Lysine/ornithine decarboxylase |
| LFY | LEAFY |
| MACE | Massive analysis of cDNA ends |
| MATE | Multidrug and toxic compound extrusion |
| MFLP | Molecular fragment length polymorphism |
| MHK10.21 | Copper amine oxidase |
| MLP31 | MAJOR LATEX PROTEIN 31 |
| MLP423 | MAJOR LATEX PROTEIN 423 |
| MWL-1 | MODIFYING WALL LIGNIN-1 |
| MWL-2 | MODIFYING WALL LIGNIN-2 |
| MYB60 | MYB transcription factor 60 |
| NACA2 | Nascent polypeptide-associated complex subunit alpha 2 |
| NPF3.1 | Protein NRT1/ PTR FAMILY 3.1-like |
| PGPS2 | CDP-diacylglycerol–glycerol-3-phosphate 3-phosphatidyltransferase 2 |
| PUP | Purine permease transporter |
| RAP2-7 | ETHYLENE RESPONSIVE TRANSCRIPTION FACTOR RAP2-7 |
| RIL | Recombinant inbred line |
| RPL40A | Ubiquitin-60S ribosomal protein L40A |
| SDR | Short-chain dehydrogenase reductase |
| SNP | Single nucleotide polymorphism |
| SOC1 | SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 |
| UGT85A2 | UDP-Glycosyltransferase 85A2 |
| UGT87A2 | UDP-Glycosyltransferase 87A2 |
References
- Święcicki, W.; Święcicki, W.K. Domestication and breeding improvement of narrow-leafed lupin (L. angustifolius L.). J. Appl. Genet. 1995, 36, 155–167. [Google Scholar]
- Landers, K.F. Vernalization responses in narrow-leafed lupin (Lupinus angustifolius) genotypes. Aust. J. Agric. Res. 1995, 46, 1011–1025. [Google Scholar] [CrossRef]
- Adhikari, K.N.; Buirchell, B.J.; Sweetingham, M.W. Length of vernalization period affects flowering time in three lupin species. Plant Breed. 2012, 131, 631–636. [Google Scholar] [CrossRef]
- Gladstones, J.; Hill, G. Selection for economic characters in Lupinus angustifolius and L. digitatus. 2. Time of flowering. Aust. J. Exp. Agric. 1969, 9, 213–220. [Google Scholar] [CrossRef]
- Wink, M.; Meißner, C.; Witte, L. Patterns of quinolizidine alkaloids in 56 species of the genus Lupinus. Phytochemistry 1995, 38, 139–153. [Google Scholar] [CrossRef]
- Bassoli, A.; Borgonovo, G.; Busnelli, G. Alkaloids and the bitter taste. In Modern Alkaloids, Fattorusso, E., Taglialatela-Scafati, O.; Wiley-VCH: Weinheim, Germany, 2007; pp. 53–72. [Google Scholar]
- Matsuura, H.N.; Fett-Neto, A.G. Plant alkaloids: Main features, toxicity, and mechanisms of action. In Plant Toxins; Gopalakrishnakone, P., Carlini, C.R., Ligabue-Braun, R., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–15. [Google Scholar]
- Gladstones, J.S. Lupins as crop plants. Field Crop Abstracts 1970, 23, 26. [Google Scholar]
- Von Sengbusch, R. Süßlupinen und Öllupinen. Die Entstehungsgeschichte einiger neuer Kulturpflanzen. Landwirtsch. Jahrbücher 1942, 91, 719–880. [Google Scholar]
- Kamel, K.A.; Święcicki, W.; Kaczmarek, Z.; Barzyk, P. Quantitative and qualitative content of alkaloids in seeds of a narrow-leafed lupin (Lupinus angustifolius L.) collection. Genet. Resour. Crop Evol. 2016, 63, 711–719. [Google Scholar] [CrossRef]
- Frick, K.M.; Kamphuis, L.G.; Siddique, K.H.; Singh, K.B.; Foley, R.C. Quinolizidine alkaloid biosynthesis in lupins and prospects for grain quality improvement. Front. Plant Sci. 2017, 8, 87. [Google Scholar] [CrossRef]
- Gladstones, J. Selection for economic characters in Lupinus angustifolius and L. digitatus. Aust. J. Exp. Agric. 1967, 7, 360–366. [Google Scholar] [CrossRef]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of two sequence-specific PCR markers linked to the le gene that reduces pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Genet. Mol. Biol. 2007, 30, 623–629. [Google Scholar] [CrossRef]
- Mikolajczyk, J. Genetic studies in Lupinus angustifolius. 2. Inheritance of some morphological characters in blue lupine. Genet. Pol. 1966, 7, 153–180. [Google Scholar]
- Li, X.; Buirchell, B.; Yan, G.; Yang, H. A molecular marker linked to the mollis gene conferring soft-seediness for marker-assisted selection applicable to a wide range of crosses in lupin (Lupinus angustifolius L.) breeding. Mol. Breed. 2012, 29, 361–370. [Google Scholar] [CrossRef]
- Nirenberg, H.I.; Feiler, U.; Hagedorn, G. Description of Colletotrichum lupini comb. nov. in modern terms. Mycologia 2002, 94, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Boersma, J.G.; You, M.; Buirchell, B.J.; Sweetingham, M.W. Development and implementation of a sequence-specific PCR marker linked to a gene conferring resistance to anthracnose disease in narrow-leafed lupin (Lupinus angustifolius L.). Mol. Breed. 2004, 14, 145–151. [Google Scholar] [CrossRef]
- Yang, H.; Renshaw, D.; Thomas, G.; Buirchell, B.; Sweetingham, M. A strategy to develop molecular markers applicable to a wide range of crosses for marker assisted selection in plant breeding: A case study on anthracnose disease resistance in lupin (Lupinus angustifolius L.). Mol. Breed. 2008, 21, 473–483. [Google Scholar] [CrossRef]
