A Combined Linkage and GWAS Analysis Identifies QTLs Linked to Soybean Seed Protein and Oil Content

 and
and
Abstract
:1. Introduction
2. Results
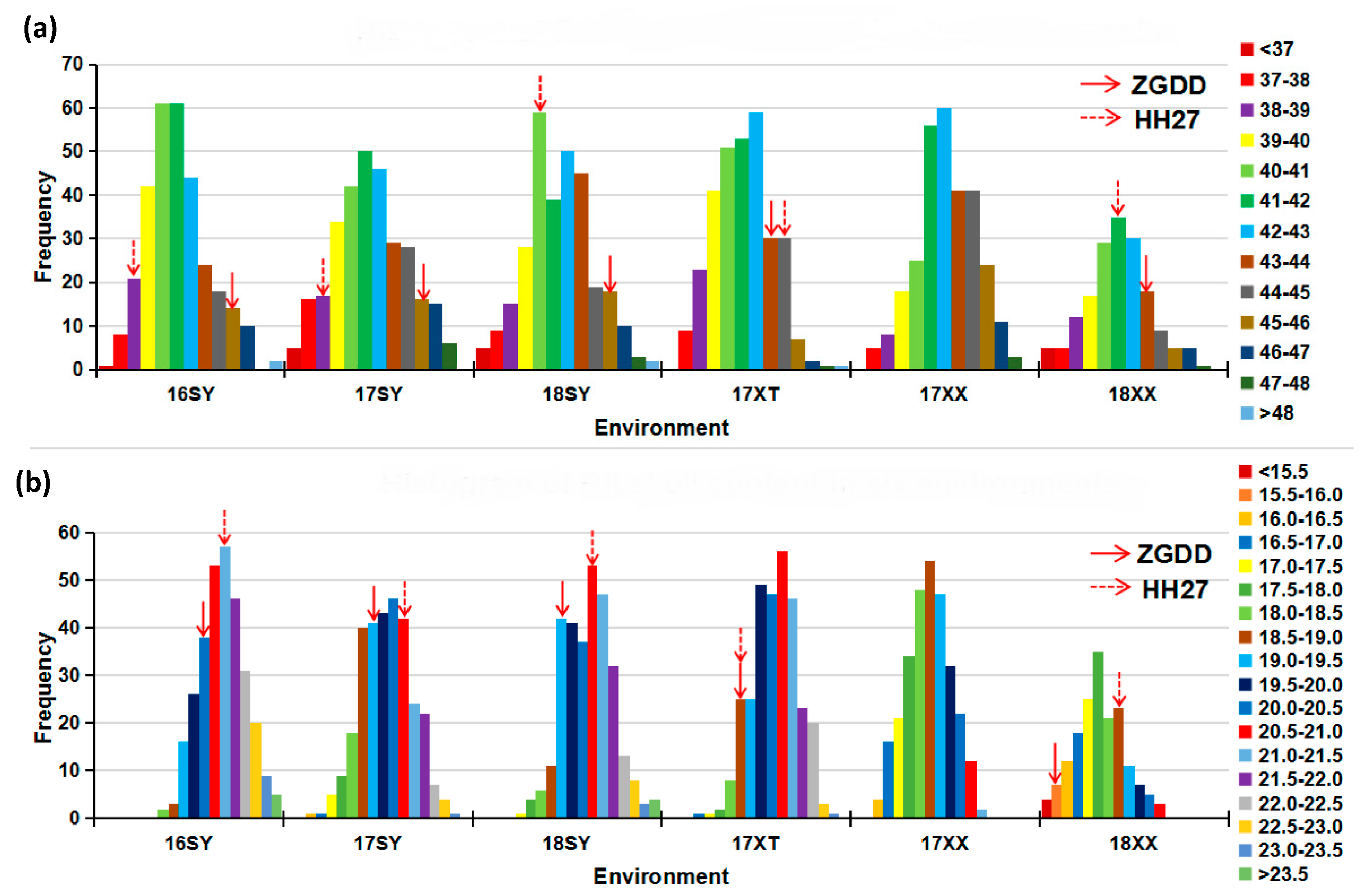
2.1. Phenotypic Variation of Protein and Oil Contents in Two Panels
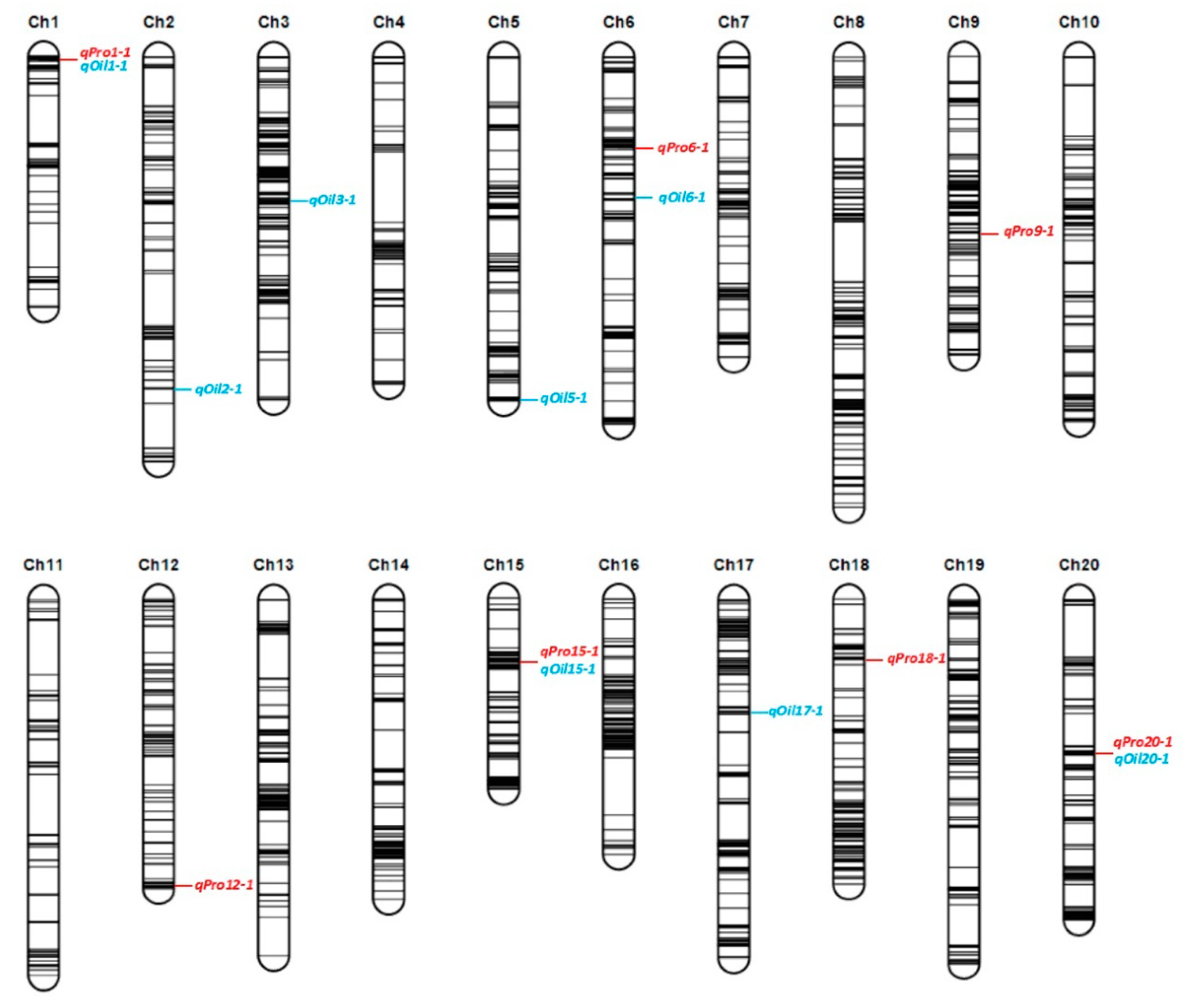
2.2. Genetic Map and QTL Analysis of Protein and Oil Contents
2.3. Genome-Wide Association Study (GWAS) Results
2.4. Co-Detected Results by Linkage Analysis and GWAS
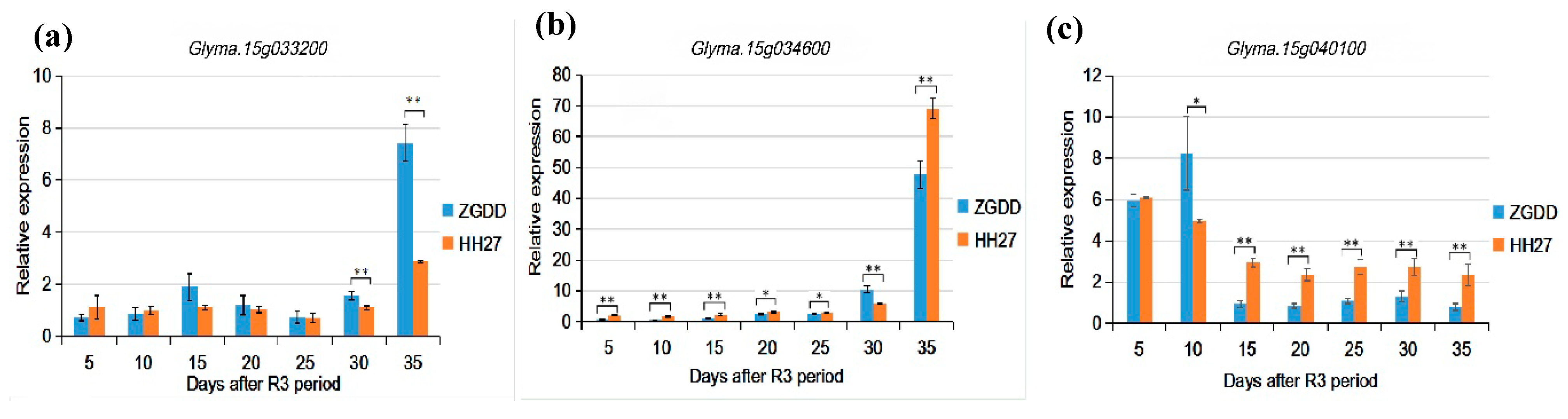
2.5. Candidate Genes and Expression Levels
3. Discussion
3.1. The Accuracy of QTL Analysis and GWAS is Improved by Using Phenotypic Data from Different Locations and Employing Ample SNP Markers
3.2. Refined QTL Intervals and SNP Loci for Protein and Oil Content Were Identified
3.3. The Candidate Genes Differentially Expressed at Late Reproductive Growth Stage between Both Parents Will Be Further Analyzed
4. Materials and Methods
4.1. Plant Materials and Field Trials
4.2. Phenotypic Data and Analysis
4.3. Genotyping and Linkage Analysis
4.4. Genotyping and GWAS
4.5. Identification and Verification of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.H.; Liu, M.F.; He, J.B.; Wang, Y.F.; Xing, G.N.; Li, Y.; Yang, S.P.; Zhao, T.J.; Gai, J.Y. Marker-assisted breeding for transgressive seed protein content in soybean [Glycine max (L.) Merr.]. Theor. Appl. Genet. 2015, 128, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Kim, K.S.; Ko, J.M.; Choi, M.S.; Kang, B.K.; Kwon, S.W.; Jun, T.H. Quantitative trait locus analysis for soybean (Glycine max) seed protein and oil concentrations using selected breeding populations. Plant Breed. 2019, 138, 95–104. [Google Scholar] [CrossRef]
- Diers, B.W.; Keim, P.; Fehr, W.R.; Shoemaker, R.C. RFLP analysis of soybean seed protein and oil content. Theor. Appl. Genet. 1992, 83, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, J.; Patil, G.B.; Sonah, H.; Deshmukh, R.K.; Vuong, T.D.; Valliyodan, B.; Nguyen, H.T. Expanding Omics Resources for Improvement of Soybean Seed Composition Traits. Front. Plant Sci. 2015, 6, 504. [Google Scholar] [CrossRef]
- Patil, G.; Vuong, T.D.; Kale, S.; Valliyodan, B.; Deshmukh, R.; Zhu, C.; Wu, X.; Bai, Y.; Yungbluth, D.; Lu, F.; et al. Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping. Plant Biotechnol. J. 2018, 16, 1939–1953. [Google Scholar] [CrossRef]
- Hartwig, E.E.; Hinson, K. Association Between Chemical Composition of Seed and Seed Yield of Soybeans1. Crop Sci. 1972, 12, 829. [Google Scholar] [CrossRef]
- Patil, G.; Mian, R.; Vuong, T.; Pantalone, V.; Song, Q.; Chen, P.; Shannon, G.J.; Carter, T.C.; Nguyen, H.T. Molecular mapping and genomics of soybean seed protein: A review and perspective for the future. Theor. Appl. Genet. 2017, 130, 1975–1991. [Google Scholar] [CrossRef]
- Sonah, H.; O’Donoughue, L.; Cober, E.; Rajcan, I.; Belzile, F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol. J. 2015, 13, 211–221. [Google Scholar] [CrossRef]
