Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
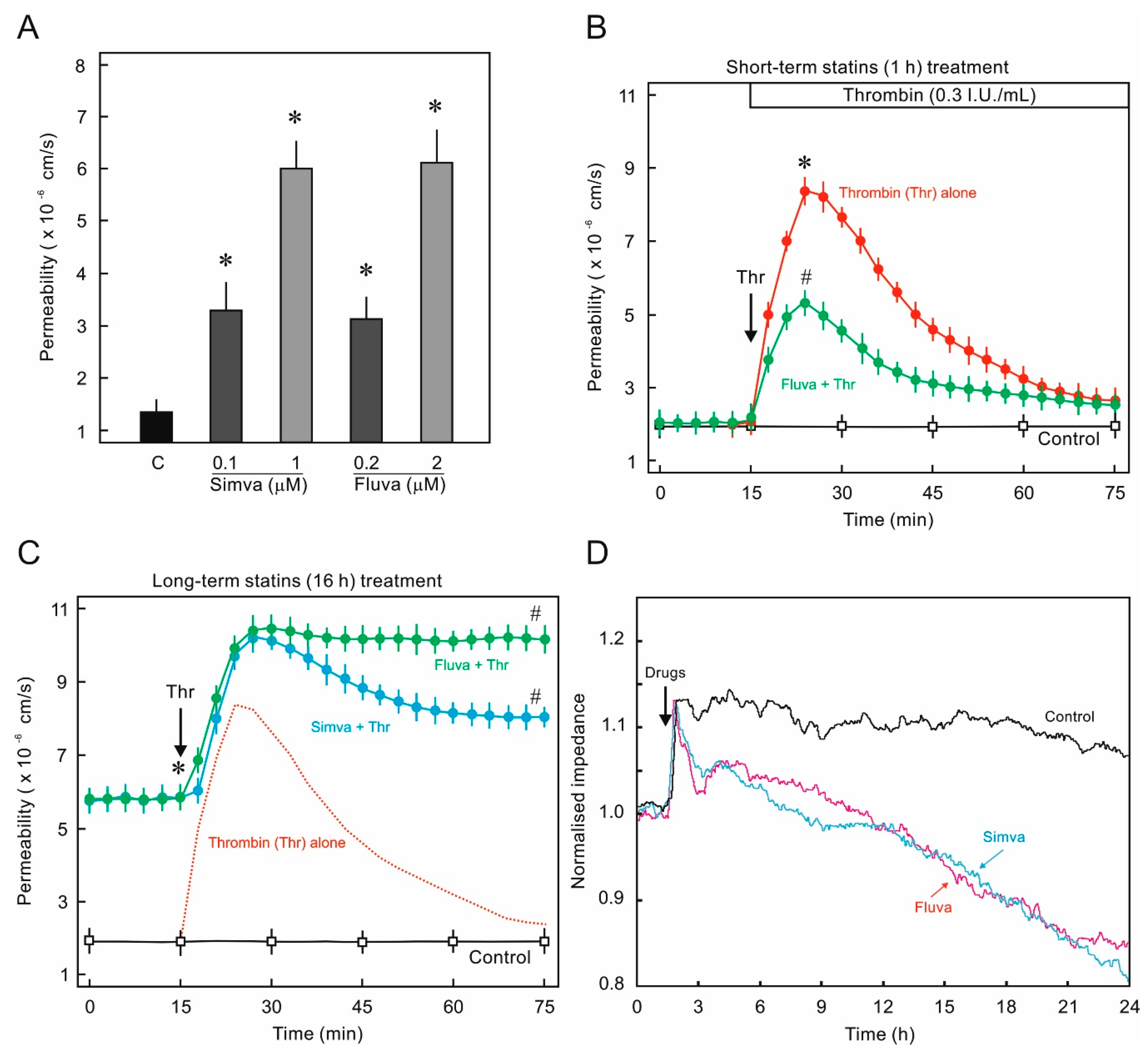
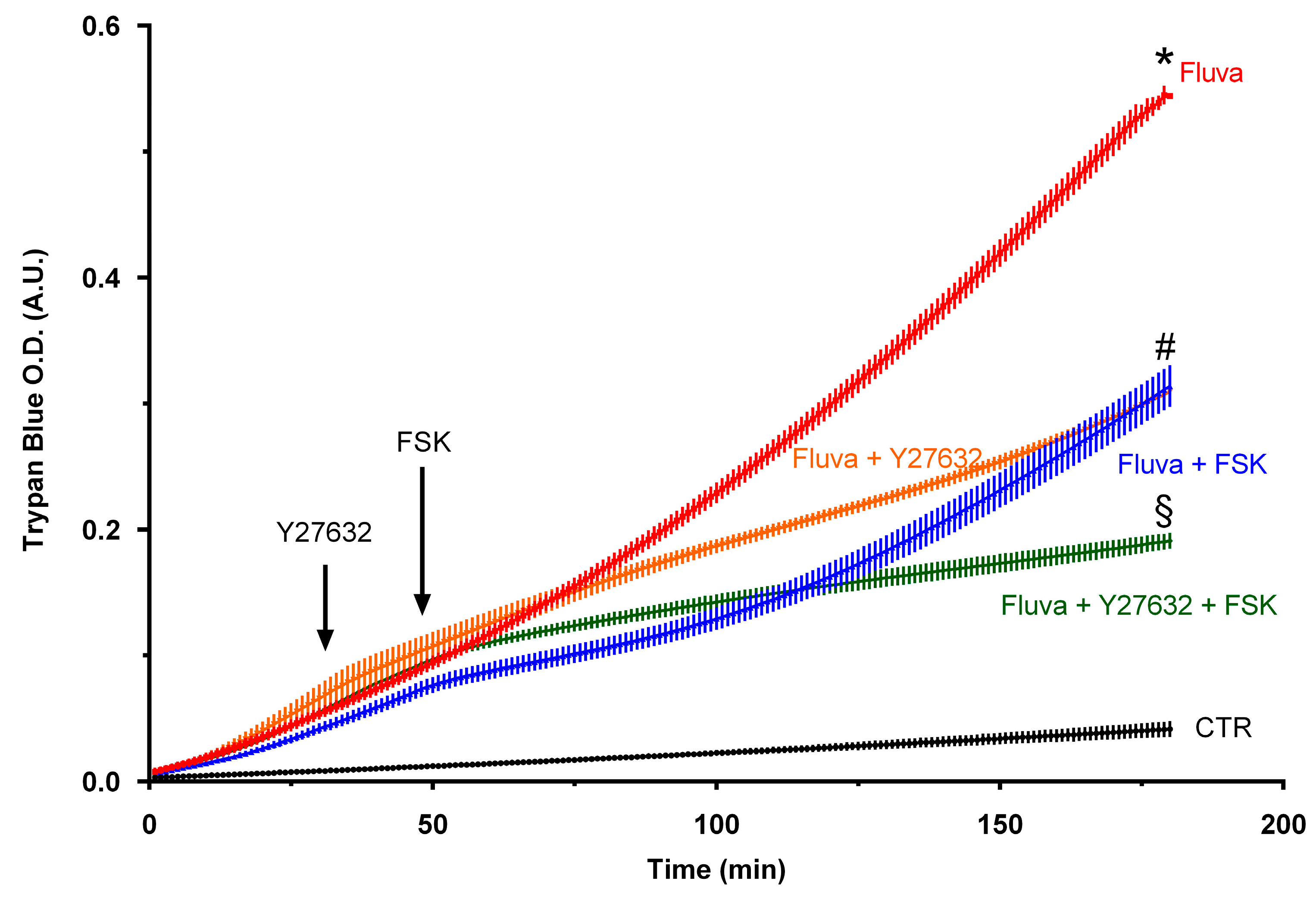
2.1. Statins Suppress Endothelial Barrier Function
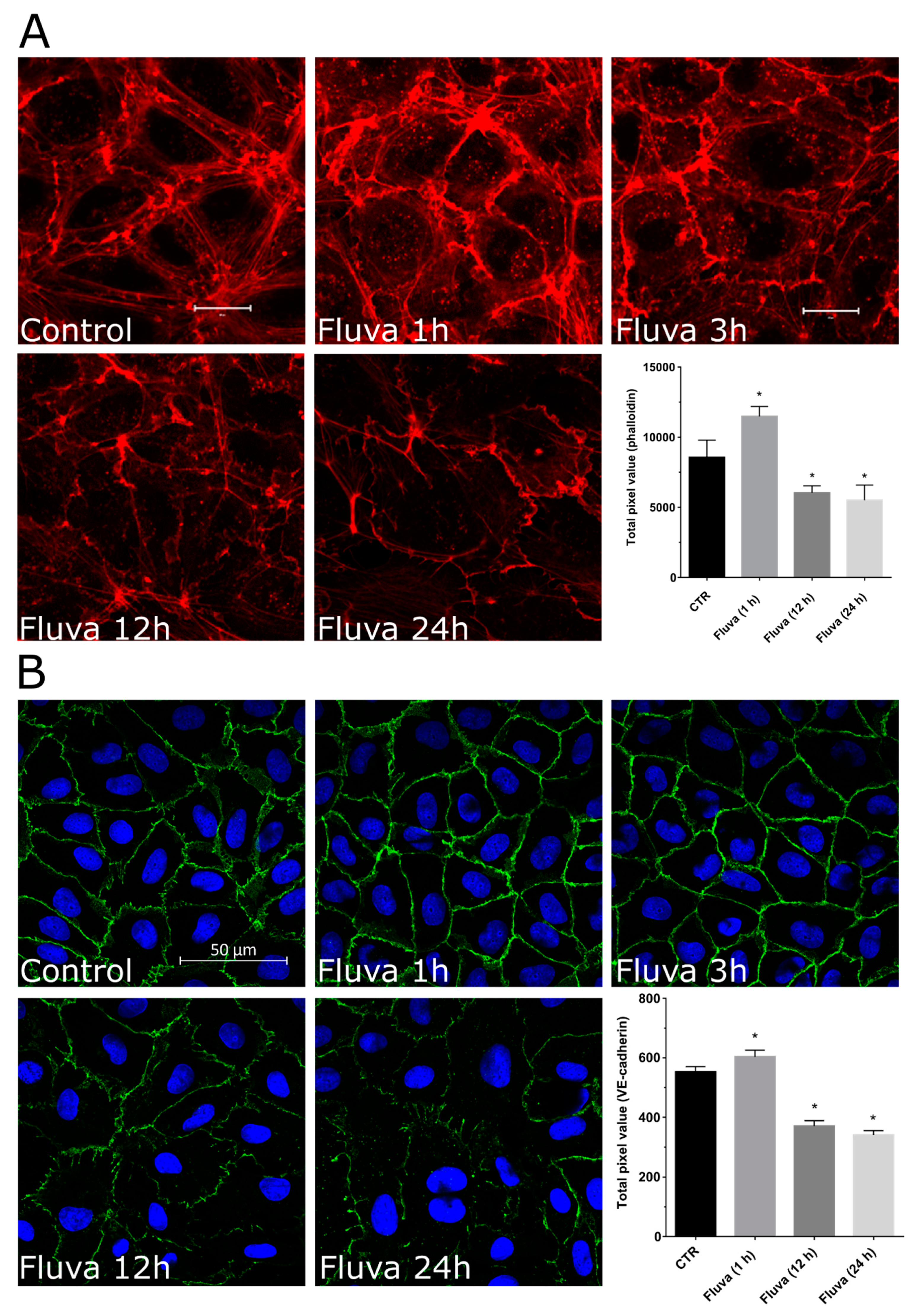
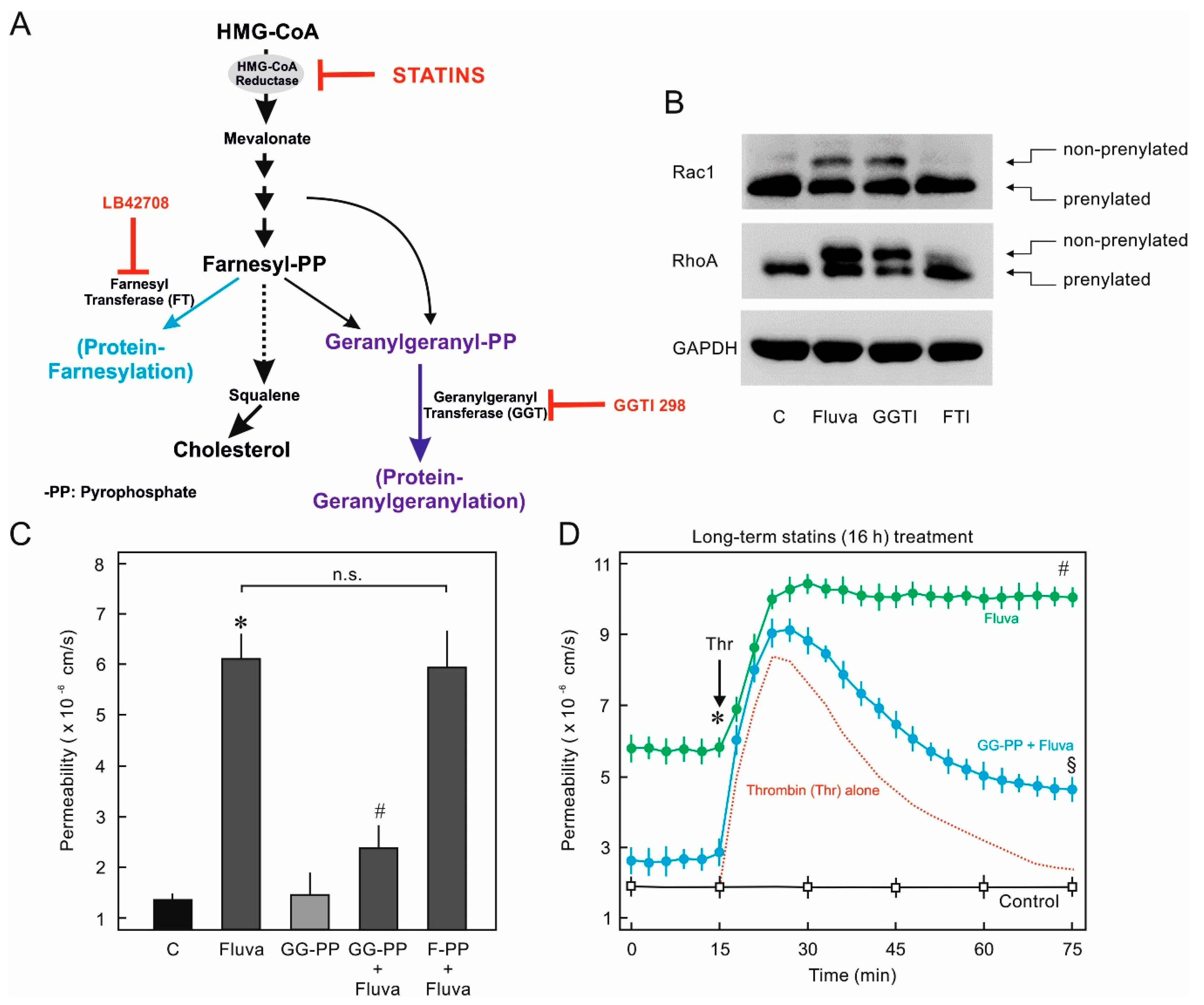
2.2. Protein Prenylation and Endothelial Barrier Properties
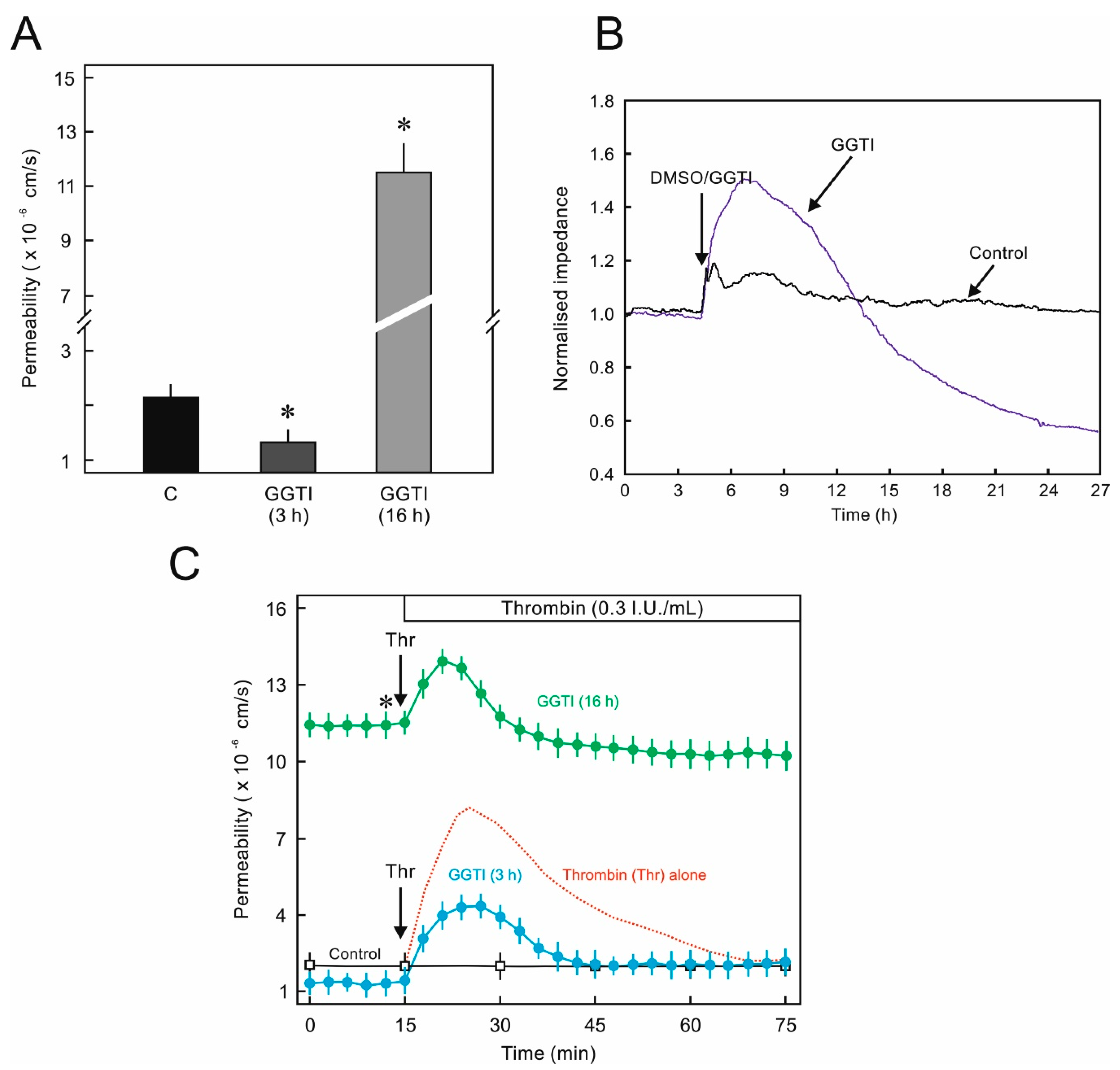
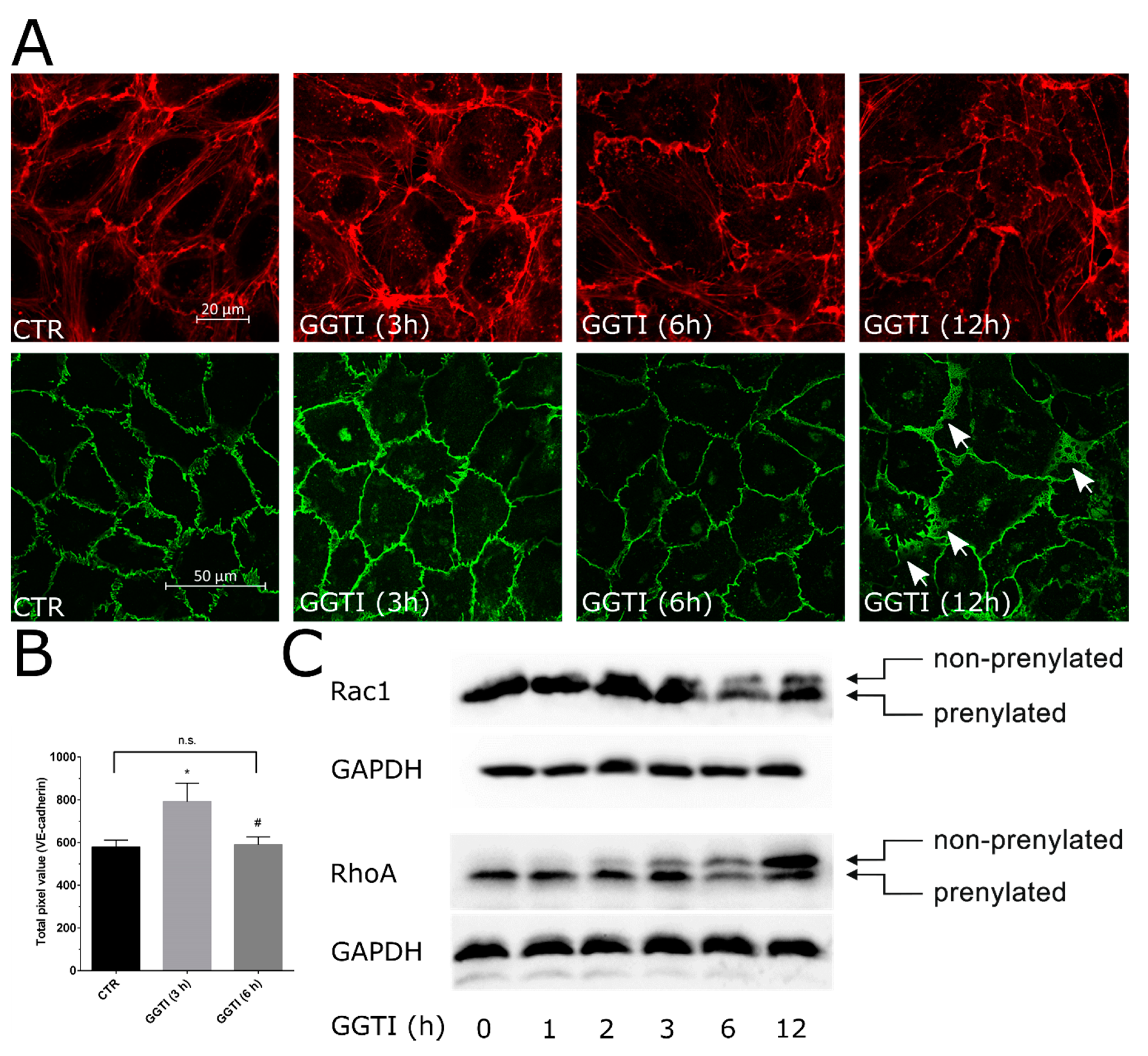
2.3. Biphasic Effect of GGT Inhibitor on EC Permeability and Cellular Junctions
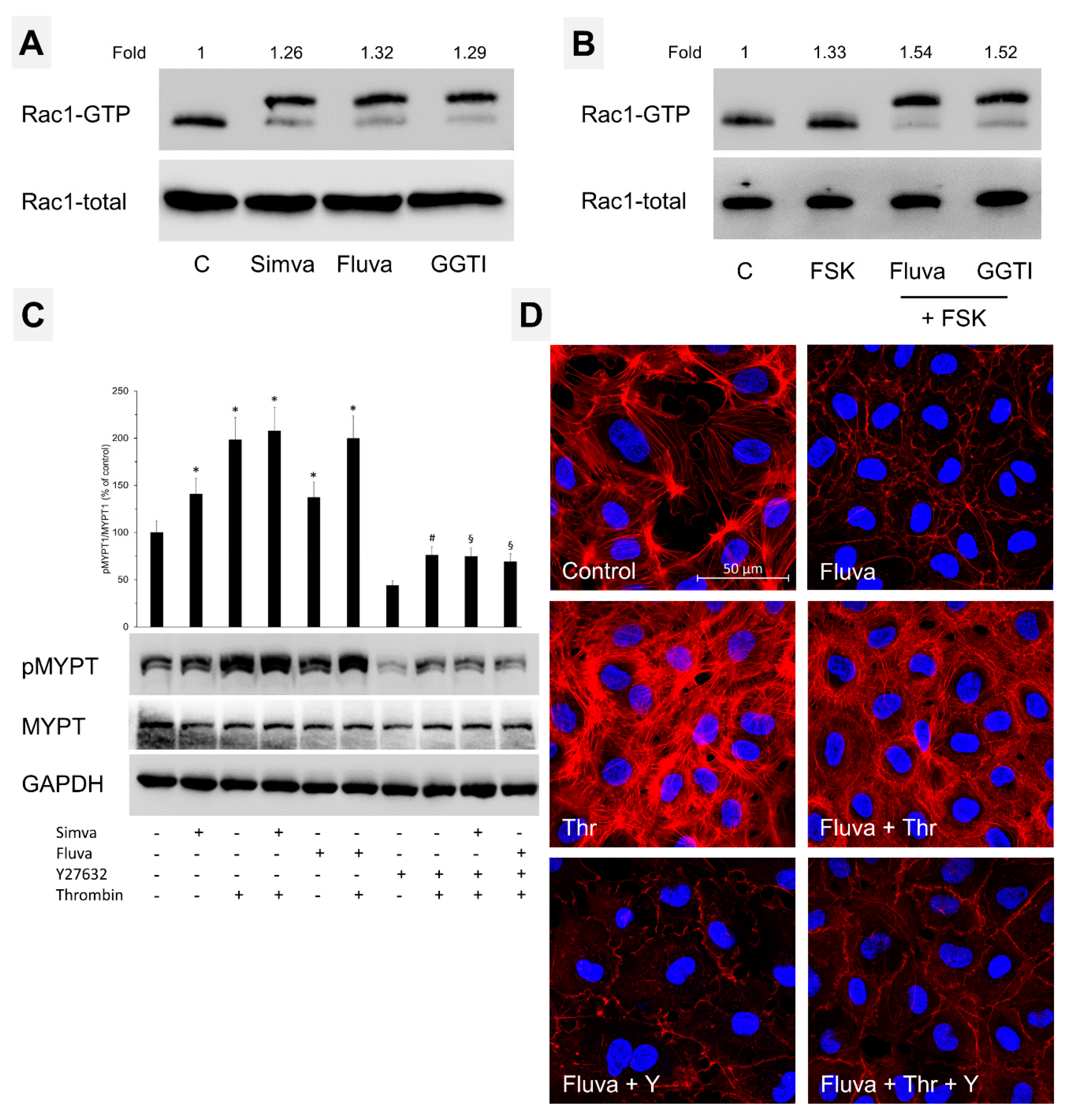
2.4. Nonprenylated Rho GTPases May Be Activated
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Experimental Protocols
4.4. Immunocytochemistry and Confocal Microscopy
4.5. Endothelial Barrier Properties
4.6. Western Blotting
4.7. GTPase Prenylation Processing Assay
4.8. Rac1 Pulldown Assay
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AJ | Adherens junctions |
| ECs | Endothelial cells |
| FPP | Farnesyl pyrophosphate |
| FT | Farnesyl transferase |
| GG-PP | Geranylgeranyl pyro phosphate |
| GGT | Geranylgeranyl transferase |