- Fischer, K.; Dieterich, R.; Nelson, M.N.; Kamphuis, L.G.; Singh, K.B.; Rotter, B.; Krezdorn, N.; Winter, P.; Wehling, P.; Ruge-Wehling, B. Characterization and mapping of LanrBo: A locus conferring anthracnose resistance in narrow-leafed lupin (Lupinus angustifolius L.). Theor. Appl. Genet. 2015, 128, 2121–2130. [Google Scholar] [CrossRef]
- Yang, H.; Sweetingham, M.W.; Cowling, W.A.; Smith, P.M.C. DNA fingerprinting based on microsatellite-anchored fragment length polymorphisms, and isolation of sequence-specific PCR markers in lupin (Lupinus angustifolius L.). Mol. Breed. 2001, 7, 203–209. [Google Scholar] [CrossRef]
- Boersma, J.G.; Pallotta, M.; Li, C.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Construction of a genetic linkage map using MFLP and identification of molecular markers linked to domestication genes in narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2005, 10, 331–344. [Google Scholar]
- Li, X.; Yang, H.; Buirchell, B.; Yan, G. Development of a DNA marker tightly linked to low-alkaloid gene iucundus in narrow-leafed lupin (Lupinus angustifolius L.) for marker-assisted selection. Crop Pasture Sci. 2011, 62, 218–224. [Google Scholar] [CrossRef]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of a sequence-specific PCR marker linked to the Ku gene which removes the vernalization requirement in narrow-leafed lupin. Plant Breed. 2007, 126, 306–309. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Yan, G. Development of a co-dominant DNA marker linked to the gene lentus conferring reduced pod shattering for marker-assisted selection in narrow-leafed lupin (Lupinus angustifolius) breeding. Plant Breed. 2012, 131, 540–544. [Google Scholar] [CrossRef]
- Boersma, J.G.; Nelson, M.N.; Sivasithamparam, K.; Yang, H.A. Development of sequence-specific PCR markers linked to the Tardus gene that reduces pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Mol. Breed. 2009, 23, 259–267. [Google Scholar] [CrossRef]
- Li, X.; Renshaw, D.; Yang, H.; Yan, G. Development of a co-dominant DNA marker tightly linked to gene tardus conferring reduced pod shattering in narrow-leafed lupin (Lupinus angustifolius L.). Euphytica 2010, 176, 49–58. [Google Scholar] [CrossRef]
- Nelson, M.N.; Phan, H.T.T.; Ellwood, S.R.; Moolhuijzen, P.M.; Hane, J.; Williams, A.; O’Lone, C.E.; Fosu-Nyarko, J.; Scobie, M.; Cakir, M.; et al. The first gene-based map of Lupinus angustifolius L.-location of domestication genes and conserved synteny with Medicago truncatula. Theor. Appl. Genet. 2006, 113, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Moolhuijzen, P.M.; Boersma, J.G.; Chudy, M.; Lesniewska, K.; Bellgard, M.; Oliver, R.P.; Swiecicki, W.; Wolko, B.; Cowling, W.A.; et al. Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume, Lotus japonicus. DNA Res. 2010, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, L.G.; Hane, J.K.; Nelson, M.N.; Gao, L.; Atkins, C.A.; Singh, K.B. Transcriptome sequencing of different narrow-leafed lupin tissue types provides a comprehensive uni-gene assembly and extensive gene-based molecular markers. Plant Biotechnol. J. 2015, 13, 14–25. [Google Scholar] [CrossRef]
- Zhou, G.; Jian, J.; Wang, P.; Li, C.; Tao, Y.; Li, X.; Renshaw, D.; Clements, J.; Sweetingham, M.; Yang, H. Construction of an ultra-high density consensus genetic map, and enhancement of the physical map from genome sequencing in Lupinus angustifolius. Theor. Appl. Genet. 2018, 131, 209–223. [Google Scholar] [CrossRef]
- Kasprzak, A.; Safár, J.; Janda, J.; Dolezel, J.; Wolko, B.; Naganowska, B. The bacterial artificial chromosome (BAC) library of the narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2006, 11, 396–407. [Google Scholar] [CrossRef]
- Gao, L.-L.; Hane, J.K.; Kamphuis, L.G.; Foley, R.; Shi, B.-J.; Atkins, C.A.; Singh, K.B. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): Construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genom. 2011, 12, 521. [Google Scholar] [CrossRef]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Muller, S.; Rotter, B.; Winter, P.; Fliser, D.; Heine, G.H. Massive analysis of cDNA Ends (MACE) and miRNA expression profiling identifies proatherogenic pathways in chronic kidney disease. Epigenetics 2014, 9, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Bojahr, J.; Nhengiwa, O.; Krezdorn, N.; Rotter, B.; Saal, B.; Ruge-Wehling, B.; Struck, C.; Winter, P. Massive analysis of cDNA ends (MACE) reveals a co-segregating candidate gene for LpPg1 stem rust resistance in perennial ryegrass (Lolium perenne). Theor. Appl. Genet. 2016, 129, 1915–1932. [Google Scholar] [CrossRef]
- Zajac, B.K.; Amendt, J.; Horres, R.; Verhoff, M.A.; Zehner, R. De novo transcriptome analysis and highly sensitive digital gene expression profiling of Calliphora vicina (Diptera: Calliphoridae) pupae using MACE (Massive Analysis of cDNA Ends). Forensic Science International: Genetics 2015, 15, 137–146. [Google Scholar] [CrossRef]
