Identification of Nanobodies against the Acute Myeloid Leukemia Marker CD33

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
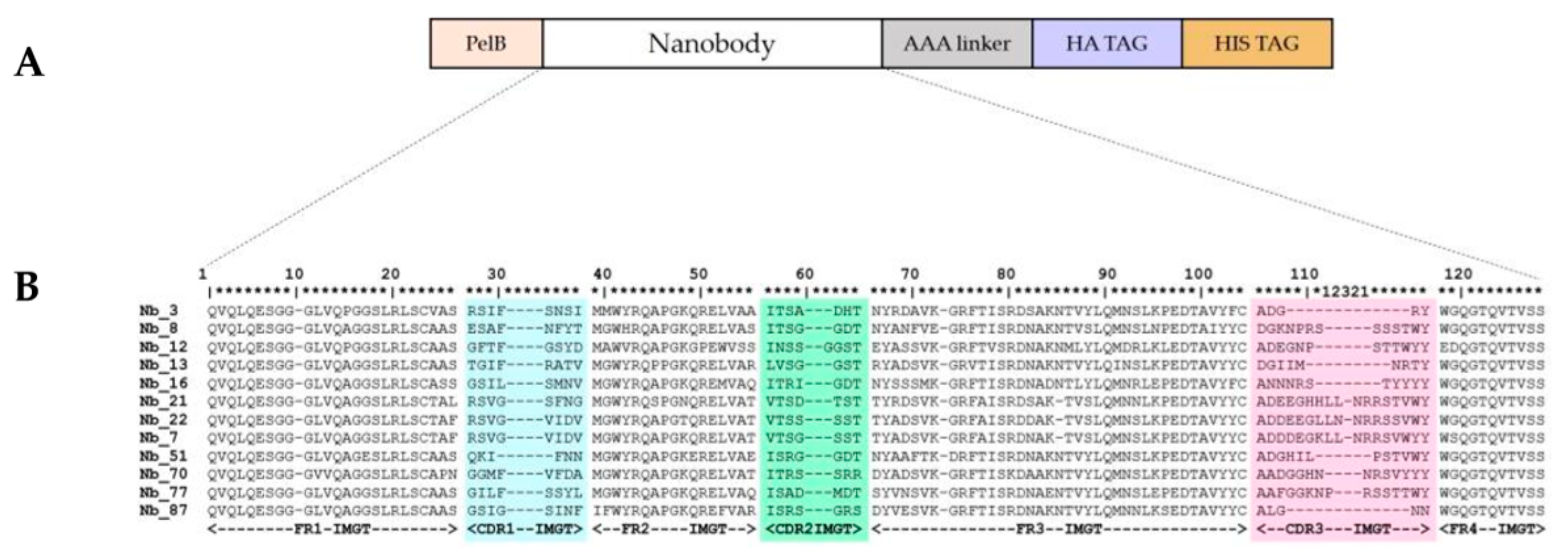
2.1. Nanobodies Were Generated against Recombinant Human CD33
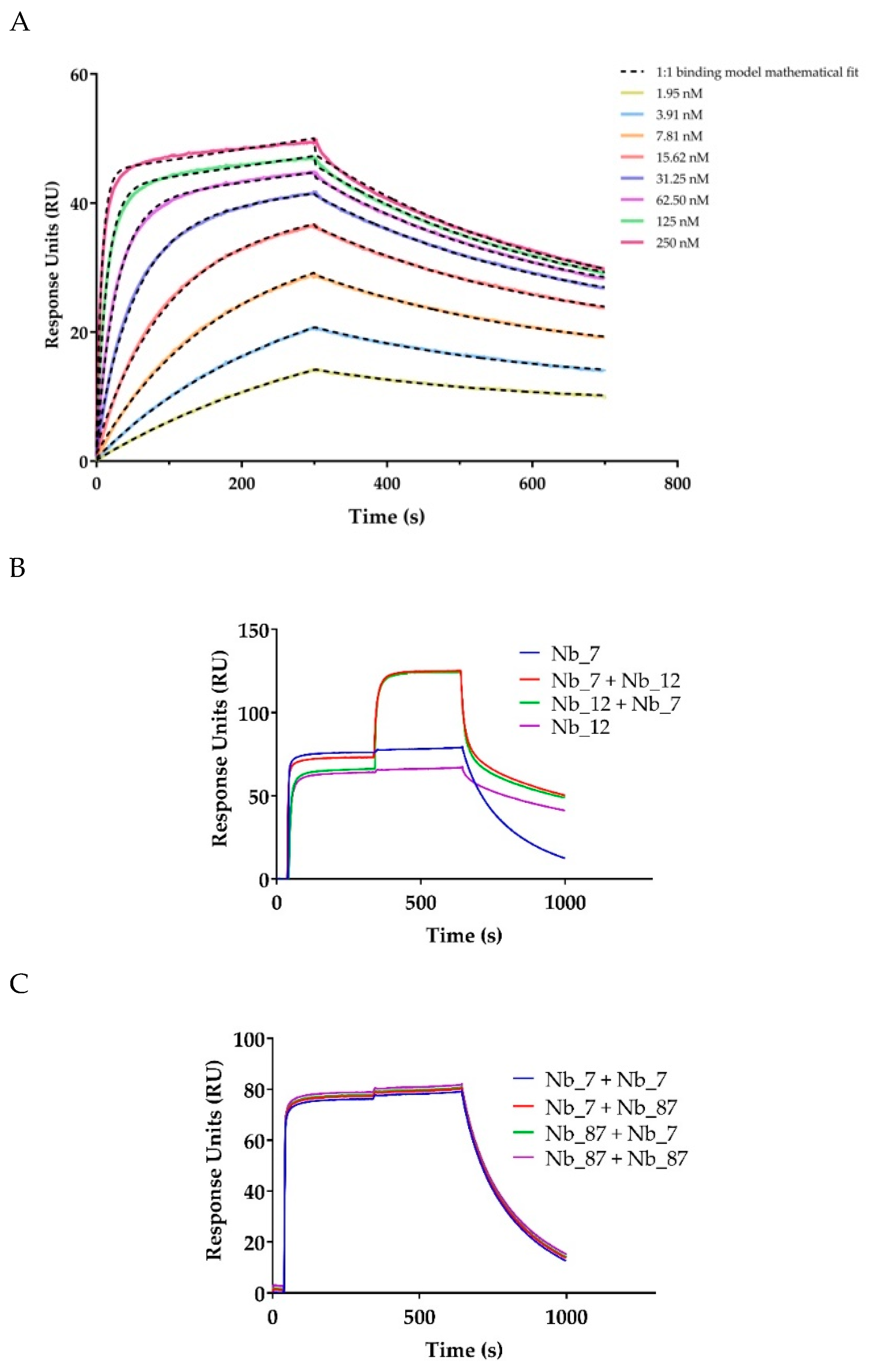
2.2. In Vitro Characterization of the Anti-CD33 Nanobodies
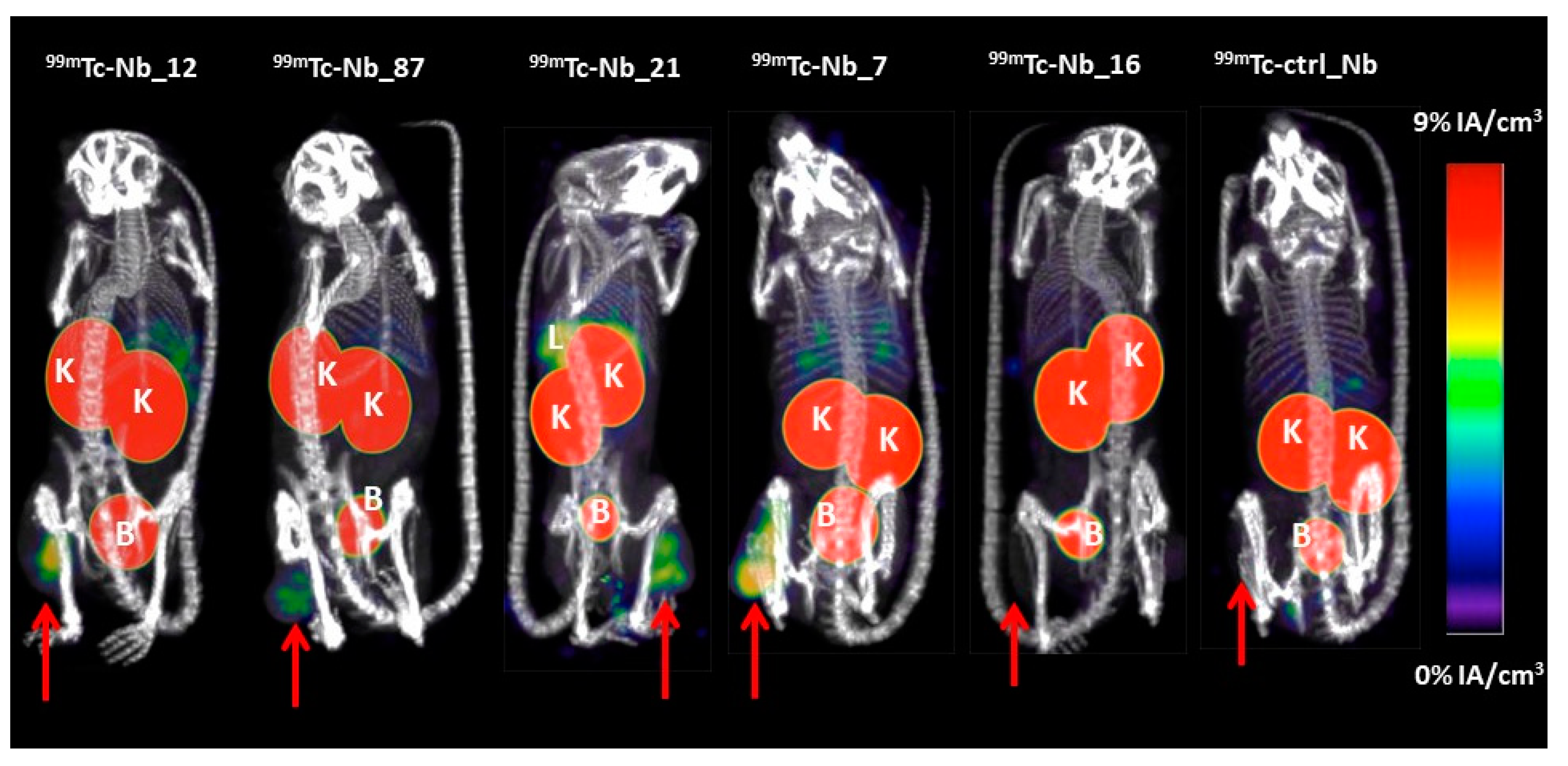
2.3. Biodistribution of 99mTc-Labeled Nbs
3. Discussion
4. Material and Methods
4.1. Anti-CD33 Nanobodies Generation, Selection, and Expression
4.2. THP-1 Maintenance and Preparation
4.3. Cell-Binding Assay
4.4. Leukemogenic Activity Assay
4.5. Surface Plasmon Resonance—Affinity Determination and Epitope Binning
4.6. Thermal Stability
4.7. Preparation of 99mTc Labeled Nbs
4.8. Animal Model
4.9. In Vivo Biodistribution of Radiolabeled Nbs
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, D.; Bray, F.; Brewster, D.H.; Mbalawa, C.G.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Ferlay, J. (Eds.) Cancer Incidence in Five Continents; International Agency for Research on Cancer: Lyon, France, 2014; Volume X. [Google Scholar]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Hansrivijit, P.; Gale, R.P.; Barrett, J.; Ciurea, S.O. Cellular therapy for acute myeloid Leukemia—Current status and future prospects. Blood Rev. 2019, 37, 1–12. [Google Scholar] [CrossRef]
- Sarkozy, C.; Gardin, C.; Gachard, N.; Merabet, F.; Turlure, P.; Malfuson, J.V.; Pautas, C.; Micol, J.B.; Thomas, X.; Quesnel, B.; et al. Outcome of older patients with acute myeloid leukemia in first relapse. Am. J. Hematol. 2013, 88, 758–764. [Google Scholar] [CrossRef]
- Buss, E.C.; Ho, A.D. Leukemia stem cells. Int. J. Cancer 2011, 129, 2328–2336. [Google Scholar] [CrossRef]
- Nagler, E.; Xavier, M.F.; Frey, N. Updates in immunotherapy for acute myeloid leukemia. Transl. Cancer Res. 2017, 6, 86–92. [Google Scholar] [CrossRef]
- Morsink, L.M.; Walter, R.B. Novel monoclonal antibody-based therapies for acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2019, 32, 116–126. [Google Scholar] [CrossRef]