- Ning, H.L.; Bai, X.L.; Li, W.B.; Xue, H.; Zhuang, X.; Li, W.X.; Liu, C.Y. Mapping QTL Protein and Oil Contents Using Population from Four-way Re-combinant Inbred Lines for Soybean (Glycine max L. Merr.). Acta Agron. Sin. 2016, 42, 1620. [Google Scholar] [CrossRef]
- Lin, Y.H.; Zhang, L.J.; Wei, L.I.; Zhang, L.F.; Ran, X.U. QTLs mapping related to protein content of soybeans. Soyb. Sci. 2010, 29, 207–209. [Google Scholar]
- Liang, H.Z.; Wang, S.; Yu, Y.; Lian, Y.; Wang, T.F.; Wei, Y.; Gong, P.T.; Liu, X.Y.; Fang, X.J. QTL mapping of isoflavone, oil and protein content in soybean. Sci. Agric. Sin. 2009, 42, 2652–2660. [Google Scholar]
- Pathan, S.M.; Vuong, T.; Clark, K.; Lee, J.D.; Shannon, J.G.; Roberts, C.A.; Ellersieck, M.R.; Burton, J.W.; Cregan, P.B.; Hyten, D.L.; et al. Genetic Mapping and Confirmation of Quantitative Trait Loci for Seed Protein and Oil Contents and Seed Weight in Soybean. Crop Sci. 2013, 53, 765. [Google Scholar] [CrossRef]
- Panthee, D.R.; Pantalone, V.R.; West, D.R.; Saxton, A.M.; Sams, C.E. Quantitative Trait Loci for Seed Protein and Oil Concentration, and Seed Size in Soybean. Crop Sci. 2005, 45, 2015. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, G.L.; Green, M.; Scott, R.A.; Song, Q.; Hyten, D.L.; Cregan, P.B. Identification and validation of quantitative trait loci for seed yield, oil and protein contents in two recombinant inbred line populations of soybean. Mol. Genet. Genom. 2014, 289, 935–949. [Google Scholar] [CrossRef]
- Hwang, E.Y.; Song, Q.; Jia, G.; Specht, J.E.; Hyten, D.L.; Costa, J.; Cregan, P.B. A genome-wide association study of seed protein and oil content in soybean. BMC Genom. 2014, 15, 1. [Google Scholar] [CrossRef]
- Qi, Z.M.; Wu, Q.; Han, X.; Sun, Y.N.; Du, X.Y.; Liu, C.Y.; Jiang, H.W.; Hu, G.H.; Chen, Q.S. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica 2011, 179, 499–514. [Google Scholar] [CrossRef]
- Warrington, C.V.; Abdel-Haleem, H.; Hyten, D.L.; Cregan, P.B.; Orf, J.H.; Killam, A.S.; Bajjalieh, N.; Li, Z.; Boerma, H.R. QTL for seed protein and amino acids in the Benning x Danbaekkong soybean population. Theor. Appl. Genet. 2015, 128, 839–850. [Google Scholar] [CrossRef]
- Wang, W.; Liu, M.; Wang, Y.; Li, X.; Cheng, S.; Shu, L.; Yu, Z.; Kong, J.; Zhao, T.; Gai, J. Characterizing Two Inter-specific Bin Maps for the Exploration of the QTLs/Genes that Confer Three Soybean Evolutionary Traits. Front. Plant Sci. 2016, 7, 242. [Google Scholar] [CrossRef]
- Yang, X.H.; Yan, J.B.; Zheng, Y.P.; Yu, J.M.; Li, J.S. Reviews of association analysis for quantitative traits in plants. Acta Agron. Sin. 2007, 33, 523–530. [Google Scholar]
- Hansen, M.; Kraft, T.; Ganestam, S.; Säll, T.; Nilsson, N.O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet. Res. 2001, 77, 61–66. [Google Scholar] [CrossRef]
- Han, S.F. Genome-Wide Association Studies for Fatty Acid Component Traits in Soybean. Master’s Thesis, Nanjing Agricultural University, Nanjing, China, 2013. [Google Scholar]