- Zhernakov, A.; Rotter, B.; Winter, P.; Borisov, A.; Tikhonovich, I.; Zhukov, V. Massive Analysis of cDNA Ends (MACE) for transcript-based marker design in pea (Pisum sativum L.). Genom. Data 2017, 11, 75–76. [Google Scholar] [CrossRef]
- Keller, M.; Consortium, S.-I.; Simm, S. The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. BMC Genom. 2018, 19, 447. [Google Scholar] [CrossRef]
- Parreira, J.R.; Balestrazzi, A.; Fevereiro, P.; Araujo, S.S. Maintaining genome integrity during seed development in Phaseolus vulgaris L.: Evidence from a transcriptomic profiling study. Genes 2018, 9, 463. [Google Scholar] [CrossRef] [Green Version]
- Oyiga, B.C.; Sharma, R.C.; Baum, M.; Ogbonnaya, F.C.; Léon, J.; Ballvora, A. Allelic variations and differential expressions detected at quantitative trait loci for salt stress tolerance in wheat. Plant Cell Environ. 2018, 41, 919–935. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, A.; Naganowska, B.; Wolko, B. Karyotyping of the narrow-leafed lupin (Lupinus angustifolius L.) by using FISH, PRINS and computer measurements of chromosomes. J. Appl. Genet. 2009, 50, 77–82. [Google Scholar] [CrossRef]
- Wyrwa, K.; Książkiewicz, M.; Szczepaniak, A.; Susek, K.; Podkowiński, J.; Naganowska, B. Integration of Lupinus angustifolius L. (narrow-leafed lupin) genome maps and comparative mapping within legumes. Chromosome Res. 2016, 24, 355–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przysiecka, Ł.; Książkiewicz, M.; Wolko, B.; Naganowska, B. Structure, expression profile and phylogenetic inference of chalcone isomerase-like genes from the narrow-leafed lupin (Lupinus angustifolius L.) genome. Front. Plant Sci. 2015, 6, 268. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Zielezinski, A.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Karlowski, W.; Wolko, B.; Naganowska, B. Remnants of the legume ancestral genome preserved in gene-rich regions: Insights from Lupinus angustifolius physical, genetic, and comparative mapping. Plant Mol. Biol. Rep. 2015, 33, 84–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Książkiewicz, M.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Majcherkiewicz, K.; Przysiecka, Ł.; Karlowski, W.; Wolko, B.; Naganowska, B. Comparative genomics of Lupinus angustifolius gene-rich regions: BAC library exploration, genetic mapping and cytogenetics. BMC Genom. 2013, 14, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leśniewska, K.; Książkiewicz, M.; Nelson, M.N.; Mahé, F.; Aïnouche, A.; Wolko, B.; Naganowska, B. Assignment of 3 genetic linkage groups to 3 chromosomes of narrow-leafed lupin. J. Hered. 2011, 102, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume cytosolic and plastid acetyl-coenzyme-A carboxylase genes differ by evolutionary patterns and selection pressure schemes acting before and after whole-genome duplications. Genes 2018, 9, 563. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.N.; Książkiewicz, M.; Rychel, S.; Besharat, N.; Taylor, C.M.; Wyrwa, K.; Jost, R.; Erskine, W.; Cowling, W.A.; Berger, J.D.; et al. The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol. 2017, 213, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Narożna, D.; Książkiewicz, M.; Przysiecka, Ł.; Króliczak, J.; Wolko, B.; Naganowska, B.; Mądrzak, C.J. Legume isoflavone synthase genes have evolved by whole-genome and local duplications yielding transcriptionally active paralogs. Plant Sci. 2017, 264, 149–167. [Google Scholar] [CrossRef]
- Książkiewicz, M.; Rychel, S.; Nelson, M.N.; Wyrwa, K.; Naganowska, B.; Wolko, B. Expansion of the phosphatidylethanolamine binding protein family in legumes: A case study of Lupinus angustifolius L. FLOWERING LOCUS T homologs, LanFTc1 and LanFTc2. BMC Genom. 2016, 17, 820. [Google Scholar]
- Boersma, J.G.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Development of a PCR marker tightly linked to mollis, the gene that controls seed dormancy in Lupinus angustifolius L. Plant Breed. 2007, 126, 612–616. [Google Scholar] [CrossRef]
- You, M.; Boersma, J.G.; Buirchell, B.J.; Sweetingham, M.W.; Siddique, K.H.M.; Yang, H. A PCR-based molecular marker applicable for marker-assisted selection for anthracnose disease resistance in lupin breeding. Cell. Mol. Biol. Lett. 2005, 10, 123–134. [Google Scholar] [PubMed]
