Underlying Histopathology Determines Response to Oxidative Stress in Cultured Human Primary Proximal Tubular Epithelial Cells

 , ,
, ,
Abstract
:1. Introduction
2. Results
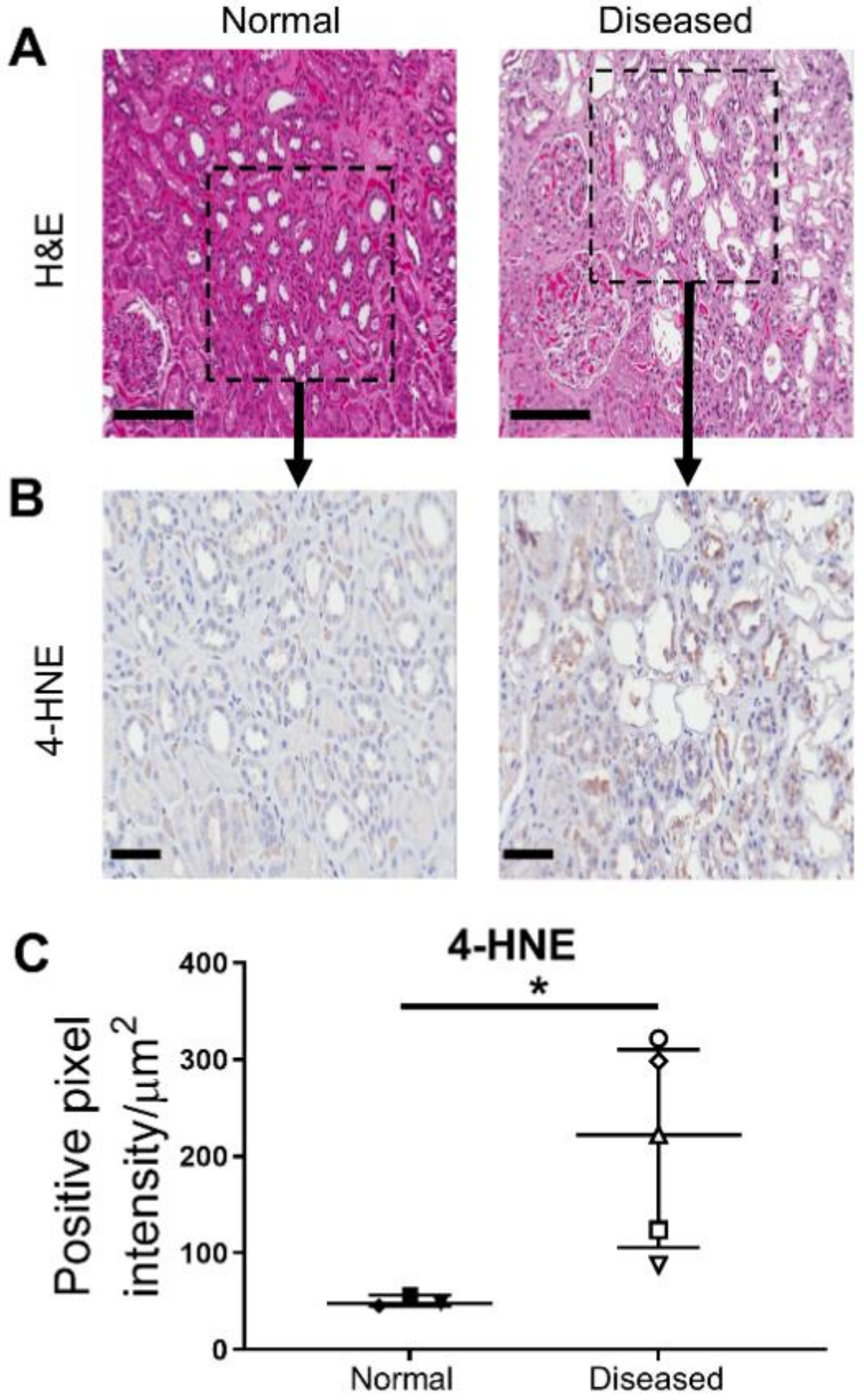
2.1. Significantly Increased Levels of Oxidative Stress in Histologically ‘Diseased’ Kidney Cortical Tissue
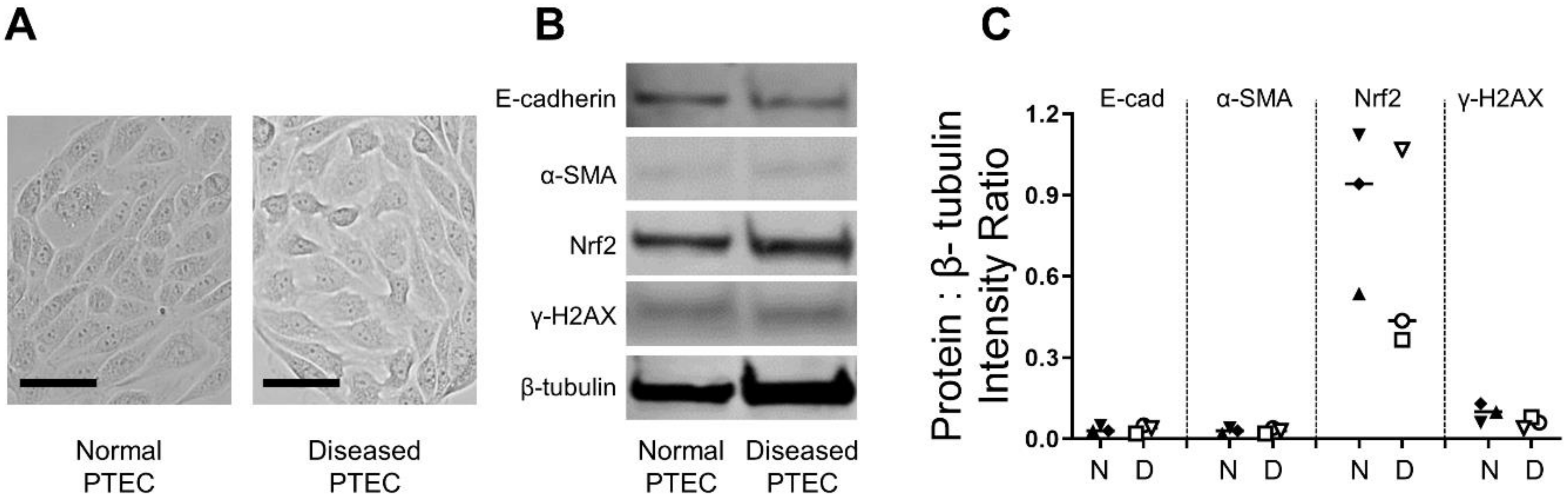
2.2. Normal and Diseased PTEC Display Comparable Morphological and Molecular Profiles
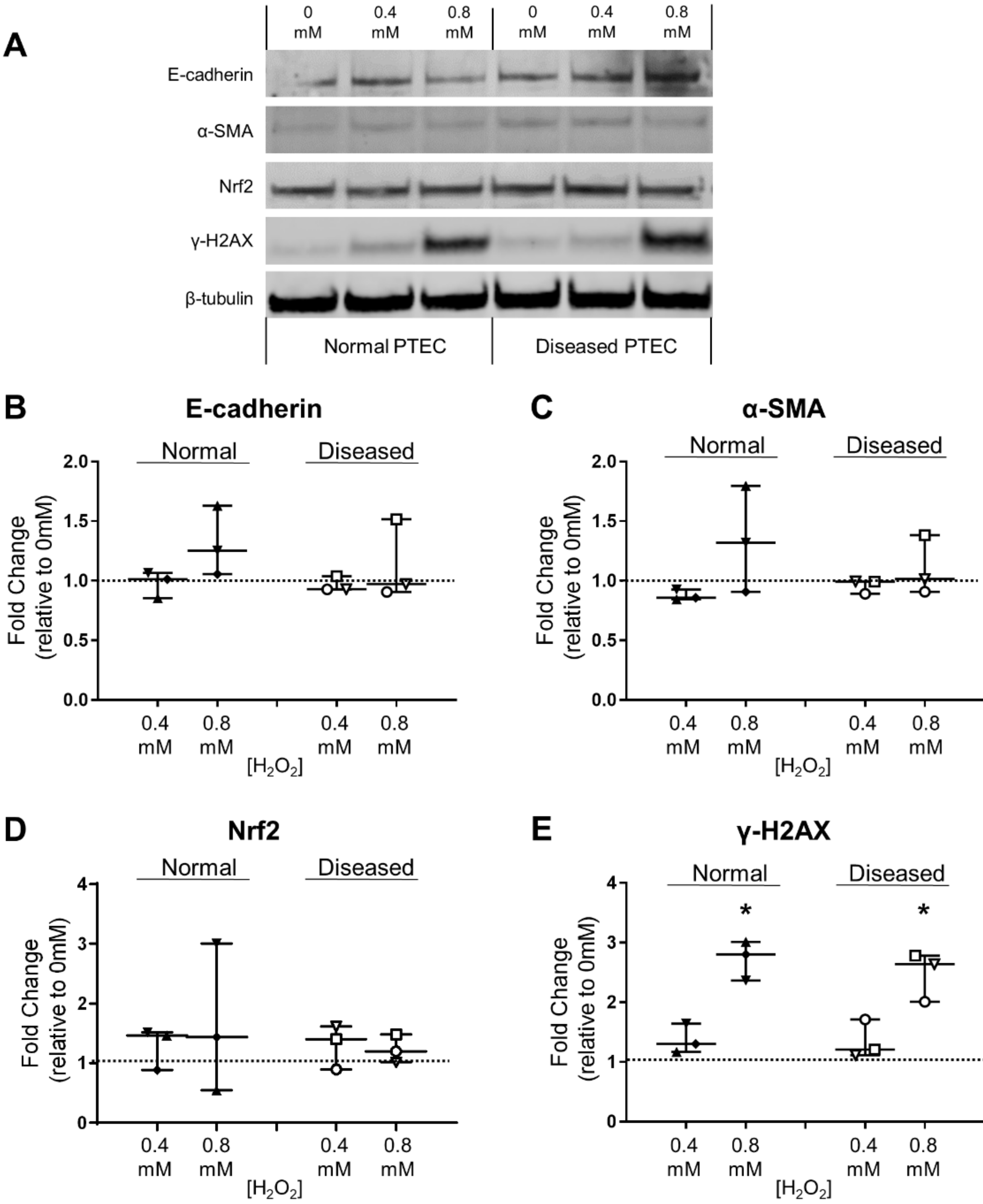
2.3. Normal and Diseased PTEC Phenotypically Respond to H2O2-Induced Oxidative Stress
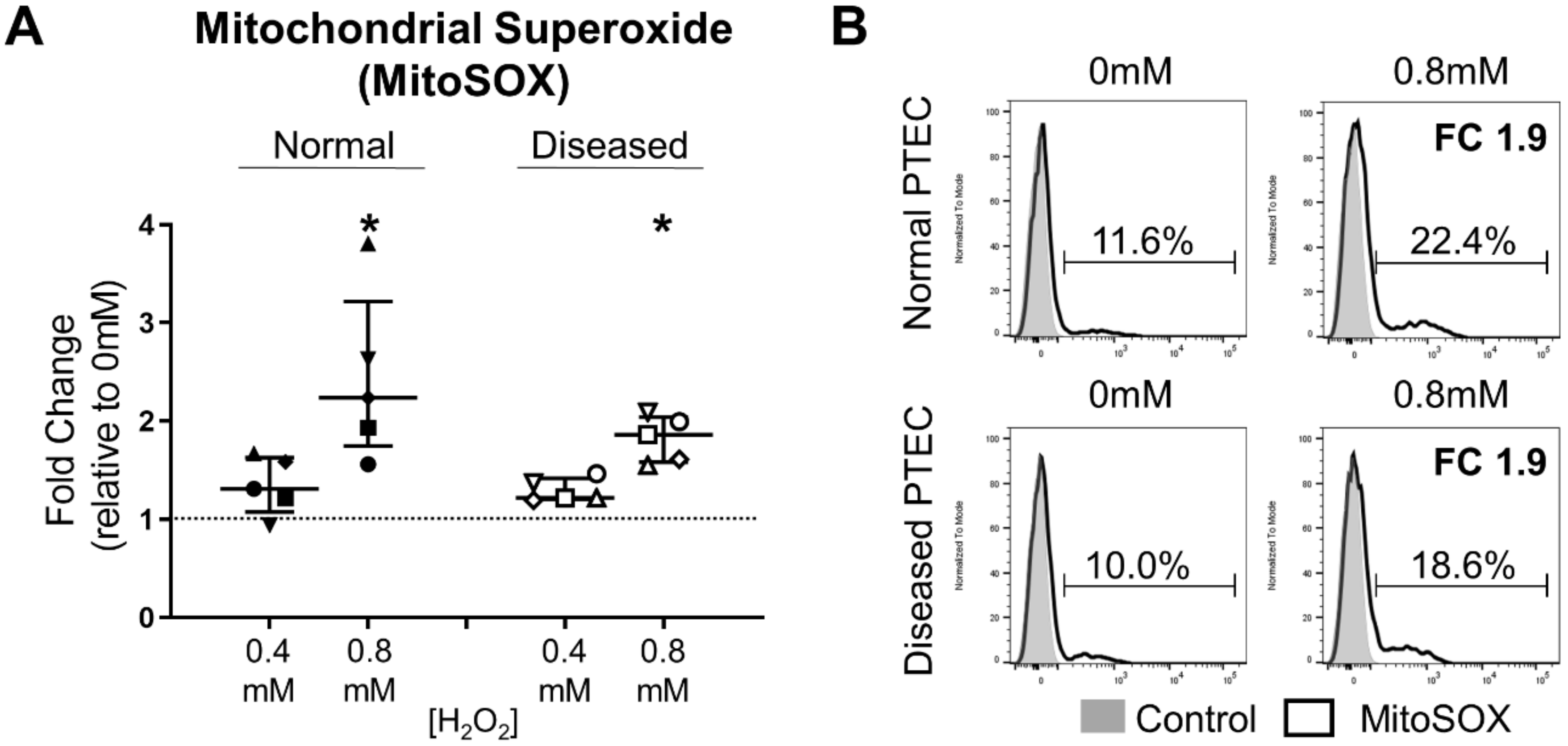
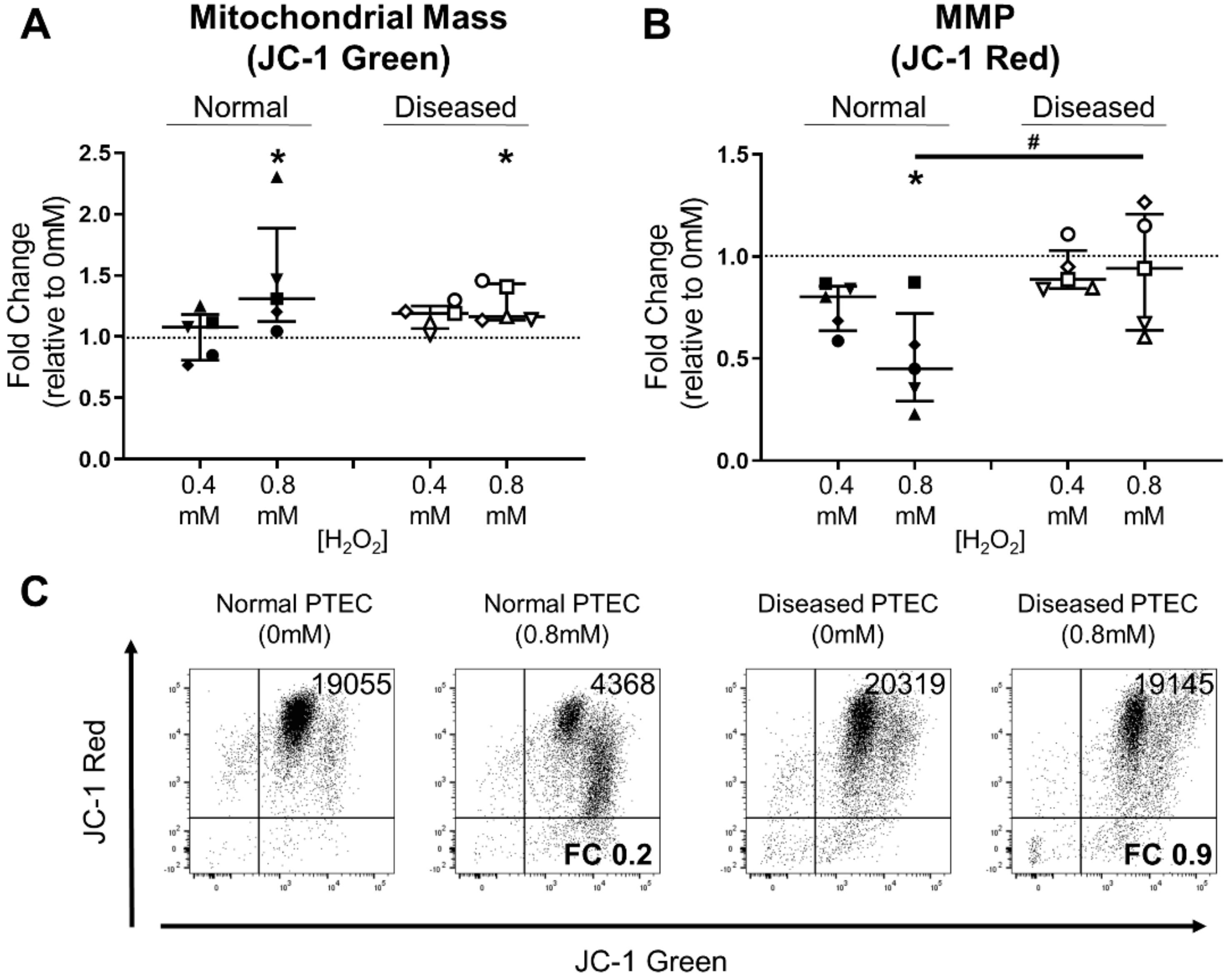
2.4. Diseased PTEC Maintain Mitochondrial Function under Oxidative Stress Conditions
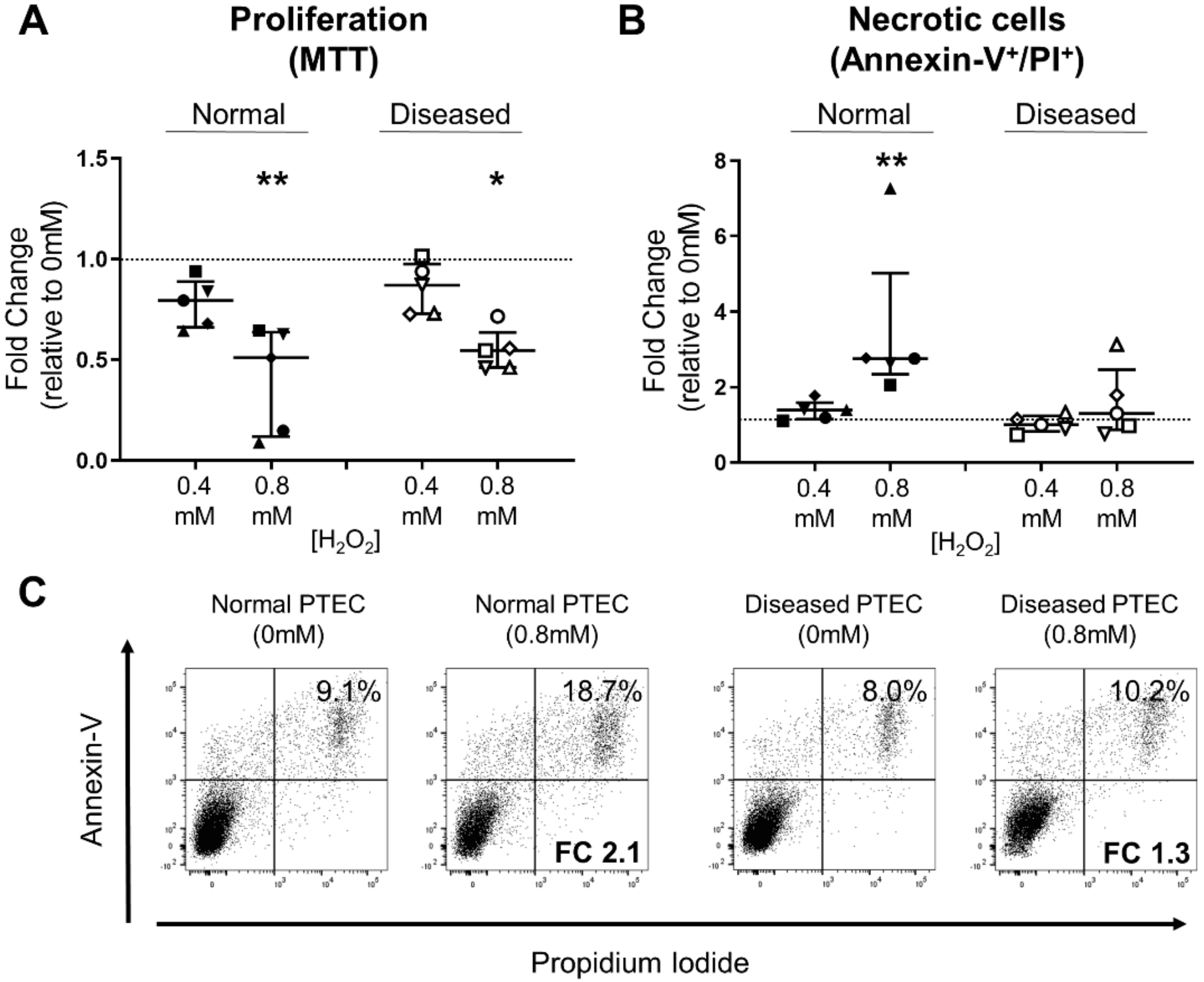
2.5. Diseased PTEC Maintain Cell Viability under Oxidative Stress Conditions
3. Discussion
4. Materials and Methods
4.1. Kidney Cortical Tissue Specimens
4.2. IHC Staining
4.3. Isolation and Culture of Human Primary PTEC
4.4. Induction of Oxidative Stress in Human Primary PTEC
4.5. Western Blotting
4.6. Mitochondrial Superoxide Detection
4.7. Assessment of Mitochondrial Changes
4.8. Annexin V/PI Viability Assay