- Zhang, J.; Wang, X.; Lu, Y.; Bhusal, S.J.; Song, Q.; Cregan, P.B.; Yen, Y.; Brown, M.; Jiang, G.L. Genome-wide Scan for Seed Composition Provides Insights into Soybean Quality Improvement and the Impacts of Domestication and Breeding. Mol. Plant 2018, 11, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Valliyodan, B.; Qiu, D.; Patil, G.; Zeng, P.; Huang, J.; Dai, L.; Chen, C.; Li, Y.; Joshi, T.; Song, L.; et al. Landscape of genomic diversity and trait discovery in soybean. Sci. Rep. 2016, 6, 23598. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Van, K.; Sung, M.; Nelson, R.; LaMantia, J.; McHale, L.K.; Mian, M.A.R. Genome-wide association study of seed protein, oil and amino acid contents in soybean from maturity groups I to IV. Theor. Appl. Genet. 2019, 132, 1639–1659. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Schultz, S.; Nelson, R.L.; Diers, B.W. Identification and fne mapping of a soybean seed protein QTL from PI 407788A on chromosome 15. Crop Sci. 2016, 56, 219–225. [Google Scholar] [CrossRef]
- Tajuddin, T. Analysis of quantitative trait loci for protein content in soybean seeds using recombinant inbred lines. J. Agron. Indones. 2005, 33, 19–24. [Google Scholar] [CrossRef]
- Van, K.; McHale, L.K. Meta-Analyses of QTLs Associated with Protein and Oil Contents and Compositions in Soybean [Glycine max (L.) Merr.] Seed. Int. J. Mol. Sci. 2017, 18, 1180. [Google Scholar] [CrossRef]
- Xu, X.; Zeng, L.; Tao, Y.; Vuong, T.; Wan, J.; Boerma, R.; Noe, J.; Li, Z.; Finnerty, S.; Pathan, S.M.; et al. Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 13469–13474. [Google Scholar] [CrossRef]
- Cao, Y.; Li, S.; Wang, Z.; Chang, F.; Kong, J.; Gai, J.; Zhao, T. Identification of Major Quantitative Trait Loci for Seed Oil Content in Soybeans by Combining Linkage and Genome-Wide Association Mapping. Front. Plant Sci. 2017, 8, 8. [Google Scholar] [CrossRef]
- Sajise, A.G.C.; Gregorio, G.B.; Kretzschmar, T.; Ismail, A.M.; Wissuwa, M.; Lee, J.S. Genetic dissection for zinc deficiency tolerance in rice using bi-parental mapping and association analysis. Theor. Appl. Genet. 2017, 130, 1903–1914. [Google Scholar]
- Lou, Q.; Chen, L.; Mei, H.; Wei, H.; Feng, F.; Wang, P.; Xia, H.; Li, T.; Luo, L. Quantitative trait locus mapping of deep rooting by linkage and association analysis in rice. J. Exp. Bot. 2015, 66, 4749–4757. [Google Scholar] [CrossRef]
- Wang, H.; Xu, S.; Fan, Y.; Liu, N.; Zhan, W.; Liu, H.; Xiao, Y.; Li, K.; Pan, Q.; Li, W.; et al. Beyond pathways: Genetic dissection of tocopherol content in maize kernels by combining linkage and association analyses. Plant Biotechnol. J. 2018, 16, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Li, D.; Luo, J.; Xiao, Y.; Liu, H.; Pan, Q.; Zhang, X.; Jin, M.; Zhao, M.; Yan, J. The genetic architecture of amino acids dissection by association and linkage analysis in maize. Plant Biotechnol. J. 2017, 15, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Sun, S.; Wu, T.T.; Liu, L.P.; Sun, X.G.; Cai, Y.P.; Li, J.C.; Xu, X.; Yuan, S.; Chen, L.; et al. Natural variation in GmPRR37 affects photoperiodic flowering and contributes to regional adaptation of soybean. Plant J. 2019. under review. [Google Scholar]