- Książkiewicz, M.; Nazzicari, N.; Yang, H.A.; Nelson, M.N.; Renshaw, D.; Rychel, S.; Ferrari, B.; Carelli, M.; Tomaszewska, M.; Stawiński, S.; et al. A high-density consensus linkage map of white lupin highlights synteny with narrow-leafed lupin and provides markers tagging key agronomic traits. Sci. Rep. 2017, 7, 15335. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.M.; Huynh, M.; Udall, J.A.; Kilian, A.; Adhikari, K.N.; Berger, J.D.; Erskine, W.; Nelson, M.N. The first genetic map for yellow lupin enables genetic dissection of adaptation traits in an orphan grain legume crop. BMC Genet. 2019, 20, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychel, S.; Książkiewicz, M. Development of gene-based molecular markers tagging low alkaloid pauper locus in white lupin (Lupinus albus L.). J. Appl. Genet. 2019, 60, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunsupa, S.; Katayama, K.; Ikeura, E.; Oikawa, A.; Toyooka, K.; Saito, K.; Yamazaki, M. Lysine decarboxylase catalyzes the first step of quinolizidine alkaloid biosynthesis and coevolved with alkaloid production in Leguminosae. Plant Cell 2012, 24, 1202–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frick, K.M.; Foley, R.C.; Kamphuis, L.G.; Siddique, K.H.M.; Garg, G.; Singh, K.B. Characterization of the genetic factors affecting quinolizidine alkaloid biosynthesis and its response to abiotic stress in narrow-leafed lupin (Lupinus angustifolius L.). Plant Cell Environ. 2018, 41, 2155–2168. [Google Scholar] [CrossRef] [Green Version]
- Kroc, M.; Koczyk, G.; Kamel, K.A.; Czepiel, K.; Fedorowicz-Strońska, O.; Krajewski, P.; Kosińska, J.; Podkowiński, J.; Wilczura, P.; Święcicki, W. Transcriptome-derived investigation of biosynthesis of quinolizidine alkaloids in narrow-leafed lupin (Lupinus angustifolius L.) highlights candidate genes linked to iucundus locus. Sci. Rep. 2019, 9, 2231. [Google Scholar] [CrossRef] [Green Version]
- Kroc, M.; Czepiel, K.; Wilczura, P.; Mokrzycka, M.; Swiecicki, W. Development and validation of a gene-targeted dCAPS marker for marker-assisted selection of low-alkaloid content in seeds of narrow-leafed lupin (Lupinus angustifolius L.). Genes 2019, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Shoji, T.; Hashimoto, T. Tobacco nicotine uptake permease regulates the expression of a key transcription factor gene in the nicotine biosynthesis pathway. Plant Physiol. 2014, 166, 2195–2204. [Google Scholar] [CrossRef] [Green Version]
- Hildreth, S.B.; Gehman, E.A.; Yang, H.; Lu, R.-H.; Ritesh, K.C.; Harich, K.C.; Yu, S.; Lin, J.; Sandoe, J.L.; Okumoto, S.; et al. Tobacco nicotine uptake permease (NUP1) affects alkaloid metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 18179–18184. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Nagy, I.; Mancinotti, D.; Otterbach, S.L.; Andersen, T.B.; Motawia, M.S.; Asp, T.; Geu-Flores, F. Transcript profiling of a bitter variety of narrow-leafed lupin to discover alkaloid biosynthetic genes. J. Exp. Bot. 2017, 68, 5527–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Kang, Y.-G.; Lee, T.; Kim, M.; Yoon, M.Y.; Lee, E.; Yang, X.; Kim, D.; Kim, Y.-J.; Lee, T.R.; et al. Comprehensive RNA sequencing and co-expression network analysis to complete the biosynthetic pathway of coumestrol, a phytoestrogen. Sci. Rep. 2019, 9, 1934. [Google Scholar] [CrossRef] [PubMed]
- Goldsbrough, A.; Belzile, F.; Yoder, J.I. Complementation of the tomato anthocyanin without (aw) mutant using the dihydroflavonol 4-reductase gene. Plant Physiol. 1994, 105, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunsupa, S.; Okada, T.; Saito, K.; Yamazaki, M. An acyltransferase-like gene obtained by differential gene expression profiles of quinolizidine alkaloid-producing and nonproducing cultivars of Lupinus angustifolius. Plant Biotechnol. 2011, 28, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. N-Methylation of quinolizidine alkaloids: An S-adenosyl-L-methionine: Cytisine N-methyltransferase from Laburnum anagyroides plants and cell cultures of L. alpinum and Cytisus canariensis. Planta 1984, 161, 339–344. [Google Scholar] [CrossRef]
- Liscombe, D.K.; Usera, A.R.; O’Connor, S.E. Homolog of tocopherol C methyltransferases catalyzes N methylation in anticancer alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18793–18798. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Smolke, C.D. Engineering biosynthesis of the anticancer alkaloid noscapine in yeast. Nat. Commun. 2016, 7, 12137. [Google Scholar] [CrossRef] [Green Version]
- Barker, S.J.; Si, P.; Hodgson, L.; Ferguson-Hunt, M.; Khentry, Y.; Krishnamurthy, P.; Averis, S.; Mebus, K.; O’Lone, C.; Dalugoda, D.; et al. Regeneration selection improves transformation efficiency in narrow-leaf lupin. Plant Cell Tissue Organ Cult. 2016, 126, 219–228. [Google Scholar] [CrossRef]
