Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA)


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
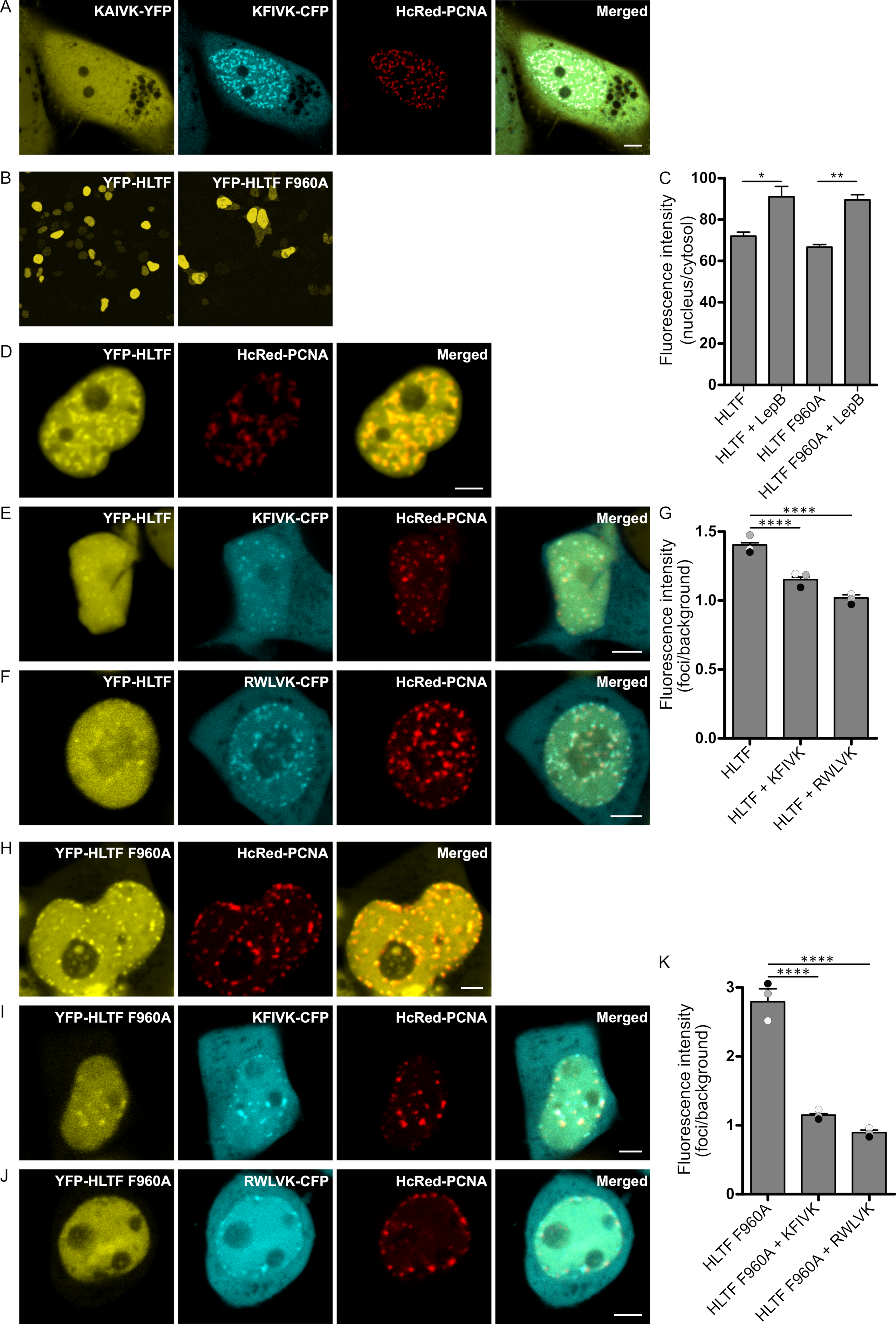
2.1. APIM in HLTF Is a Functional PCNA Interacting Motif
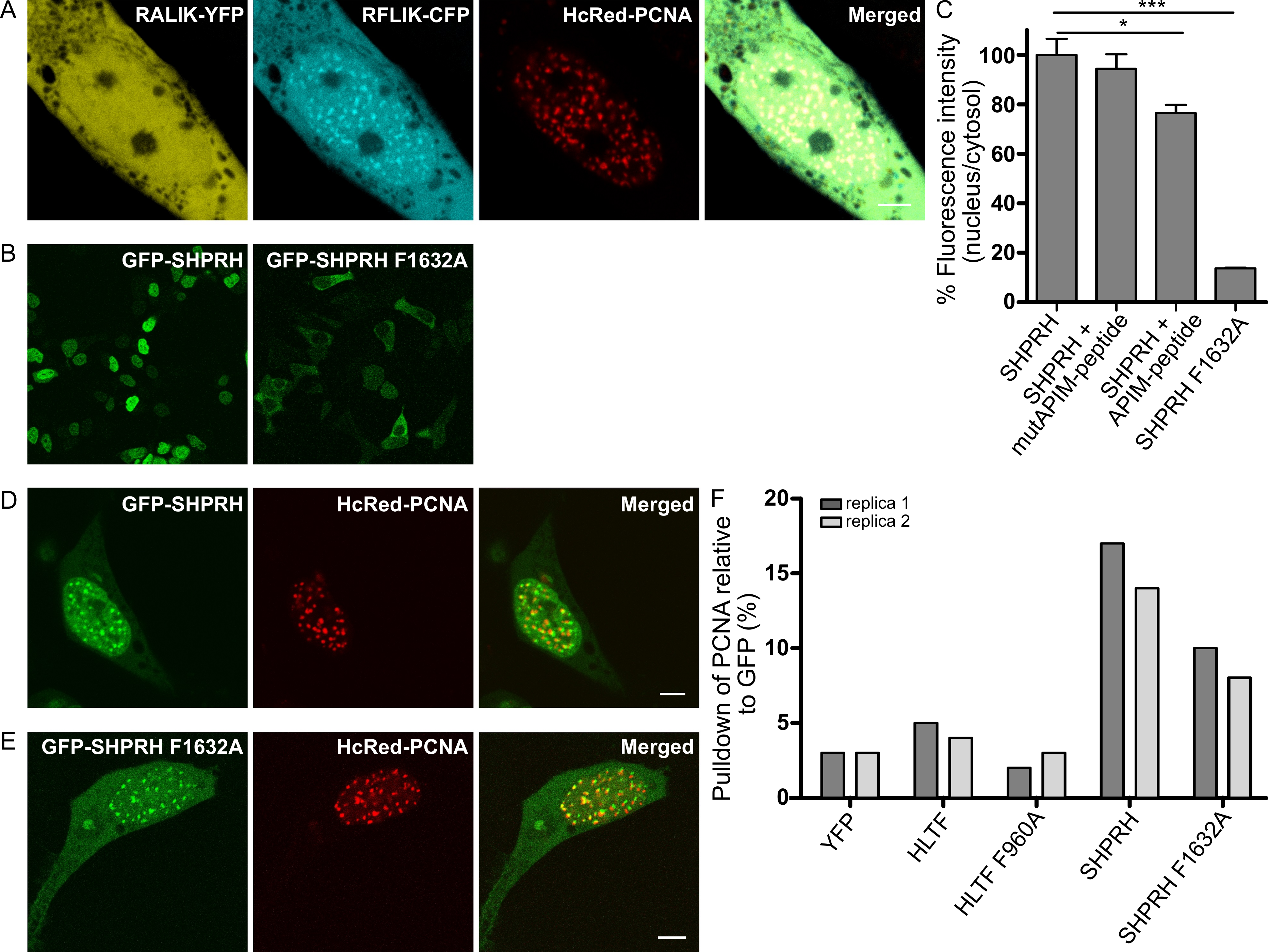
2.2. Nuclear Localization of SHPRH Depends on Its Interaction with PCNA
2.3. APIM in SHPRH and HLTF Is Required for Maximal Pull Down of PCNA After DNA Damage
2.4. Direct Interaction with PCNA Is Important for the Regulation of DDT by HLTF and SHPRH
2.5. Direct Binding of HLTF to PCNA Is Important for Error-Free DDT and/or DNA Repair
2.6. SHPRH Overexpression Stimulates Error-Prone TLS
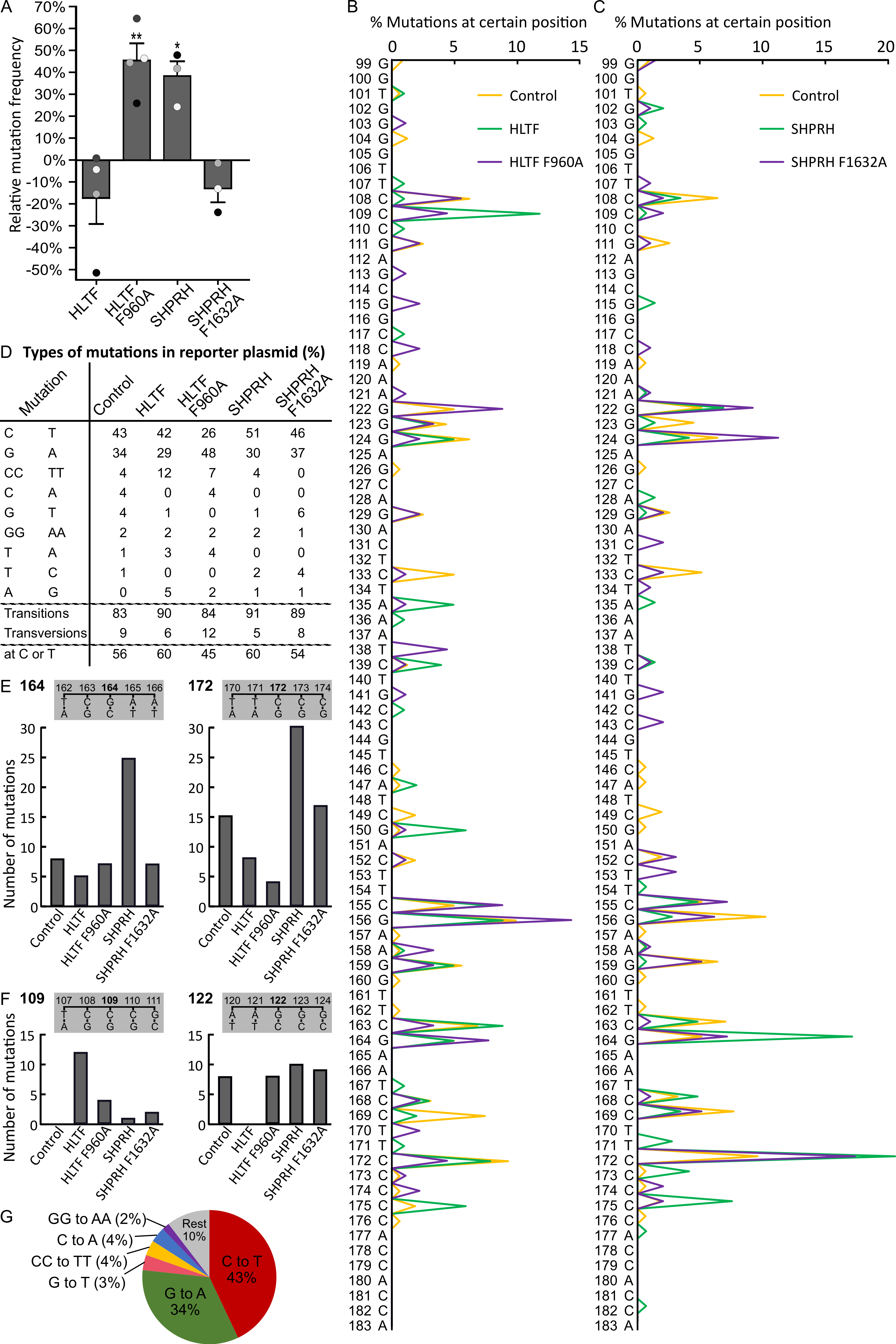
2.7. Reduced Level of Putative Transcribed Strand Mutations after Overexpression of HLTF and SHRPH
3. Material and Methods
3.1. Expression Constructs
3.2. Cell Lines
3.3. SupF Assay
3.4. Imaging
3.5. Measurement of Fluorescence Intensities After APIM-Peptide or Leptomycin B Treatment
3.6. Preparation of Cross-Linked Cell Extracts
3.7. Immunoprecipitation
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| APIMPCNA | AlkB homolog 2 PCNA interacting motifProliferating cell nuclear antigen |
| CPD | Cyclo-pyrimidine dimer |
| DDT | DNA damage tolerance |
| HLTF | Helicase-like transcription factor |
| MMS | Methyl methanesulfonate |
| NER | Nucleotide excision repair |
| SHPRH | SNF2 histone-linker PHD and RING finger domain-containing helicase |
| TLS | Translesion synthesis |
| TS | Template switch |
| XPV | Xeroderma Pigmentosum Variant |
| 6-4PPs | (6-4)photoproducts |
References
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Yoon, J.H.; Lee, C.S.; O’Connor, T.R.; Yasui, A.; Pfeifer, G.P. The DNA damage spectrum produced by simulated sunlight. J. Mol. Biol. 2000, 299, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Formation and processing of UV photoproducts: Effects of DNA sequence and chromatin environment. Photochem. Photobiol. 1997, 65, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Sancar, A. DNA excision repair: Where do all the dimers go? Cell Cycle 2012, 11, 2997–3002. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Riou, L.; Zeng, L.; Chevallier-Lagente, O.; Stary, A.; Nikaido, O.; Taieb, A.; Weeda, G.; Mezzina, M.; Sarasin, A. The relative expression of mutated XPB genes results in xeroderma pigmentosum/Cockayne’s syndrome or trichothiodystrophy cellular phenotypes. Hum. Mol. Genet. 1999, 8, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Garinis, G.A.; Mitchell, J.R.; Moorhouse, M.J.; Hanada, K.; de Waard, H.; Vandeputte, D.; Jans, J.; Brand, K.; Smid, M.; van der Spek, P.J.; et al. Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J. 2005, 24, 3952–3962. [Google Scholar] [CrossRef] [Green Version]
- Cordonnier, A.M.; Lehmann, A.R.; Fuchs, R.P. Impaired translesion synthesis in xeroderma pigmentosum variant extracts. Mol. Cell. Biol. 1999, 19, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Inui, H.; Oh, K.S.; Nadem, C.; Ueda, T.; Khan, S.G.; Metin, A.; Gozukara, E.; Emmert, S.; Slor, H.; Busch, D.B.; et al. Xeroderma pigmentosum-variant patients from America, Europe, and Asia. J. Investig. Derm. 2008, 128, 2055–2068. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, K.H.; Lee, M.M.; Andrews, A.D.; Lambert, W.C. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Derm. 1994, 130, 1018–1021. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Lee, M.M.; Scotto, J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch. Derm. 1987, 123, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Menck, C.F.; Munford, V. DNA repair diseases: What do they tell us about cancer and aging? Genet. Mol. Biol. 2014, 37, 220–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, W.; Baxley, R.M.; Moldovan, G.L.; Bielinsky, A.K. Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, Y.; Suzuki, M.; Kawai, H.; Hishiki, A.; Hashimoto, H.; Masutani, C.; Hishida, T.; Suzuki, F.; Kamiya, K. En bloc transfer of polyubiquitin chains to PCNA in vitro is mediated by two different human E2-E3 pairs. Nucleic Acids Res. 2012, 40, 10394–10407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, D.A.; Greer, B.H.; Eichman, B.F. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J. Biol. Chem. 2018, 293, 8484–8494. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.R.; Zeman, M.K.; Chen, J.Y.; Yee, M.C.; Cimprich, K.A. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 2011, 42, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Chung, S.W.; Kim, J.H.; Kim, J.W.; Oh, J.S.; Kim, S.; Song, S.Y.; Park, J.; Kim, D.H. Promoter methylation of helicase-like transcription factor is associated with the early stages of gastric cancer with family history. Ann. Oncol. 2006, 17, 657–662. [Google Scholar] [CrossRef]
- Capouillez, A.; Noel, J.C.; Arafa, M.; Arcolia, V.; Mouallif, M.; Guenin, S.; Delvenne, P.; Belayew, A.; Saussez, S. Expression of the helicase-like transcription factor and its variants during carcinogenesis of the uterine cervix: Implications for tumour progression. Histopathology 2011, 58, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene 2018, 37, 1805. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; Chen, W.D.; Shen, L.; Smiraglia, D.; Olechnowicz, J.; Ravi, L.; Kasturi, L.; Myeroff, L.; Plass, C.; Parsons, R.; et al. HLTF gene silencing in human colon cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 4562–4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E. PCNA binding through a conserved motif. Bioessays 1998, 20, 195–199. [Google Scholar] [CrossRef]
- Olaisen, C.; Kvitvang, H.F.N.; Lee, S.; Almaas, E.; Bruheim, P.; Drablos, F.; Otterlei, M. The role of PCNA as a scaffold protein in cellular signaling is functionally conserved between yeast and humans. FEBS Open Bio 2018, 8, 1135–1145. [Google Scholar] [CrossRef]
- Muller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Vatsveen, T.K.; Ro, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef] [Green Version]
- Bacquin, A.; Pouvelle, C.; Siaud, N.; Perderiset, M.; Salome-Desnoulez, S.; Tellier-Lebegue, C.; Lopez, B.; Charbonnier, J.B.; Kannouche, P.L. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013, 41, 6501–6513. [Google Scholar] [CrossRef]