- Hu, M.X.; Wan, C.W. The effect of different ecogeographic enviroment on the seed quality of soybeans in China. Soybean Sci. 1990, 9, 39–49. [Google Scholar]
- Zu, S.H. Tne agroclimatic analysis on tne oil content of soybean and its geographical distribution in Heilongjiang province. Soybean Sci. 1983, 2, 266–276. [Google Scholar]
- Song, W.; Yang, R.; Wu, T.; Wu, C.; Sun, S.; Zhang, S.; Jiang, B.; Tian, S.; Liu, X.; Han, T. Analyzing the Effects of Climate Factors on Soybean Protein, Oil Contents, and Composition by Extensive and High-Density Sampling in China. J. Agric. Food Chem. 2016, 64, 4121–4130. [Google Scholar] [CrossRef]
- Kale, S.M.; Jaganathan, D.; Ruperao, P.; Chen, C.; Punna, R.; Kudapa, H.; Thudi, M.; Roorkiwal, M.; Katta, M.A.; Doddamani, D.; et al. Prioritization of candidate genes in “QTL-hotspot” region for drought tolerance in chickpea (Cicer arietinum L.). Sci. Rep. 2015, 5, 15296. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Wang, D.; Shannon, G.; Zeng, A.; Orazaly, M.; Wu, C. Identification and mapping of stable QTL for protein content in soybean seeds. Mol. Breed. 2015, 35, 92. [Google Scholar] [CrossRef]
- Reinprecht, Y.; Poysa, V.W.; Yu, K.; Rajcan, I.; Ablett, G.R.; Pauls, K.P. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 2006, 49, 1510–1527. [Google Scholar] [CrossRef]
- Lu, W.; Wen, Z.; Li, H.; Yuan, D.; Li, J.; Zhang, H.; Huang, Z.; Cui, S.; Du, W. Identification of the quantitative trait loci (QTL) underlying water soluble protein content in soybean. Theor. Appl. Genet. 2013, 126, 425–433. [Google Scholar] [CrossRef]
- Bandillo, N.; Jarquin, D.; Song, Q.; Nelson, R.; Cregan, P.; Specht, J.; Lorenz, A. A Population Structure and Genome-Wide Association Analysis on the USDA Soybean Germplasm Collection. Plant Genome 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Priolli, R.H.G.; Campos, J.B.; Stabellini, N.S.; Pinheiro, J.B.; Vello, N.A. Association mapping of oil content and fatty acid components in soybean. Euphytica 2015, 203, 83–96. [Google Scholar] [CrossRef]
- Yu, Z.R.; Huang, X.T. Modern Biochemistry; Chemical Industry Press: Beijing, China, 2001; pp. 238–246. [Google Scholar]
- Zhu, Y.; Li, Y.; Zheng, X. Modern Molecular Bology, 3rd ed.; Higher Education Press: Beijing, China, 2010; pp. 126–153. [Google Scholar]
- Yu, F.Y.; Xin, X.J.; Zhang, D.J.; Zhou, S.Q.; Qiu, H.M. Dynamic accumulation of dry matter, oil and protein in soybean seed. Res. Agric. Mod. 2009, 30, 637–640. [Google Scholar]
- Qiu, L.; Wang, J.; Meng, Q. A preliminary study on accumulation characteristics of protein and fat in developing soybean seeds. Sci. Agric. Sin. 1990, 23, 28–32. [Google Scholar]