- Rao, A.V.; Schmader, K. Monoclonal antibodies as targeted therapy in hematologic malignancies in older adults. Am. J. Geriatr. Pharm. 2007, 5, 247–262. [Google Scholar] [CrossRef]
- McMillan, S.J.; Crocker, P.R. CD33-related sialic-acid-binding immunoglobulin-like lectins in health and disease. Carbohydr. Res. 2008, 343, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Jamil, M.O.; Al-Obaidi, M.; Pereboeva, L.; Salzman, D.; Erba, H.P.; Lamb, L.S.; Bhatia, R.; Mineishi, S.; Di Stasi, A. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor T-cells for acute myeloid leukemia. PLoS ONE 2016, 11, e0166891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, F.; Elsadek, B. Clinical impact of serum proteins on drug delivery. J. Control. Release 2012, 161, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Rahbarizadeh, F.; Ahmadvand, D.; Sharifzadeh, Z. Nanobody: An old concept and new vehicle for immunotargeting. Immunol. Invest. 2011, 40, 299–338. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Bajyana Songa, E.; Bendahman, N.; Hamers, R. Naturally occuring anitbodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; Xavier, C.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert Opin. Drug Deliv. 2014, 11, 1939–1954. [Google Scholar] [CrossRef]
- Krasniqi, A.; D’Huyvetter, M.; Xavier, C.; Van der Jeught, K.; Muyldermans, S.; Van Der Heyden, J.; Lahoutte, T.; Tavernier, J.; Devoogdt, N. Theranostic Radiolabeled Anti-CD20 sdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2017, 16, 2828–2840. [Google Scholar] [CrossRef] [Green Version]
- D’Huyvetter, M.; Vincke, C.; Xavier, C.; Aerts, A.; Impens, N.; Baatout, S.; De Raeve, H.; Muyldermans, S.; Caveliers, V.; Devoogdt, N.; et al. Targeted Radionuclide Therapy with A 177 Lu-labeled Anti-HER2 Nanobody. Theranostics 2014, 4, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Vincke, C.; Gutiérrez, C.; Wernery, U.; Devoogdt, N.; Hassanzadeh-ghassabeh, G.; Muyldermans, S. Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. In Methods in Molecular Biology—Antibody Engineering; Chames, P., Ed.; Humana Press: Totowa, NJ, USA, 2012; Volume 907, pp. 145–176. [Google Scholar]