| HUVEC | Human umbilical vein endothelial cells |
| HMG CoA reductase | 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase |
References
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Corada, M.; Caveda, L.; Breviario, F.; Ayalon, O.; Geiger, B.; Dejana, E. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J. Cell Biol. 1995, 129, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciak-Stothard, B.; Tsang, L.Y.; Haworth, S.G. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L749–L760. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Tsang, L.Y.; Paleolog, E.; Hall, S.M.; Haworth, S.G. Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1173–L1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouret-Craviari, V.; Boquet, P.; Pouyssegur, J.; Van Obberghen-Schilling, E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol. Biol. Cell 1998, 9, 2639–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciak-Stothard, B.; Entwistle, A.; Garg, R.; Ridley, A.J. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J. Cell. Physiol. 1998, 176, 150–165. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Potempa, S.; Eichholtz, T.; Ridley, A.J. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 2001, 114, 1343–1355. [Google Scholar]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Casey, P.J. Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2014, 2, 12175–12187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broman, M.T.; Mehta, D.; Malik, A.B. Cdc42 regulates the restoration of endothelial adherens junctions and permeability. Trends Cardiovasc. Med. 2007, 17, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martin, L.; Marcos-Ramiro, B.; Bigarella, C.L.; Graupera, M.; Cain, R.J.; Reglero-Real, N.; Jimenez, A.; Cernuda-Morollon, E.; Correas, I.; Cox, S.; et al. Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e90–e102. [Google Scholar] [CrossRef] [Green Version]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Van Buul, J.D.; Geerts, D.; Huveneers, S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell Adhes. Migr. 2014, 8, 108–124. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, M.; Liao, J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Palsuledesai, C.C.; Distefano, M.D. Protein prenylation: Enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Boissonneault, G.A.; Hennig, B.; Ouyang, C.M. Oxysterols, cholesterol biosynthesis, and vascular endothelial cell monolayer barrier function. Proc. Soc. Exp. Biol. Med. 1991, 196, 338–343. [Google Scholar] [CrossRef]
- Van Nieuw Amerongen, G.P.; Vermeer, M.A.; Negre-Aminou, P.; Lankelma, J.; Emeis, J.J.; van Hinsbergh, V.W. Simvastatin improves disturbed endothelial barrier function. Circulation 2000, 102, 2803–2809. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, J.R.; Dudek, S.M.; Birukov, K.G.; Ye, S.Q.; Grigoryev, D.N.; Girgis, R.E.; Garcia, J.G. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am. J. Respir. Cell Mol. Biol. 2004, 30, 662–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, J.R.; Barnard, J.W.; Grigoryev, D.N.; Ma, S.F.; Tuder, R.M.; Garcia, J.G. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1026–L1032. [Google Scholar] [CrossRef] [PubMed]
- Van de Visse, E.P.; van der Heijden, M.; Verheij, J.; van Nieuw Amerongen, G.P.; van Hinsbergh, V.W.; Girbes, A.R.; Groeneveld, A.B. Effect of prior statin therapy on capillary permeability in the lungs after cardiac or vascular surgery. Eur. Respir. J. 2006, 27, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef]
- Zhang, H.; Plutzky, J.; Skentzos, S.; Morrison, F.; Mar, P.; Shubina, M.; Turchin, A. Discontinuation of statins in routine care settings: A cohort study. Ann. Intern. Med. 2013, 158, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.Y.S. Clinical quiz: An old lady with oedema of all four limbs. Hong Kong, J. Emerg. Med. 2009, 16, 179–181. [Google Scholar] [CrossRef]
- Nisly, S.A.; Kara, A.; Knight, T.B. Simvastatin: A risk factor for angioedema? J. Pharm. Technol. 2013, 29, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Kusama, T.; Mukai, M.; Tatsuta, M.; Matsumoto, Y.; Nakamura, H.; Inoue, M. Selective inhibition of cancer cell invasion by a geranylgeranyltransferase-I inhibitor. Clin. Exp. Metastasis 2003, 20, 561–567. [Google Scholar] [CrossRef]