- Bruening, W.P.; Egli, D.B. Accumulation of Nitrogen and Dry Matter by Soybean Seeds with Genetic Differences in Protein Concentration. Crop Sci. 2007, 47, 359–366. [Google Scholar]
- Zhang, J.W.; Han, F.X.; Sun, J.M.; Han, G.Z.; Yu, S.X.; Yu, F.K.; Yan, S.R.; Yang, H. Genetic variation of protein and fat content in soybean mini core collections. J. Plant Genet. Resour. 2014, 15, 405–410. [Google Scholar]
- Nyquist, W.E.; Baker, R. Estimation of heritability and prediction of selection response in plant populations. Crit. Rev. Plant Sci. 1991, 10, 235–322. [Google Scholar] [CrossRef]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef]
- Stam, P. Construction of integrated genetic linkage maps by means of a new computer package: JoinMap. Plant J. 1993, 3, 739–744. [Google Scholar] [CrossRef]
- Wang, J.K. Inclusive Composite Interval Mapping of Quantitative Trait Genes. Acta Agron. Sin. 2009, 35, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J. Mixed model approaches of mapping genes for complex quantitative traits. J. Zhejiang Univ. Sci. B 1999, 33, 327–335. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, W.S. LOWESS: A Program for Smoothing Scatterplots by Robust Locally Weighted Regression. Am. Stat. 1981, 35, 54. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Zhao, X.; Jiang, H.; Feng, L.; Qu, Y.; Teng, W.; Qiu, L.; Zheng, H.; Han, Y.; Li, W. Genome-wide association and transcriptional studies reveal novel genes for unsaturated fatty acid synthesis in a panel of soybean accessions. BMC Genom. 2019, 20, 68. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Trait | Environment a | Parents | Means (%) | Variance | Range (%) | CV c (%) | Skewness | Kurtosis | F Value of Variance Analysis | H2d | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HH27 b (%) | ZGDD b (%) | Genotype (G) | Environment (E) | G*E | ||||||||||
| RILs g | Protein | 16SY | 38.55 | 45.33 | 41.54 ± 0.12 | 4.52 | 36.29~48.64 | 5.12 | 0.49 | 0.11 | 14.33 ***,f | 47.63 *** | 2.75 *** | 0.83 |
| 17SY | 38.94 | 45.17 | 41.89 ± 0.14 | 6.33 | 35.90~48.00 | 6.00 | 0.15 | −0.46 | ||||||
| 18SY | 40.15 | 45.01 | 41.93 ± 0.14 | 5.65 | 33.48~48.50 | 5.67 | 0.07 | 0.20 | ||||||
| 17XT | 44.65 | 43.68 | 41.58 ± 0.11 | 4.03 | 37.35~48.38 | 4.83 | 0.16 | −0.17 | ||||||
| 17XX | NA e | NA | 42.62 ± 0.12 | 4.14 | 37.53~47.29 | 4.77 | −0.10 | −0.34 | ||||||
| 18XX | 41.74 | 43.57 | 41.48 ± 0.17 | 5.14 | 33.19~47.23 | 5.46 | −0.18 | 0.72 | ||||||
| Oil | 16SY | 21.40 | 20.21 | 21.13 ± 0.06 | 1.17 | 18.13~24.38 | 5.11 | 0.08 | −0.08 | 18.27 *** | 1533.15 *** | 2.57 *** | 0.87 | |
| 17SY | 20.53 | 19.13 | 19.93 ± 0.07 | 1.47 | 16.12~23.17 | 6.09 | 0.00 | −0.18 | ||||||
| 18SY | 20.73 | 19.43 | 20.54 ± 0.07 | 1.37 | 17.49~24.26 | 5.69 | 0.16 | 0.04 | ||||||
| 17XT | 18.83 | 18.91 | 20.38 ± 0.06 | 1.18 | 16.76~23.14 | 5.32 | −0.20 | −0.14 | ||||||
| 17XX | NA | NA | 18.71 ± 0.06 | 1.12 | 16.19~21.44 | 5.65 | −0.04 | −0.49 | ||||||