- Yang, T.; Chaudhuri, S.; Yang, L.; Du, L.; Poovaiah, B.W. A calcium/calmodulin-regulated member of the receptor-like kinase family confers cold tolerance in plants. J. Biol. Chem. 2010, 285, 7119–7126. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Hyun, Y.; Park, J.-Y.; Park, M.-J.; Park, M.-K.; Kim, M.D.; Kim, H.-J.; Lee, M.H.; Moon, J.; Lee, I.; et al. A genetic link between cold responses and flowering time through FVE in Arabidopsis thaliana. Nat. Genet. 2004, 36, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Jung, J.-H.; Park, C.-M. INDUCER OF CBF EXPRESSION 1 integrates cold signals into FLOWERING LOCUS C-mediated flowering pathways in Arabidopsis. Plant J. 2015, 84, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Lee, H.; Jeon, J.; Park, H.; Kim, J.; Noh, Y.S.; Lee, I. Crosstalk between cold response and flowering in Arabidopsis is mediated through the flowering-time gene SOC1 and its upstream negative regulator FLC. Plant Cell 2009, 21, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Gilmour, S.J.; Grumet, R.; Thomashow, M.F. CBF-dependent and CBF-independent regulatory pathways contribute to the differences in freezing tolerance and cold-regulated gene expression of two Arabidopsis ecotypes locally adapted to sites in Sweden and Italy. PLoS ONE 2018, 13, e0207723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, W.; Zhang, L.; Liang, S.; Jones, R.L.; Lu, Y.T. A tobacco calcium/calmodulin-binding protein kinase functions as a negative regulator of flowering. J. Biol. Chem. 2004, 279, 31483–31494. [Google Scholar] [CrossRef] [Green Version]
- Hecht, V.; Foucher, F.; Ferrandiz, C.; Macknight, R.; Navarro, C.; Morin, J.; Vardy, M.E.; Ellis, N.; Beltran, J.P.; Rameau, C.; et al. Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 2005, 137, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, K.; Kurotani, A.; Iida, K.; Kuromori, T.; Shinozaki, K.; Sakurai, T. RARGE II: An integrated phenotype database of Arabidopsis mutant traits using a controlled vocabulary. Plant Cell Physiol. 2014, 55, e4. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Balasubramanian, V.K.; Chopra, R.; Mendu, V. The photoperiodic flowering time regulator FKF1 negatively regulates cellulose biosynthesis. Plant Physiol. 2019, 180, 2240–2253. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Jin, S.-H.; Hu, H.-Q.; Sun, Y.-G.; Wang, Y.-W.; Han, P.; Hou, B.-K. UGT87A2, an Arabidopsis glycosyltransferase, regulates flowering time via FLOWERING LOCUS C. New Phytol. 2012, 194, 666–675. [Google Scholar] [CrossRef]
- Taylor, C.M.; Kamphuis, L.G.; Zhang, W.; Garg, G.; Berger, J.D.; Mousavi-Derazmahalleh, M.; Bayer, P.E.; Edwards, D.; Singh, K.B.; Cowling, W.A.; et al. INDEL variation in the regulatory region of the major flowering time gene LanFTc1 is associated with vernalization response and flowering time in narrow-leafed lupin (Lupinus angustifolius L.). Plant Cell Environ. 2019, 42, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Abe, M.; Kobayashi, Y.; Yamamoto, S.; Daimon, Y.; Yamaguchi, A.; Ikeda, Y.; Ichinoki, H.; Notaguchi, M.; Goto, K.; Araki, T. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 2005, 309, 1052–1056. [Google Scholar] [CrossRef]
- Andrés, F.; Romera-Branchat, M.; Martínez-Gallegos, R.; Patel, V.; Schneeberger, K.; Jang, S.; Altmüller, J.; Nürnberg, P.; Coupland, G. Floral induction in Arabidopsis by FLOWERING LOCUS T requires direct repression of BLADE-ON-PETIOLE genes by the homeodomain protein PENNYWISE. Plant Physiol. 2015, 169, 2187–2199. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson-Sánchez, D.; Martínez-López, S.; Navarro-Cartagena, S.; Jover-Gil, S.; Micol, J.L. Members of the DEAL subfamily of the DUF1218 gene family are required for bilateral symmetry but not for dorsoventrality in Arabidopsis leaves. New Phytol. 2018, 217, 1307–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, S.; Wei, H.; Milne, J.; Page, G.P.; Somerville, C.R. Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc. Natl. Acad. Sci. USA 2005, 102, 8633–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubeda-Tomas, S.; Edvardsson, E.; Eland, C.; Singh, S.K.; Zadik, D.; Aspeborg, H.; Gorzsàs, A.; Teeri, T.T.; Sundberg, B.; Persson, P.; et al. Genomic-assisted identification of genes involved in secondary growth in Arabidopsis utilising transcript profiling of poplar wood-forming tissues. Physiol. Plant. 2007, 129, 415–428. [Google Scholar] [CrossRef]
- Mewalal, R.; Mizrachi, E.; Coetzee, B.; Mansfield, S.D.; Myburg, A.A. The Arabidopsis Domain of Unknown Function 1218 (DUF1218) containing proteins, MODIFYING WALL LIGNIN-1 and 2 (At1g31720/MWL-1 and At4g19370/MWL-2) function redundantly to alter secondary cell wall lignin content. PLoS ONE 2016, 11, e0150254. [Google Scholar] [CrossRef]
- Miyauchi, H.; Moriyama, S.; Kusakizako, T.; Kumazaki, K.; Nakane, T.; Yamashita, K.; Hirata, K.; Dohmae, N.; Nishizawa, T.; Ito, K.; et al. Structural basis for xenobiotic extrusion by eukaryotic MATE transporter. Nat. Commun. 2017, 8, 1633. [Google Scholar] [CrossRef]
- Upadhyay, N.; Kar, D.; Deepak Mahajan, B.; Nanda, S.; Rahiman, R.; Panchakshari, N.A.; Bhagavatula, L.; Datta, S. The multitasking abilities of MATE transporters in plants. J. Exp. Bot. 2019, 70, 4643–4656. [Google Scholar] [CrossRef]
- Yang, X.; Xia, X.; Zhang, Z.; Nong, B.; Zeng, Y.; Wu, Y.; Xiong, F.; Zhang, Y.; Liang, H.; Pan, Y.; et al. Identification of anthocyanin biosynthesis genes in rice pericarp using PCAMP. Plant Biotechnol. J. 2019, 17, 1700–1702. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Y.; Liu, H.; Kang, L.; Geng, J.; Gai, Y.; Ding, Y.; Sun, H.; Li, Y. Identification and expression analysis of MATE genes involved in flavonoid transport in blueberry plants. PLoS ONE 2015, 10, e0118578. [Google Scholar] [CrossRef] [Green Version]