4.9. Cell Proliferation Measurements
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AKI | Acute kidney injury |
| BSA | Bovine serum albumin |
| CKD | Chronic kidney disease |
| eGFR | Estimated glomerular filtration rate |
| DM | Defined medium |
| DMSO | Dimethyl sulfoxide |
| EMT | Epithelial-to-mesenchymal transition |
| H&E | Hematoxylin and eosin |
| 4-HNE | 4-Hydroxynonenal |
| HO-1 | Heme oxygenase-1 |
| H2O2 | Hydrogen peroxide |
| IHC | Immunohistochemical |
| JC-1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazolcarbocyanine iodide |
| MFI | Median fluorescence intensity |
| MMP | Mitochondrial membrane potential |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PAGE | Polyacrylamide gel electrophoresis |
| PI | Propidium iodide |
| PTEC | Proximal tubular epithelial cells |
| RCC | Renal cell carcinoma |
| ROS | Reactive oxygen species |
| α-SMA | α-smooth muscle actin |
References
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Hommos, M.S.; Glassock, R.J.; Rule, A.D. Structural and Functional Changes in Human Kidneys with Healthy Aging. J. Am. Soc. Nephrol. 2017, 28, 2838–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pract. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Percy, C.J.; Power, D.; Gobe, G.C. Renal ageing: Changes in the cellular mechanism of energy metabolism and oxidant handling. Nephrology 2008, 13, 147–152. [Google Scholar] [CrossRef]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D., Jr.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar] [CrossRef] [Green Version]
- Reers, M.; Smith, T.W.; Chen, L.B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.L.; Ding, Y.; Alexander, L.D.; Bao, C.; Al-Khalili, O.K.; Simonson, M.; Eaton, D.C.; Douglas, J.G. Oxidative signaling in renal epithelium: Critical role of cytosolic phospholipase A2 and p38(SAPK). Free Radic. Biol. Med. 2006, 41, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Small, D.M.; Bennett, N.C.; Roy, S.; Gabrielli, B.G.; Johnson, D.W.; Gobe, G.C. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron Exp. Nephrol. 2012, 122, 123–130. [Google Scholar] [CrossRef]
- Hou, X.; Xiao, H.; Zhang, Y.; Zeng, X.; Huang, M.; Chen, X.; Birnbaumer, L.; Liao, Y. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018, 9, 1015. [Google Scholar] [CrossRef]