- Fu, D.; Samson, L.D.; Hubscher, U.; van Loon, B. The interaction between ALKBH2 DNA repair enzyme and PCNA is direct, mediated by the hydrophobic pocket of PCNA and perturbed in naturally-occurring ALKBH2 variants. DNA Repair 2015, 35, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Sebesta, M.; Cooper, C.D.O.; Ariza, A.; Carnie, C.J.; Ahel, D. Structural insights into the function of ZRANB3 in replication stress response. Nat. Commun. 2017, 8, 15847. [Google Scholar] [CrossRef] [Green Version]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gederaas, O.A.; Sogaard, C.D.; Viset, T.; Bachke, S.; Bruheim, P.; Arum, C.J.; Otterlei, M. Increased Anticancer Efficacy of Intravesical Mitomycin C Therapy when Combined with a PCNA Targeting Peptide. Transl. Oncol. 2014, 7, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogaard, C.K.; Blindheim, A.; Rost, L.M.; Petrovic, V.; Nepal, A.; Bachke, S.; Liabakk, N.B.; Gederaas, O.A.; Viset, T.; Arum, C.J.; et al. “Two hits—one stone”; increased efficacy of cisplatin-based therapies by targeting PCNA’s role in both DNA repair and cellular signaling. Oncotarget 2018, 9, 32448–32465. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, C.K.; Moestue, S.A.; Rye, M.B.; Kim, J.; Nepal, A.; Liabakk, N.B.; Bachke, S.; Bathen, T.F.; Otterlei, M.; Hill, D.K. APIM-peptide targeting PCNA improves the efficacy of docetaxel treatment in the TRAMP mouse model of prostate cancer. Oncotarget 2018, 9, 11752–11766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattah, F.J.; Hara, K.; Fattah, K.R.; Yang, C.; Wu, N.; Warrington, R.; Chen, D.J.; Zhou, P.; Boothman, D.A.; Yu, H. The transcription factor TFII-I promotes DNA translesion synthesis and genomic stability. PLoS Genet. 2014, 10, e1004419. [Google Scholar] [CrossRef]
- Raeder, S.B.; Nepal, A.; Bjoras, K.O.; Seelinger, M.; Kolve, R.S.; Nedal, A.; Muller, R.; Otterlei, M. APIM-Mediated REV3L(-)PCNA Interaction Important for Error Free TLS Over UV-Induced DNA Lesions in Human Cells. Int. J. Mol. Sci. 2018, 20. [Google Scholar] [CrossRef] [Green Version]