| 18XX | 18.86 | 15.77 | 17.81 ± 0.09 | 1.39 | 15.13~20.90 | 6.62 | 0.17 | −0.09 | ||||||
| Accessions | Protein | 18SY | - | - | 42.02 ± 0.23 | 12.42 | 33.40~51.33 | 8.39 | 0.19 | −0.45 | 20.28 *** | 3.94 ** | 3.15 *** | 0.86 |
| 17XT | - | - | 42.21 ± 0.17 | 7.08 | 35.51~49.22 | 6.30 | −0.01 | −0.33 | ||||||
| 17XX | - | - | 42.10 ± 0.20 | 8.83 | 36.10~48.83 | 7.06 | 0.22 | −0.66 | ||||||
| 18XX | - | - | 42.46 ± 0.21 | 8.58 | 30.68~49.84 | 6.90 | −0.51 | 1.01 | ||||||
| Oil | 18SY | - | - | 20.73 ± 0.11 | 2.84 | 15.65~23.94 | 8.12 | −0.54 | −0.33 | 27.54 *** | 535.37 *** | 2.95 *** | 0.90 | |
| 17XT | - | - | 21.14 ± 0.09 | 2.04 | 17.78~25.35 | 6.75 | 0.10 | −0.25 | ||||||
| 17XX | - | - | 20.10 ± 0.10 | 2.31 | 16.14~23.53 | 7.56 | −0.28 | −0.48 | ||||||
| 18XX | - | - | 19.20 ± 0.12 | 2.63 | 15.29~22.93 | 8.44 | −0.03 | −0.54 | ||||||
| Trait | QTL Name | Chr. (LG) a | Method b | Location (cM) | Marker Interval (cM) | Physical Region (bp) | LOD/F Value c | PVE (%) d | Additive Effect e | Environment f | Reference g |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein | qPro1-1 | 1 (D1a) | ICIM | 1 | 0~1.5 | 1488983~1566969 | 2.90~6.49 | 2.73~5.52 | −0.41~−0.49 | 1, 3 | Seed protein 3-4 |
| MCIM | 7.5 | 6.5~8.4 | 2605140~2852655 | 5.00 | - | −0.24 | - | ||||
| qPro6-1 | 6 (C2) | ICIM | 33 | 32.5~33.5 | 5836780~5931027 | 4.93 | 4.17 | 0.42 | 1 | cqSeed protein-005, Seed protein 30-5 | |
| MCIM | 32.1 | 31.7~32.2 | 5609477~5632020 | 5.10 | - | 0.26 | - | ||||
| qPro9-1 | 9 (K) | ICIM | 62~68 | 59.5 ~70.5 | 38117239~41020511 | 3.63~8.96 | 3.43~8.40 | 0.38~0.41 | 1, 2, 4 | Seed protein 33-3, Seed protein 34-6 | |
| MCIM | 61.7 | 60.7~62.7 | 38117239~39894925 | 10.40 | - | 0.42 | - | ||||
| qPro12-1 | 12 (H) | ICIM | 105~107 | 103.5~107 | 38776571~39867556 | 3.64~3.78 | 3.05~3.58 | 0.36~0.46 | 1, 3 | Seed protein 6-1 | |
| qPro15-1 | 15 (E) | ICIM | 23~26 | 22.5~26.5 | 2691560~3476238 | 9.00~19.05 | 13.40~17.81 | 0.79~0.89 | 3, 4, 5 | Seed protein 30-3 | |
| MCIM | 26.2 | 26.1~26.3 | 3311604~3350307 | 26.20 | - | 0.52 | - | ||||
| qPro18-1 | 18 (G) | ICIM | 22 | 21.5~22.5 | 5577815~5618246 | 6.06 | 5.80 | −0.59 | 3 | Seed protein 47-6 | |
| MCIM | 22.3 | 22.0~23.3 | 5618246~5979842 | 4.80 | - | −0.24 | - | ||||
| qPro20-1 | 20 (I) | ICIM | 54~61 | 48.5~62.5 | 34734798~37115770 | 6.14~8.62 | 7.24~9.39 | 0.56~0.75 | 1, 2, 3 | Seed protein 26-5, Seed protein 34-11 | |
| MCIM | 58.7 | 57.7~59.7 | 36089907~37115770 | 15.30 | - | 0.34 | - | ||||
| Oil | qOil1-1 | 1 (D1a) | ICIM | 1~10 | 0~14.5 | 1488983~3316074 | 2.85~2.99 | 1.56~1.67 | 0.14~0.17 | 1, 3 | Seed oil 23-2 |
| qOil2-1 | 2 (D1b) | ICIM | 121 | 116.5~126.5 | 43783867~45442501 | 2.56 | 3.64 | 0.19 | 5 | cqSeed oil-014, Seed oil 39-6 | |
| MCIM | 112.8 | 111.8~113.1 | 42545649~43226016 | 5.40 | - | 0.14 | - | ||||
| qOil3-1 | 3 (N) | ICIM | 52~54 | 50.5 ~55.5 | 33430615~34447425 | 2.62~2.72 | 1.86~2.78 | 0.15~0.21 | 2, 4 | Seed oil 43-30 | |