- Muyldermans, S.; Atarhouch, T.; Saldanha, J.; Barbosa, J.A.R.G.; Hamers, R. Sequence and structure of vh domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. Des. Sel. 1994, 7, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, M.P.; Pommié, C.; Ruiz, M.; Giudicelli, V.; Foulquier, E.; Truong, L.; Thouvenin-Contet, V.; Lefranc, G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 2003, 27, 55–77. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Ghahroudi, M.A.; Decanniere, K.; Loris, R.; Kinne, J.; Wyns, L.; Muyldermans, S. Strong in vivo maturation compensates for structurally restricted H3 loops in antibody repertoires. J. Biol. Chem. 2005, 280, 14114–14121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchouate Gainkam, L.O.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Xavier, C.; Boerman, O.; Muyldermans, S.; Bossuyt, A.; Lahoutte, T. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol. Imaging 2011, 6, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, M.; D’Huyvetter, M.; Lahoutte, T.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Kronenberger, P.; Wernery, U.; Muyldermans, S.; Devoogdt, N.; et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia 2014, 28, 444–447. [Google Scholar] [CrossRef]
- Thein, M.S.; Ershler, W.B.; Jemal, A.; Yates, J.W.; Baer, M.R. Outcome of older patients with acute myeloid leukemia: An Analysis of SEER Data over 3 Decades. Cancer 2013, 119, 2720–2727. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.M.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, 2646–2654. [Google Scholar] [CrossRef] [Green Version]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [Green Version]
- Evazalipour, M.; D’Huyvetter, M.; Tehrani, B.S.; Abolhassani, M.; Omidfar, K.; Abdoli, S.; Arezumand, R.; Morovvati, H.; Lahoutte, T.; Muyldermans, S.; et al. Generation and characterization of nanobodies targeting PSMA for molecular imaging of prostate cancer. Contrast Media Mol. Imaging 2014, 9, 211–220. [Google Scholar] [CrossRef]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K. Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaneycken, I.; Devoogdt, N.; Van Gassen, N.; Vincke, C.; Xavier, C.; Wernery, U.; Muyldermans, S.; Lahoutte, T.; Caveliers, V. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011, 25, 2433–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Schütze, K.; Petry, K.; Hambach, J.; Schuster, N.; Fumey, W.; Schriewer, L.; Röckendorf, J.; Menzel, S.; Albrecht, B.; Haag, F.; et al. CD38-specific biparatopic heavy chain antibodies display potent complement-dependent cytotoxicity against multiple myeloma cells. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J. Targeting of the CD33-calicheamicin immunoconjugate mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2000, 97, 3197–3205. [Google Scholar] [CrossRef] [PubMed]
- Garfin, P.M.; Feldman, E.J. Antibody-Based Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2016, 11, 545–552. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; De Vos, J.; Xavier, C.; Pruszynski, M.; Sterckx, Y.G.J.; Massa, S.; Raes, G.; Caveliers, V.; Zalutsky, M.R.; Lahoutte, T.; et al. 131I-labeled anti-HER2 camelid sdAb as a theranostic tool in cancer treatment. Clin. Cancer Res. 2017, 23, 6616–6628. [Google Scholar] [CrossRef] [Green Version]
- Füchtner, F.; Zessin, J.; Mäding, P.; Wuest, F. Aspects of 6-[18F]fluoro-L-DOPA preparation. Deuterochloroform as a substitute solvent for Freon 11. Nuklearmedizin 2008, 47, 62–64. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Hu, P.; Wu, D.; Chen, W.; Ying, T.; Zhu, Z.; Dimitrov, D.S.; Dropulic, B.; Orentas, R.J. A unique human immunoglobulin heavy chain variable domain-only CD33 CAR for the treatment of acute myeloid leukemia. Front. Oncol. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Petrov, J.C.; Wada, M.; Pinz, K.G.; Yan, L.E.; Chen, K.H.; Shuai, X.; Liu, H.; Chen, X.; Leung, L.H.; Salman, H.; et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia 2018, 32, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Brogdon, J.; June, C.H.; Loew, A.; Maus, M.; Scholler, J. Treatment of Cancer using Humanized Anti-CD19 Chimeric Antigen Receptor. Patent Application US2014/0271635A1, 18 September 2014. [Google Scholar]