- Lin, M.; Li, L.; Zhang, Y.; Zheng, L.; Xu, M.; Rong, R.; Zhu, T. Baicalin ameliorates H2O2 induced cytotoxicity in HK-2 cells through the inhibition of ER stress and the activation of Nrf2 signaling. Int. J. Mol. Sci. 2014, 15, 12507–12522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreucci, M.; Fuiano, G.; Presta, P.; Lucisano, G.; Leone, F.; Fuiano, L.; Bisesti, V.; Esposito, P.; Russo, D.; Memoli, B.; et al. Downregulation of cell survival signalling pathways and increased cell damage in hydrogen peroxide-treated human renal proximal tubular cells by alpha-erythropoietin. Cell Prolif. 2009, 42, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Shin, J.W.; Lee, E.H.; Moon, Y.; Seo, Y.J.; Kim, J.Y.; Kim, J.U. Protective effects of propofol against hydrogen peroxide-induced oxidative stress in human kidney proximal tubular cells. Korean J. Anesthesiol. 2012, 63, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Tan, X.; Chen, Y.; Zhang, X. Postconditioning protects renal fibrosis by attenuating oxidative stress-induced mitochondrial injury. Nephrol. Dial. Transplant. 2017, 32, 1628–1636. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.Y.; Huang, T.P.; Yang, W.C.; Chen, Z.P.; Yang, A.H.; Mu, W.; Nikolic-Paterson, D.J.; Atkins, R.C.; Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998, 54, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.T.; Lee, H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pang, L.; Zhang, Y.; Lin, J.; Zhou, H. Fenofibrate Improved Interstitial Fibrosis of Renal Allograft through Inhibited Epithelial-Mesenchymal Transition Induced by Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol.-Ren. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef] [PubMed]
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal. 2018, 12, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colavitti, R.; Finkel, T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef]
- Eirin, A.; Lerman, A.; Lerman, L.O. The Emerging Role of Mitochondrial Targeting in Kidney Disease. Handb. Exp. Pharmacol. 2017, 240, 229–250. [Google Scholar] [CrossRef] [Green Version]
- Glynne, P.A.; Evans, T.J. Inflammatory cytokines induce apoptotic and necrotic cell shedding from human proximal tubular epithelial cell monolayers. Kidney Int. 1999, 55, 2573–2597. [Google Scholar] [CrossRef]