- Dunford, J.E.; Rogers, M.J.; Ebetino, F.H.; Phipps, R.J.; Coxon, F.P. Inhibition of protein prenylation by bisphosphonates causes sustained activation of Rac, Cdc42, and Rho GTPases. J. Bone Miner. Res. 2006, 21, 684–694. [Google Scholar] [CrossRef]
- Virtanen, S.S.; Sandholm, J.; Yegutkin, G.; Kalervo Vaananen, H.; Harkonen, P.L. Inhibition of GGTase-I and FTase disrupts cytoskeletal organization of human PC-3 prostate cancer cells. Cell Biol. Int. 2010, 34, 815–826. [Google Scholar] [CrossRef]
- Khan, O.M.; Ibrahim, M.X.; Jonsson, I.M.; Karlsson, C.; Liu, M.; Sjogren, A.K.; Olofsson, F.J.; Brisslert, M.; Andersson, S.; Ohlsson, C.; et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Investig. 2011, 121, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.M.; Akula, M.K.; Skalen, K.; Karlsson, C.; Stahlman, M.; Young, S.G.; Boren, J.; Bergo, M.O. Targeting GGTase-I activates RHOA, increases macrophage reverse cholesterol transport, and reduces atherosclerosis in mice. Circulation 2013, 127, 782–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, C.E.; Lum, H.; Schaphorst, K.L.; Verin, A.D.; Garcia, J.G. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium 2000, 7, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Spindler, V.; Werthmann, R.C.; Bunemann, M.; Waschke, J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J. Cell. Physiol. 2009, 220, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ponce, A.; Chanez Paredes, S.; Castro Ochoa, K.F.; Schnoor, M. Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family. Tissue Barriers 2016, 4, e1228439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mariscal, A.; Li, H.; Pedersen, E.; Peyrollier, K.; Ryan, K.M.; Stanley, A.; Quondamatteo, F.; Brakebusch, C. Loss of RhoA promotes skin tumor formation and invasion by upregulation of RhoB. Oncogene 2018, 37, 847–860. [Google Scholar] [CrossRef]
- Aslam, M.; Pfeil, U.; Gündüz, D.; Rafiq, A.; Kummer, W.; Piper, H.M.; Noll, T. Intermedin (adrenomedullin2) stabilises endothelial barrier and antagonises thrombin-induced barrier failure. Br. J. Pharmacol. 2012, 165, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.M.; Rowell, C.; Ackermann, K.; Kowalczyk, J.J.; Lewis, M.D. Peptidomimetic inhibitors of Ras farnesylation and function in whole cells. J. Biol. Chem. 1993, 268, 18415–18418. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Gündüz, D.; Hamm, C.W. Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. Int. J. Mol. Sci. 2020, 21, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010002
Aslam M, Troidl C, Tanislav C, Rohrbach S, Gündüz D, Hamm CW. Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. International Journal of Molecular Sciences. 2020; 21(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010002
Chicago/Turabian StyleAslam, Muhammad, Christian Troidl, Christian Tanislav, Susanne Rohrbach, Dursun Gündüz, and Christian W. Hamm. 2020. "Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function" International Journal of Molecular Sciences 21, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010002