- Krasniqi, A.; D’Huyvetter, M.; Devoogdt, N.; Frejd, F.Y.; Sörensen, J.; Orlova, A.; Keyaerts, M.; Tolmachev, V. Same-day imaging using small proteins: Clinical experience and translational prospects in oncology. J. Nucl. Med. 2018, 59, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Debie, P.; Lafont, C.; Defrise, M.; Hansen, I.; van Willigen, D.M.; van Leeuwen, F.W.B.; Gijsbers, R.; D’Huyvetter, M.; Devoogdt, N.; Lahoutte, T.; et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J. Control. Release 2020, 317, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegt, E.; Jong, M.D.e.; Wetzels, J.F.M.; Masereeuw, R.; Melis, M.; Oyen, W.J.G.; Gotthardt, M.; Boerman, O.C. Renal toxicity of radiolabeled peptides and antibody fragments: Mechanisms, impact on radionuclide therapy, and strategies for prevention. J. Nucl. Med. 2010, 51, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, M.; Barone, R.; Krenning, E.; Bernard, B.; Melis, M.; Visser, T.; Gekle, M.; Willnow, T.E.; Walrand, S.; Jamar, F.; et al. Megalin is essential for renal proximal tubule reabsorption of 111in-DTPA-octreotide. J. Nucl. Med. 2005, 46, 1696–1700. [Google Scholar]
- Pelosi, E.; Castelli, G.; Testa, U. Targeting LSCs through membrane antigens selectively or preferentially expressed on these cells. Blood Cells Mol. Dis. 2015, 55, 336–346. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef] [Green Version]
- Sargur Madabushi, S.; Brooks, J.; Zuro, D.; Kumar, B.; Sanchez, J.; Echavarria Parra, L.; Orellana, M.; Vishwasrao, P.; Nair, I.; Chea, J.; et al. ImmunoPET, [64Cu] Cu-DOTA-anti-CD33 PET-CT, imaging of an AML Xenograft model. Clin. Cancer Res. 2019, 25, 7463–7474. [Google Scholar]
- Moonens, K.; Gideonsson, P.; Subedi, S.; Bugaytsova, J.; Romaõ, E.; Mendez, M.; Nordén, J.; Fallah, M.; Rakhimova, L.; Shevtsova, A.; et al. Structural Insights into Polymorphic ABO Glycan Binding by Helicobacter pylori. Cell Host Microbe 2016, 19, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Boivin, S.; Kozak, S.; Meijers, R. Optimization of protein purification and characterization using Thermofluor screens. Protein Eng. Des. Sel. 2013, 91, 192–206. [Google Scholar] [CrossRef]