- Kassianos, A.J.; Sampangi, S.; Wang, X.; Roper, K.E.; Beagley, K.; Healy, H.; Wilkinson, R. Human proximal tubule epithelial cells modulate autologous dendritic cell function. Nephrol. Dial. Transplant. 2013, 28, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Rajesh, M.; Yoshihiro, K.; Hasko, G.; Pacher, P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 2007, 358, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, M.; Anderson, B.O.; Caldwell, E.; Sedghinasab, M.; Paty, P.B.; Hockenbery, D.M. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J. Cell Biol. 1997, 138, 449–469. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Years)/Sex (M/F) | Primary Disease | eGFR | Coexisting Pathology in Nontumor Renal Parenchyma (% Diseased Cortical Area) |
|---|---|---|---|---|
| Normal PTEC (n = 5) | ||||
| 1 | 64/F | Clear cell RCC (Grade 2) | 72 | Nil |
| 2 | 58/M | Clear cell RCC (Grade 2) | 77 | Nil |
| 3 | 65/F | Clear cell RCC (Grade 2) | 81 | Nil |
| 4 | 61/M | Clear cell RCC (Grade 2) | 88 | Nil |
| 5 | 45/M | Clear cell RCC (Grade 2) | 90 | Nil |
| Diseased PTEC (n = 5) | ||||
| 1 | 53/M | Chromophobe RCC | 75 | Glomerulosclerosis (5–10%) |
| 2 | 56/F | Chromophobe RCC | 90 | Glomerulosclerosis (5–10%) |
| 3 | 52/F | Clear cell RCC (Grade 2) | 90 | Arteriosclerosis (25–30%) |