- Ellis, T.J.; Field, D.L. Repeated gains in yellow and anthocyanin pigmentation in flower colour transitions in the Antirrhineae. Ann. Bot. 2016, 117, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Shiono, K.; Ando, M.; Nishiuchi, S.; Takahashi, H.; Watanabe, K.; Nakamura, M.; Matsuo, Y.; Yasuno, N.; Yamanouchi, U.; Fujimoto, M.; et al. RCN1/OsABCG5, an ATP-binding cassette (ABC) transporter, is required for hypodermal suberization of roots in rice (Oryza sativa). Plant J. 2014, 80, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, R.; Smolka, U.; Altmann, S.; Eschen-Lippold, L.; Senning, M.; Sonnewald, S.; Weigel, B.; Frolova, N.; Strehmel, N.; Hause, G.; et al. The ABC transporter ABCG1 is required for suberin formation in potato tuber periderm. Plant cell 2014, 26, 3403–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro, S.; Lee, Y.; Tohge, T.; Sudre, D.; Osorio, S.; Park, J.; Bovet, L.; Lee, Y.; Geldner, N.; Fernie, A.R.; et al. AtABCG29 is a monolignol transporter involved in lignin biosynthesis. Curr. Biol. 2012, 22, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Sibout, R.; Höfte, H. Plant Cell Biology: The ABC of monolignol transport. Curr. Biol. 2012, 22, R533–R535. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, M.; Fleck, A.T.; Biedermann, E.; Ngo, N.S.; Schreiber, L.; Schenk, M.K. An ABC transporter is involved in the silicon-induced formation of Casparian Bands in the exodermis of rice. Front. Plant Sci. 2017, 8, 671. [Google Scholar] [CrossRef]
- Forbes, I.; Wells, H.D. Hard and soft seededness in blue lupine, Lupinus angustifolius L.: Inheritance and phenotype classification. Crop Sci. 1968, 8, 195–197. [Google Scholar] [CrossRef]
- Miao, Z.H.; Fortune, J.A.; Gallagher, J. Anatomical structure and nutritive value of lupin seed coats. Aust. J. Agric. Res. 2001, 52, 985–993. [Google Scholar] [CrossRef]
- Smýkal, P.; Vernoud, V.; Blair, M.W.; Soukup, A.; Thompson, R.D. The role of the testa during development and in establishment of dormancy of the legume seed. Front. Plant Sci. 2014, 5, 351. [Google Scholar]
- Silvestro, D.; Andersen, T.G.; Schaller, H.; Jensen, P.E. Plant sterol metabolism. Δ(7)-Sterol-C5-desaturase (STE1/DWARF7), Δ(5,7)-sterol-Δ(7)-reductase (DWARF5) and Δ(24)-sterol-Δ(24)-reductase (DIMINUTO/DWARF1) show multiple subcellular localizations in Arabidopsis thaliana (Heynh) L. PLoS ONE 2013, 8, e56429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBolt, S.; Scheible, W.-R.; Schrick, K.; Auer, M.; Beisson, F.; Bischoff, V.; Bouvier-Navé, P.; Carroll, A.; Hematy, K.; Li, Y.; et al. Mutations in UDP-Glucose:sterol glucosyltransferase in Arabidopsis cause transparent testa phenotype and suberization defect in seeds. Plant Physiol. 2009, 151, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loubéry, S.; De Giorgi, J.; Utz-Pugin, A.; Demonsais, L.; Lopez-Molina, L. A maternally deposited endosperm cuticle contributes to the physiological defects of transparent testa seeds. Plant Physiol. 2018, 177, 1218–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.E.J.; Frew, T.J.; Murray, S.R.; Hofer, J.M.I.; Jacobs, J.M.E.; et al. Identification of Mendel’s white flower character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, X.-B.; Shi, Y.-F.; Wang, H.-M.; Feng, B.-H.; Li, X.-H.; Huang, Q.-N.; Song, L.-X.; Guo, D.; He, Y.; et al. A point mutation in an F-Box domain-containing protein is responsible for brown hull phenotype in rice. Rice Sci. 2016, 23, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhernakov, A.I.; Shtark, O.Y.; Kulaeva, O.A.; Fedorina, J.V.; Afonin, A.M.; Kitaeva, A.B.; Tsyganov, V.E.; Afonso-Grunz, F.; Hoffmeier, K.; Rotter, B.; et al. Mapping-by-sequencing using NGS-based 3’-MACE-Seq reveals a new mutant allele of the essential nodulation gene Sym33 (IPD3) in pea (Pisum sativum L.). Peer J. 2019, 7, e6662. [Google Scholar] [CrossRef]
- Berger, J.D.; Buirchell, B.J.; Luckett, D.J.; Nelson, M.N. Domestication bottlenecks limit genetic diversity and constrain adaptation in narrow-leafed lupin (Lupinus angustifolius L.). Theor. Appl. Genet. 2012, 124, 637–652. [Google Scholar] [CrossRef]
- Cowling, W.A. Pedigrees and characteristics of narrow-leafed lupin cultivars released in Australia from 1967 to 1998. Bull. Agric. West. Aust. 1999, 4365, 4–11. [Google Scholar]
- Cowling, W.A.; Buirchell, B.J.; Falk, D.E. A model for incorporating novel alleles from the primary gene pool into elite crop breeding programs while reselecting major genes for domestication or adaptation. Crop Pasture Sci. 2009, 60, 1009–1015. [Google Scholar] [CrossRef]