- Xavier, C.; Devoogdt, N.; Hernot, S.; Vaneyken, I.; D’Huyvetter, M.; De Vos, J.; Massa, S.; Lahoutte, T.; Caveliers, V. Site-Specific Labeling of His-Tagged Nanobodies with 99mTc: A Practical Guide. In Single Domain Antibodies: Methods and Protocols; Saerens, D., Muyldermans, S., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 911, pp. 485–490. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanobody | ka (M−1·s−1) | kd (s−1) | KD (M) | Tm (°C) |
|---|---|---|---|---|
| Nb_7 | 1.1 × 106 | 6.1 × 10−3 | 5.5 × 10−9 | 54.47 ± 0.05 |
| Nb_12 | 5.4 × 105 | 2.1 × 10−3 | 3.9 × 10−9 | 58.00 ± 0.23 |
| Nb_16 | 1.3 × 104 | 3.4 × 10−3 | 2.6 × 10−7 | 57.80 ± 0.03 |
| Nb_21 | 1.3 × 106 | 9.1 × 10−3 | 7.0 × 10−9 | 52.67 ± 0.04 |
| Nb_22 | 1.5 × 106 | 1.9 × 10−3 | 1.3 × 10−8 | 58.22 ± 0.03 |
| Nb_87 | 9.3 × 106 | 7.7 × 10−3 | 8.1 × 10−8 | 67.80 ± 0.08 |
| αBabA Nb_19 * | 2.8 × 106 | 1.2 × 10−3 | 0.4 × 10−9 | 61.75 ± 0.14 |
| Nanobody | Nb_7 | Nb_12 | Nb_16 | Nb_21 | Nb_22 | Nb_87 |
|---|---|---|---|---|---|---|
| Nb_7 | competitive | non-competitive | non-competitive | competitive | competitive | competitive |
| Nb_12 | competitive | competitive | non-competitive | non-competitive | non-competitive | non-competitive |
| Nb_16 | competitive | competitive | competitive | non-competitive | non-competitive | non-competitive |
| Nb_21 | competitive | competitive | competitive | competitive | competitive | competitive |
| Nb_22 | competitive | competitive | competitive | competitive | competitive | competitive |
| Nb_87 | competitive | competitive | competitive | competitive | competitive | competitive |
| Nb_7 | Nb_12 | Nb_16 | Nb_21 | Nb_87 | ctrl_Nb | |
|---|---|---|---|---|---|---|
| Blood | 0.70 ± 0.18 | 0.46 ± 0.06 | 0.62 ± 0.07 | 0.73 ± 0.14 | 0.78 ± 0.07 | 0.57 ± 0.09 |
| Heart | 0.32 ± 0.05 | 0.23 ± 0.15 | 0.31 ± 0.04 | 0.50 ± 0.13 | 0.37 ± 0.03 | 0.25 ± 0.06 |
| Lungs | 0.94 ± 0.24 | 0.80 ± 0.31 | 0.81 ± 0.08 | 1.40 ± 0.20 | 1.23 ± 0.07 | 0.78 ± 0.04 |