| 4 | 60/M | Clear cell RCC (Grade 4) | 75 | Arteriosclerosis (10–20%) |
| 5 | 53/F | Chromophobe RCC | 76 | Tubular atrophy (10–20%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A.; Wang, X.; Giuliani, K.T.K.; Nag, P.; Grivei, A.; Ungerer, J.; Hoy, W.; Healy, H.; Gobe, G.; Kassianos, A.J. Underlying Histopathology Determines Response to Oxidative Stress in Cultured Human Primary Proximal Tubular Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020560
Khan MA, Wang X, Giuliani KTK, Nag P, Grivei A, Ungerer J, Hoy W, Healy H, Gobe G, Kassianos AJ. Underlying Histopathology Determines Response to Oxidative Stress in Cultured Human Primary Proximal Tubular Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(2):560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020560
Chicago/Turabian StyleKhan, Muhammad Ali, Xiangju Wang, Kurt T.K. Giuliani, Purba Nag, Anca Grivei, Jacobus Ungerer, Wendy Hoy, Helen Healy, Glenda Gobe, and Andrew J. Kassianos. 2020. "Underlying Histopathology Determines Response to Oxidative Stress in Cultured Human Primary Proximal Tubular Epithelial Cells" International Journal of Molecular Sciences 21, no. 2: 560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020560