- Mousavi-Derazmahalleh, M.; Bayer, P.E.; Nevado, B.; Hurgobin, B.; Filatov, D.; Kilian, A.; Kamphuis, L.G.; Singh, K.B.; Berger, J.D.; Hane, J.K.; et al. Exploring the genetic and adaptive diversity of a pan-Mediterranean crop wild relative: Narrow-leafed lupin. Theor. Appl. Genet. 2018, 131, 887–901. [Google Scholar] [CrossRef] [Green Version]
- Mousavi-Derazmahalleh, M.; Nevado, B.; Bayer, P.E.; Filatov, D.A.; Hane, J.K.; Edwards, D.; Erskine, W.; Nelson, M.N. The western Mediterranean region provided the founder population of domesticated narrow-leafed lupin. Theor. Appl. Genet. 2018, 131, 2543–2554. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, A.; Ausubel, F.M. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 1993, 4, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Neff, M.M.; Neff, J.D.; Chory, J.; Pepper, A.E. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: Experimental applications in Arabidopsis thaliana genetics. Plant J. 1998, 14, 387–392. [Google Scholar] [CrossRef]
- Neff, M.M.; Turk, E.; Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 2002, 18, 613–615. [Google Scholar] [CrossRef]
- Thiel, T.; Kota, R.; Grosse, I.; Stein, N.; Graner, A. SNP2CAPS: A SNP and INDEL analysis tool for CAPS marker development. Nucleic Acids Res. 2004, 32, e5. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Number of Markers | Number of Loci | Number of Genes | Length (cM) | Number of Scaffolds |
|---|---|---|---|---|---|
| NLL-01 | 289 | 120 | 251 | 156.38 | 11 |
| NLL-02 | 207 | 73 | 185 | 132.78 | 21 |
| NLL-03 | 204 | 77 | 169 | 115.02 | 22 |
| NLL-04 | 193 | 95 | 157 | 126.93 | 8 |
| NLL-05 | 144 | 70 | 130 | 101.29 | 10 |
| NLL-06 | 304 | 119 | 267 | 144.66 | 10 |
| NLL-07 | 191 | 85 | 155 | 104.25 | 10 |
| NLL-08 | 231 | 101 | 194 | 119.29 | 8 |
| NLL-09 | 192 | 93 | 169 | 140.98 | 10 |
| NLL-10 | 154 | 75 | 121 | 85.64 | 6 |
| NLL-11 | 269 | 107 | 216 | 119.57 | 8 |
| NLL-12 | 271 | 88 | 220 | 85.86 | 15 |
| NLL-13 | 230 | 81 | 200 | 78.5 | 11 |
| NLL-14 | 160 | 76 | 121 | 87.5 | 10 |
| NLL-15 | 248 | 87 | 207 | 86.65 | 10 |
| NLL-16 | 240 | 96 | 204 | 99.42 | 9 |
| NLL-17 | 227 | 70 | 176 | 91.57 | 6 |
| NLL-18 | 196 | 77 | 151 | 91.34 | 9 |
| NLL-19 | 144 | 60 | 122 | 92.08 | 10 |
| NLL-20 | 199 | 85 | 169 | 103.92 | 9 |
| Total | 4309 | 1735 | 3590 | 2163.63 | 209 |
| Domestication Trait | Genes with eQTL 1 Peak | Mean eQTL Peak LOD Value | Maximum eQTL Peak LOD Value | Genes with cis Genomic Positions | Genes with trans Genomic Positions | Genes in Unassigned Scaffolds |
|---|---|---|---|---|---|---|
| iucundus | 61 | 14.77 | 37.70 | 11 | 45 | 5 |
| Ku | 25 | 11.20 | 40.97 | 11 | 11 | 3 |
| leucospermus | 9 | 13.18 | 35.34 | 5 | 3 | 1 |
| lentus | 6 | 16.36 | 42.97 | 5 | 1 | 0 |
| tardus | 2 | 25.05 | 31.82 | 2 | 0 | 0 |
| Lanr1 | 4 | 10.20 | 13.48 | 4 | 0 | 0 |
| mollis | 1 | 9.59 | 9.59 | 0 | 1 | 0 |
| Protein | Group | Peak cM | Peak LOD | PVE 1 % | Association Based on t-Student Test | Protein Annotation |
|---|---|---|---|---|---|---|
| OIW21347.1 | NLL-07 | 40.2 | 37.7 | 84.2 | −0.82 | voltage-gated potassium channel subunit beta 1, KAB1 |
| OIV96299.1 | NLL-07 | 40.1 | 37.3 | 73.4 | 0.81 | lysine/ornithine decarboxylase, LDC |
| OIV96574.1 | NLL-07 | 40.1 | 35.4 | 60.5 | 0.84 | purine permease 1, PUP1 |
| OIW10551.1 | NLL-07 | 39.6 | 29.4 | 67.2 | 0.80 | MLP-like protein 423, MLP423 |
| OIV89004.1 | NLL-07 | 39.6 | 29.2 | 56.5 | 0.78 | ethylene-responsive transcription factor, RAP2-7 |
| OIW02927.1 | NLL-07 | 39.6 | 27.3 | 69.9 | 0.69 | amino acid permease, AAP |
| OIW07732.1 | NLL-07 | 41.3 | 26.3 | 81.9 | −0.73 | uncharacterized protein |
| OIW13431.1 | NLL-07 | 40.2 | 25.9 | 59.3 | 0.78 | dihydroflavonol 4-reductase, DFR |
| OIV89008.1 | NLL-07 | 40.2 | 25.8 | 66.2 | −0.77 | Fe superoxide dismutase 2, FSD2 |
| OIV90042.1 | NLL-07 | 39.6 | 24.6 | 59.6 | 0.72 | aspartate kinase 1, AK1 |
| OIV95196.1 | NLL-07 | 39.6 | 24.3 | 57.2 | 0.77 | HXXXD-type ACYL-TRANSFERASE, LaAT |
| OIW13432.1 | NLL-07 | 40.1 | 24.0 | 50.2 | 0.77 | dihydroflavonol 4-reductase, DFR |
| OIW03412.1 | NLL-07 | 40.1 | 23.5 | 50.7 | 0.75 | homeobox protein knotted-1-like, KNAT1 |
| OIW15620.1 | NLL-07 | 40.2 | 22.7 | 56.6 | 0.67 | uncharacterized protein |
| OIW20548.1 | NLL-07 | 40.1 | 21.7 | 50.3 | 0.72 | diaminopimelate decarboxylase 1, DAPDC1 |
| OIW02909.1 | NLL-07 | 40.1 | 20.4 | 53.3 | 0.72 | cytochrome P450, CYP71B23 |
| OIV89669.1 | NLL-07 | 39.6 | 20.4 | 48.0 | 0.70 | cinnamoyl-CoA reductase 1, CCR1 |
| OIV96948.1 | NLL-07 | 40.1 | 19.5 | 38.5 | 0.73 | copper amine oxidase, MHK10.21 |
| OIW20507.1 | NLL-07 | 40.1 | 18.3 | 40.1 | 0.70 | anthocyanidin reductase, BAN |
| OIW20661.1 | NLL-07 | 41.3 | 18.0 | 49.4 | 0.70 | 4-hydroxy-tetrahydrodipicolinate synthase, DHDPS |
| OIV97872.1 | NLL-07 | 39.6 | 17.2 | 43.5 | 0.72 | purine permease 1, PUP1 |
| OIV93156.1 | NLL-07 | 40.8 | 14.9 | 45.1 | 0.64 | LL-diaminopimelate aminotransferase, DapL |