| Liver | 0.92 ± 0.07 | 1.22 ± 0.38 | 1.02 ± 0.34 | 3.42 ± 0.65 | 1.50 ± 0.35 | 0.57 ± 0.05 |
| Spleen | 0.39 ± 0.06 | 0.50 ± 0.53 | 0.36 ± 0.11 | 2.28 ± 0.82 | 0.44 ± 0.02 | 0.28 ± 0.01 |
| Stomach | 0.39 ± 0.15 | 0.57 ± 0.52 | 0.38 ± 0.07 | 0.48 ± 0.10 | 0.37 ± 0.11 | 1.02 ± 0.63 |
| Intestines | 0.48 ± 0.10 | 0.36 ± 0.09 | 0.46 ± 0.19 | 0.97 ± 0.24 | 0.58 ± 0.12 | 0.37 ± 0.13 |
| Kidneys | 232.04 ± 15.83 | 177.47 ± 12.14 | 211.16 ± 64.88 | 197.04 ± 25.96 | 249.11 ± 37.07 | 163.68 ± 4.78 |
| Muscle | 0.15 ± 0.03 | 0.16 ± 0.06 | 0.16 ± 0.04 | 0.16 ± 0.04 | 0.27 ± 0.13 | 0.12 ± 0.01 |
| Bone | 0.15 ± 0.03 | 0.17 ± 0.05 | 0.13 ± 0.02 | 0.49 ± 0.31 | 0.22 ± 0.00 | 0.11 ± 0.00 |
| Tumor | 2.53 ± 0.69 | 1.91 ± 0.64 | 0.70 ± 0.13 | 2.28 ± 0.56 | 2.34 ± 0.79 | 0.16 ± 0.04 |
| Tumor/Blood | 3.66 ± 0.94 | 4.15 ± 0.69 | 1.12 ± 0.09 | 3.11 ± 0.46 | 3.02 ± 1.08 | 0.27 ± 0.03 |
| Tumor/Muscle | 17.57 ± 5.13 | 12.56 ± 4.43 | 4.39 ± 0.88 | 14.03 ± 3.08 | 9.20 ± 3.51 | 1.31 ± 0.28 |
| Tumor/Bone | 16.37 ± 1.95 | 11.42 ± 3.60 | 5.36 ± 1.12 | 5.35 ± 1.66 | 10.64 ± 3.68 | 1.46 ± 0.43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romão, E.; Krasniqi, A.; Maes, L.; Vandenbrande, C.; Sterckx, Y.G.-J.; Stijlemans, B.; Vincke, C.; Devoogdt, N.; Muyldermans, S. Identification of Nanobodies against the Acute Myeloid Leukemia Marker CD33. Int. J. Mol. Sci. 2020, 21, 310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010310
Romão E, Krasniqi A, Maes L, Vandenbrande C, Sterckx YG-J, Stijlemans B, Vincke C, Devoogdt N, Muyldermans S. Identification of Nanobodies against the Acute Myeloid Leukemia Marker CD33. International Journal of Molecular Sciences. 2020; 21(1):310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010310
Chicago/Turabian StyleRomão, Ema, Ahmet Krasniqi, Laila Maes, Camille Vandenbrande, Yann G.-J. Sterckx, Benoit Stijlemans, Cécile Vincke, Nick Devoogdt, and Serge Muyldermans. 2020. "Identification of Nanobodies against the Acute Myeloid Leukemia Marker CD33" International Journal of Molecular Sciences 21, no. 1: 310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010310