| qOil5-1 | 5 (A1) | ICIM | 117~126 | 116.5~126 | 40003403~41813079 | 3.89~35.35 | 7.04~23.98 | −0.27~−0.63 | 1, 2, 3, 4, 5, 6 | Seed oil 39-1, Seed oil 35-2, Seed oil 13-1 | |
| MCIM | 125.9 | 124.9~126.4 | 40566361~41813079 | 25.10 | - | −0.40 | - | ||||
| qOil6-1 | 6 (C2) | ICIM | 50~52 | 44.5~52.5 | 8313637~9652882 | 3.24~4.09 | 4.16~6.68 | −0.26~-0.34 | 2, 6 | cqSeed oil-016 | |
| qOil15-1 | 15 (E) | ICIM | 26 | 25.5~26.5 | 2691560~3240013 | 19.25 | 15.97 | −0.44 | 4 | cqSeed oil-007, Seed oil 2-3 | |
| MCIM | 26.2 | 26.1~26.3 | 3311604~3350307 | 28.40 | - | −0.28 | - | ||||
| qOil17-1 | 17 (D2) | ICIM | 41 | 39.5~41.5 | 7100839~8674575 | 3.19 | 1.72 | 0.18 | 3 | Seed oil 23-3 | |
| MCIM | 45.1 | 44.1~46.1 | 7453724~9120650 | 5.30 | - | 0.13 | - | ||||
| qOil20-1 | 20 (I) | ICIM | 56~62 | 51.5~62.5 | 34734798~37115770 | 3.93~5.20 | 2.30~2.87 | −0.20 | 1, 3 | Seed oil 27-4, Seed oil 24-6 |
| Chr. a | Trait | Method b | Environment c | Markers Interval (cM)/SNP Number d | SNP Loci Region/Location (bp) | LOD/F Value e | PVE (%) f | Additive Effect g |
|---|---|---|---|---|---|---|---|---|
| 2 | Oil | ICIM | 5 | 116.5~126.5 | 43783867~45442501 | 2.56 | 3.64 | 0.19 |
| GWAS | 4 | 1 | 45017225 | - | - | - | ||
| 6 | Protein | ICIM | 1 | 32.5~33.5 | 5836780~5931027 | 4.93 | 4.17 | 0.42 |
| MCIM | - | 31.7~32.2 | 5609477~5632020 | 5.10 | - | 0.26 | ||
| Oil | GWAS | 3, 5 | 2 | 5713084~5992538 | - | - | - | |
| 9 | Protein | ICIM | 1, 2, 4 | 59.5~70.5 | 38117239~41020511 | 3.63~8.96 | 3.43~8.40 | 0.38~0.41 |
| MCIM | - | 60.7~62.7 | 38117239~39894925 | 10.40 | - | 0.42 | ||
| Oil | GWAS | 6 | 1 | 40301013 | - | - | - | |
| 20 | Protein | ICIM | 1, 2, 3 | 48.5~62.5 | 34734798~37115770 | 6.14~8.62 | 7.24~9.39 | 0.56~0.75 |
| MCIM | - | 57.7~59.7 | 36089907~37115770 | 15.30 | - | 0.34 | ||
| GWAS | 5, 6 | 5 | 34990940~35578946 | - | - | - | ||
| Oil | ICIM | 1, 3 | 51.5~62.5 | 34734798~37115770 | 3.93~5.20 | 2.30~2.87 | −0.20 | |
| GWAS | 5 | 4 | 34801441~35512580 | - | - | - |
| Trait | Gene | Start (bp) | Stop (bp) | Annotation |
|---|---|---|---|---|
| Oil | Glyma.15g034100 | 2722009 | 2727957 | acyltransferase activity, diacylglycerol and triacylglycerol biosynthesis |
| Glyma.15g034400 | 2740960 | 2746344 | aldehyde dehydrogenase family, aldehyde dehydrogenase [NAD (P)+] activity | |
| Glyma.15g034600 | 2765299 | 2770528 | drug transmembrane transport, associated with the transport of citric acid and malic acid | |
| Glyma.15g042500 | 3339156 | 3341447 | fatty acid, lipid biosynthetic process, transferase activity, 3-oxoacyl-[acyl-carrier-protein] synthase activity | |
| Glyma.20g107600 | 35025241 | 35029762 | Arabidopsis phospholipase-like protein, regulation of gene expression | |
| Glyma.20g107800 | 35038552 | 35042864 | hydroxypyruvate reductase, glycerate dehydrogenase, glyoxylate reductase, NADP activity | |
| Glyma.20g108800 | 35116048 | 35118928 | mitochondrial pyruvate transmembrane transport | |