- Olaisen, C.; Muller, R.; Nedal, A.; Otterlei, M. PCNA-interacting peptides reduce Akt phosphorylation and TLR-mediated cytokine secretion suggesting a role of PCNA in cellular signaling. Cell Signal. 2015, 27, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Prakash, L.; Prakash, S. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18219–18224. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, K.; Kawazu, M.; Koya, J.; Yoshimi, A.; Masamoto, Y.; Maki, H.; Toya, T.; Kobayashi, T.; Nannya, Y.; Arai, S.; et al. A germline HLTF mutation in familial MDS induces DNA damage accumulation through impaired PCNA polyubiquitination. Leukemia 2019, 33, 1773–1782. [Google Scholar] [CrossRef]
- Masuda, Y.; Mitsuyuki, S.; Kanao, R.; Hishiki, A.; Hashimoto, H.; Masutani, C. Regulation of HLTF-mediated PCNA polyubiquitination by RFC and PCNA monoubiquitination levels determines choice of damage tolerance pathway. Nucleic Acids Res. 2018, 46, 11340–11356. [Google Scholar] [CrossRef]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Besaratinia, A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem. Photobiol. Sci. 2012, 11, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef]
- Choi, J.H.; Pfeifer, G.P. The role of DNA polymerase eta in UV mutational spectra. DNA Repair 2005, 4, 211–220. [Google Scholar] [CrossRef]
- Aas, P.A.; Otterlei, M.; Falnes, P.O.; Vagbo, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjoras, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 42 1, 859–863. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seelinger, M.; Otterlei, M. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int. J. Mol. Sci. 2020, 21, 693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030693
Seelinger M, Otterlei M. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). International Journal of Molecular Sciences. 2020; 21(3):693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030693
Chicago/Turabian StyleSeelinger, Mareike, and Marit Otterlei. 2020. "Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA)" International Journal of Molecular Sciences 21, no. 3: 693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030693