| OIV97100.1 | NLL-07 | 39.6 | 13.9 | 44.4 | 0.64 | MYB transcription factor 34, MYB34 |
| OIW07643.1 | NLL-07 | 39.0 | 13.7 | 36.3 | 0.64 | 4-hydroxy-tetrahydrodipicolinate synthase, DHDPS |
| OIV89772.1 | NLL-07 | 39.6 | 13.3 | 40.7 | 0.51 | pentatricopeptide repeat-containing protein |
| OIW10549.1 | NLL-07 | 40.2 | 12.7 | 38.4 | 0.57 | MLP-like protein 423, MLP423 |
| OIV96820.1 | NLL-07 | 40.2 | 11.8 | 30.5 | 0.57 | glutamate synthase 1, GLT1 |
| OIW21355.1 | NLL-07 | 39.6 | 11.8 | 32.3 | 0.56 | carboxylesterase 1, CXE1 |
| OIW10098.1 | NLL-07 | 40.1 | 11.8 | 28.3 | 0.53 | aspartate-semialdehyde dehydrogenase, ASDH |
| OIW04462.1 | NLL-07 | 39.6 | 11.7 | 30.8 | 0.57 | VQ motif-containing protein |
| OIW10550.1 | NLL-07 | 40.1 | 10.2 | 14.9 | 0.74 | MLP-like protein 423, MLP423 |
| OIW20088.1 | NLL-07 | 39.6 | 9.6 | 27.4 | 0.53 | short-chain dehydrogenase reductase, SDR |
| OIV89148.1 | NLL-07 | 41.3 | 8.2 | 22.4 | 0.51 | MLP-like protein 31, MLP31 |
| OIW02362.1 | NLL-07 | 39.6 | 7.9 | 21.4 | −0.55 | DMR6-like OXYGENASE 2, 2OG |
| Protein | Group | Peak cM | Peak LOD | PVE 1 % | Association Based on t-Student Test | Protein Annotation |
|---|---|---|---|---|---|---|
| OIW03171.1 | NLL-10 | 50.8 | 41.0 | 92.1 | 0.85 | calcium/calmodulin-regulated receptor-like kinase 1, CRLK1 |
| OIW20567.1 | NLL-10 | 49.7 | 38.9 | 65.1 | −0.89 | uncharacterized protein |
| OIV96743.1 | NLL-10 | 51.4 | 18.4 | 53.5 | −0.71 | galacturonosyltransferase 10-like, GAUT10 |
| OIW03144.1 | NLL-10 | 51.3 | 16.7 | 45.2 | 0.62 | MHM17-10, AT5G56980 |
| OIW03199.1 | NLL-10 | 48.4 | 16.4 | 50.5 | 0.69 | general transcription factor IIH subunit 2, GTF2H2 |
| OIW03193.1 | NLL-10 | 50.3 | 10.6 | 31.5 | −0.53 | pentatricopeptide repeat-containing protein |
| OIW20134.1 | NLL-10 | 49.7 | 9.7 | 28.9 | 0.52 | UDP-Glycosyltransferase 85A2, UGT85A2 |
| OIV89838.1 | NLL-10 | 51.4 | 9.6 | 30.6 | 0.51 | reticulon family protein |
| OIW19675.1 | NLL-10 | 50.3 | 8.9 | 22.6 | −0.56 | MADS-box transcription factor AGAMOUS-LIKE 8, AGL8 |
| OIW03269.1 | NLL-10 | 52.0 | 8.5 | 24.6 | 0.51 | MYB transcription factor 60, MYB60 |
| OIV92673.1 | NLL-10 | 50.3 | 7.4 | 22.2 | 0.53 | protein FD-like, FD |
| OIW03334.1 | NLL-10 | 49.1 | 7.0 | 26.7 | −0.51 | flowering locus protein T, LanFTc1 |
| Trait | Protein | Group | Peak cM | Peak LOD | PVE 1 % | Association Based on t-Student Test | Protein Annotation |
|---|---|---|---|---|---|---|---|
| Lanr1 | OIW02433.1 | NLL-11 | 41.7 | 13.5 | 38.0 | 0.57 | adenine nucleotide alpha hydrolases-like |
| Lanr1 | OIW02411.1 | NLL-11 | 41.6 | 9.0 | 29.0 | −0.51 | galactosyltransferase family protein |
| le | OIW06948.1 | NLL-08 | 75.2 | 43.0 | 75.8 | 0.76 | fiber protein Fb34, DUF1218 |
| le | OIW06960.1 | NLL-08 | 73.3 | 17.4 | 45.8 | −0.66 | MATE efflux family protein DETOXIFICATION14, DTX14 |
| le | OIW06846.1 | NLL-08 | 75.2 | 13.0 | 38.6 | 0.56 | CDP-diacylglycerol–glycerol-3-phosphate 3-phosphatidyltransferase 2, PGPS2 |
| leuc | OIW21684.1 | NLL-03 | 59.1 | 35.3 | 75.1 | −0.86 | ubiquitin-60S ribosomal protein L40A, RPL40A |
| leuc | OIW15321.1 | NLL-03 | 59.6 | 20.8 | 53.4 | 0.76 | nascent polypeptide-associated complex subunit alpha 2, NACA2 |
| leuc | OIW15287.1 | NLL-03 | 59.1 | 14.7 | 40.4 | 0.59 | F-box/WD repeat-containing protein |
| leuc | OIV97389.1 | NLL-03 | 56.1 | 13.1 | 36.6 | −0.54 | protein NRT1/ PTR FAMILY 3.1-like, NPF3.1 |
| leuc | OIV89020.1 | NLL-03 | 59.1 | 10.4 | 28.6 | 0.57 | GATA type zinc finger transcription factor, WLIM2a |
| mol | OIW15058.1 | NLL-17 | 79.0 | 9.6 | 34.6 | −0.50 | FAM32A, 7-dehydrocholesterol reductase, DWARF5 |
| ta | OIW17837.1 | NLL-01 | 34.3 | 31.8 | 70.9 | −0.79 | BolA-like family protein 2, BolA2 |
| ta | OIW17820.1 | NLL-01 | 33.1 | 18.3 | 44.6 | 0.66 | G-family ATP-binding ABC transporter 5, ABCG5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plewiński, P.; Książkiewicz, M.; Rychel-Bielska, S.; Rudy, E.; Wolko, B. Candidate Domestication-Related Genes Revealed by Expression Quantitative Trait Loci Mapping of Narrow-Leafed Lupin (Lupinus angustifolius L.). Int. J. Mol. Sci. 2019, 20, 5670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225670
Plewiński P, Książkiewicz M, Rychel-Bielska S, Rudy E, Wolko B. Candidate Domestication-Related Genes Revealed by Expression Quantitative Trait Loci Mapping of Narrow-Leafed Lupin (Lupinus angustifolius L.). International Journal of Molecular Sciences. 2019; 20(22):5670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225670
Chicago/Turabian StylePlewiński, Piotr, Michał Książkiewicz, Sandra Rychel-Bielska, Elżbieta Rudy, and Bogdan Wolko. 2019. "Candidate Domestication-Related Genes Revealed by Expression Quantitative Trait Loci Mapping of Narrow-Leafed Lupin (Lupinus angustifolius L.)" International Journal of Molecular Sciences 20, no. 22: 5670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225670