| Glyma.20g109900 | 35222837 | 35228540 | lipid metabolic process, steroid biosynthetic process, mevalonate pathway | |
| Glyma.20g110000 | 35229423 | 35231861 | acetyltransferase activity | |
| Glyma.20g111000 | 35315630 | 35319063 | fatty acid desaturase, lipid metabolic process | |
| Protein | Glyma.15g033200 | 2656030 | 2657795 | structural constituent of ribosome, 28S ribosomal protein |
| Glyma.15g039000 | 3068347 | 3075209 | 60S ribosomal protein | |
| Glyma.15g040100 | 3164697 | 3168839 | ACT domain-containing protein, metabolic process like protein synthesis and degradation. | |
| Glyma.15g041500 | 3255042 | 3256599 | ribosomal large subunit assembly, 60S ribosomal protein L23 | |
| Glyma.15g042300 | 3307111 | 3308840 | structural constituent of ribosome, 60S ribosomal protein L35 | |
| Glyma.20g103200 | 34605252 | 34609867 | tryptophan biosynthetic process, anthranilate synthase activity | |
| Glyma.20g105300 | 34757381 | 34771672 | ACT-like protein, serine/threonine kinase family protein | |
| Glyma.20g106200 | 34862155 | 34865242 | amino acid transmembrane transport | |
| Glyma.20g106900 | 34962043 | 34967985 | translation initiation factor 3 (IF-3) family protein | |
| Glyma.20g109600 | 35200934 | 35205885 | ubiquitin-dependent protein catabolic process, proteasome complex, proteolysis activity | |
| Glyma.20g110100 | 35232344 | 35233758 | nutrient reservoir activity, cupins superfamily protein, storage protein | |
| Glyma.20g110400 | 35261156 | 35268971 | ACT domain-containing protein, metabolic process like protein synthesis and degradation. | |
| Glyma.20g111900 | 35396205 | 35400722 | cationic amino acid transporter, amino acid transmembrane transporter activity | |
| Protein/Oil | Glyma.20g106800 | 34935548 | 34940516 | protein dephosphorylation, phosphatase activity, pyruvate dehydrogenase |
| Glyma.20g110200 | 35235204 | 35239070 | lipoate biosynthetic, radical SAM superfamily protein, transferase activity |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Wu, T.; Wang, L.; Jiang, B.; Zhen, C.; Yuan, S.; Hou, W.; Wu, C.; Han, T.; Sun, S. A Combined Linkage and GWAS Analysis Identifies QTLs Linked to Soybean Seed Protein and Oil Content. Int. J. Mol. Sci. 2019, 20, 5915. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235915
Zhang T, Wu T, Wang L, Jiang B, Zhen C, Yuan S, Hou W, Wu C, Han T, Sun S. A Combined Linkage and GWAS Analysis Identifies QTLs Linked to Soybean Seed Protein and Oil Content. International Journal of Molecular Sciences. 2019; 20(23):5915. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235915
Chicago/Turabian StyleZhang, Tengfei, Tingting Wu, Liwei Wang, Bingjun Jiang, Caixin Zhen, Shan Yuan, Wensheng Hou, Cunxiang Wu, Tianfu Han, and Shi Sun. 2019. "A Combined Linkage and GWAS Analysis Identifies QTLs Linked to Soybean Seed Protein and Oil Content" International Journal of Molecular Sciences 20, no. 23: 5915. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235915