C-Terminal Domain of the Human Zinc Transporter hZnT8 Is Structurally Indistinguishable from Its Disease Risk Variant (R325W)

 , , and
, , and
Abstract
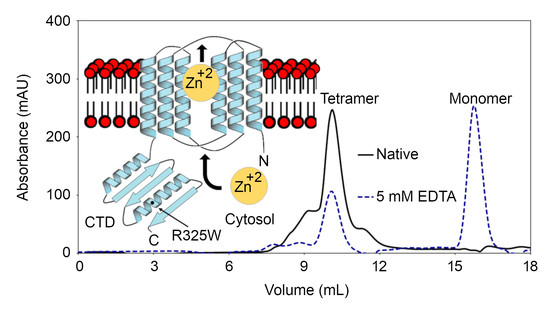
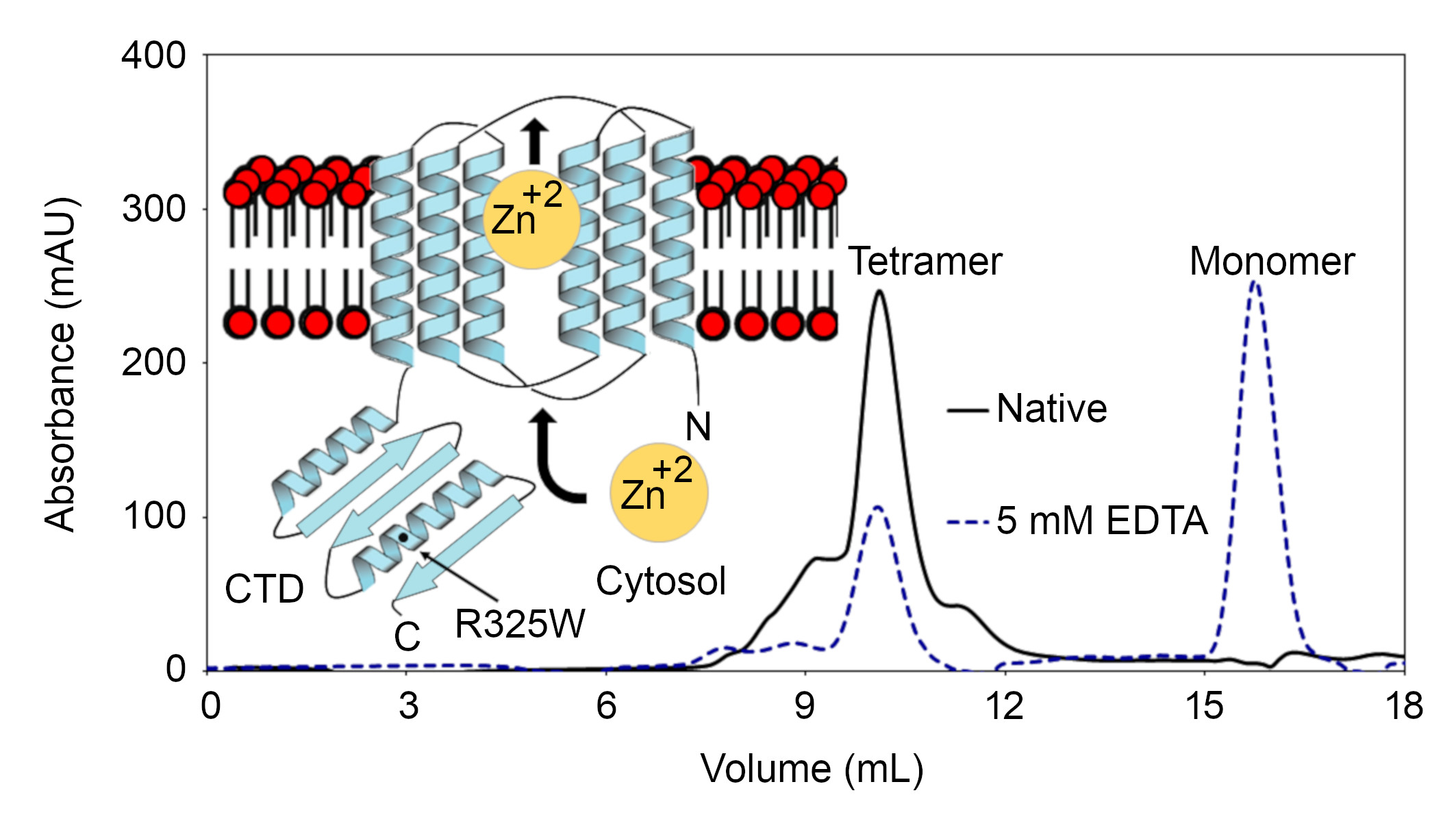
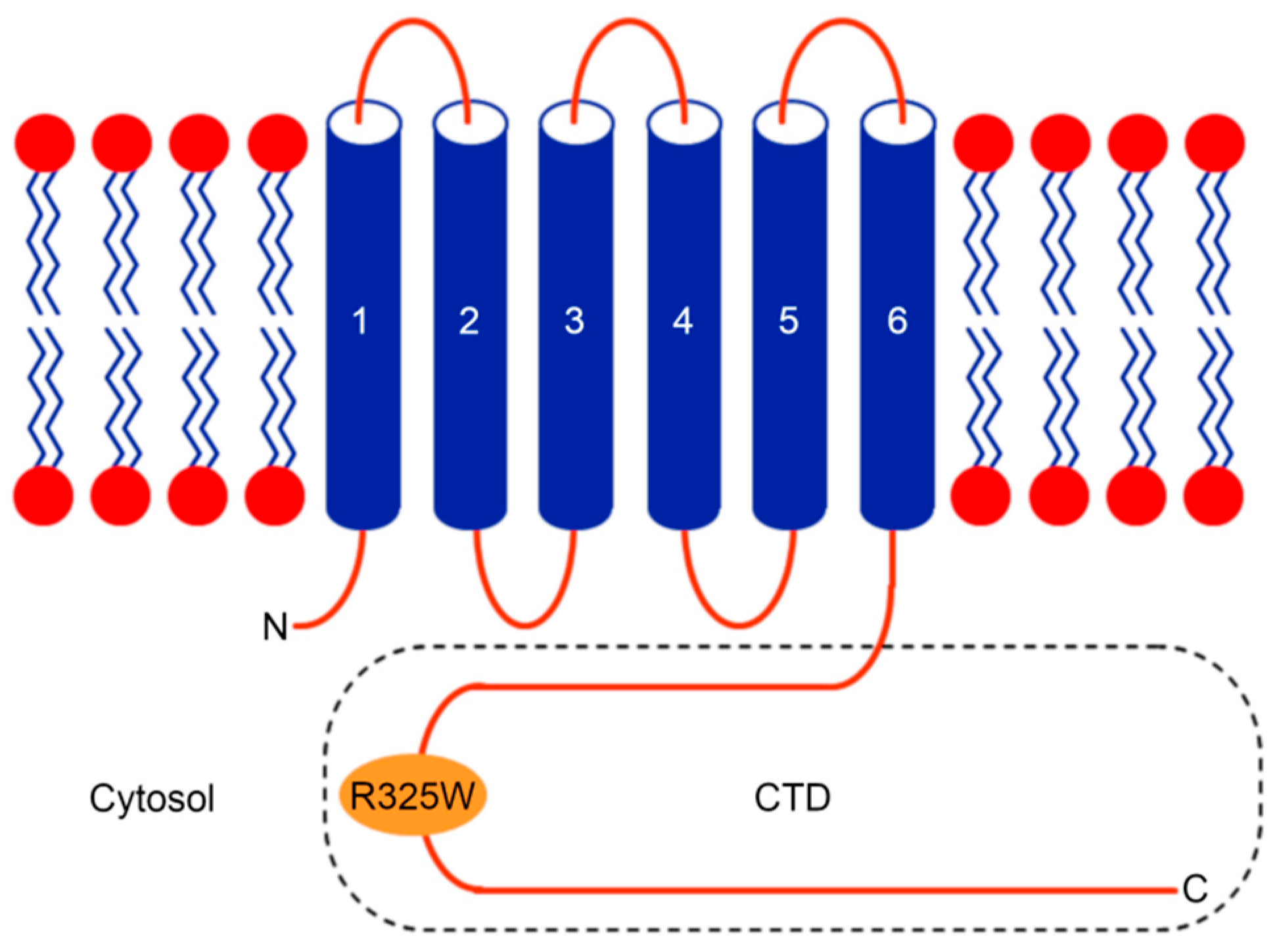
:
1. Introduction
2. Results
2.1. The Maltose-Binding Protein (MBP) Tag Improves the Solubility of the CTDs
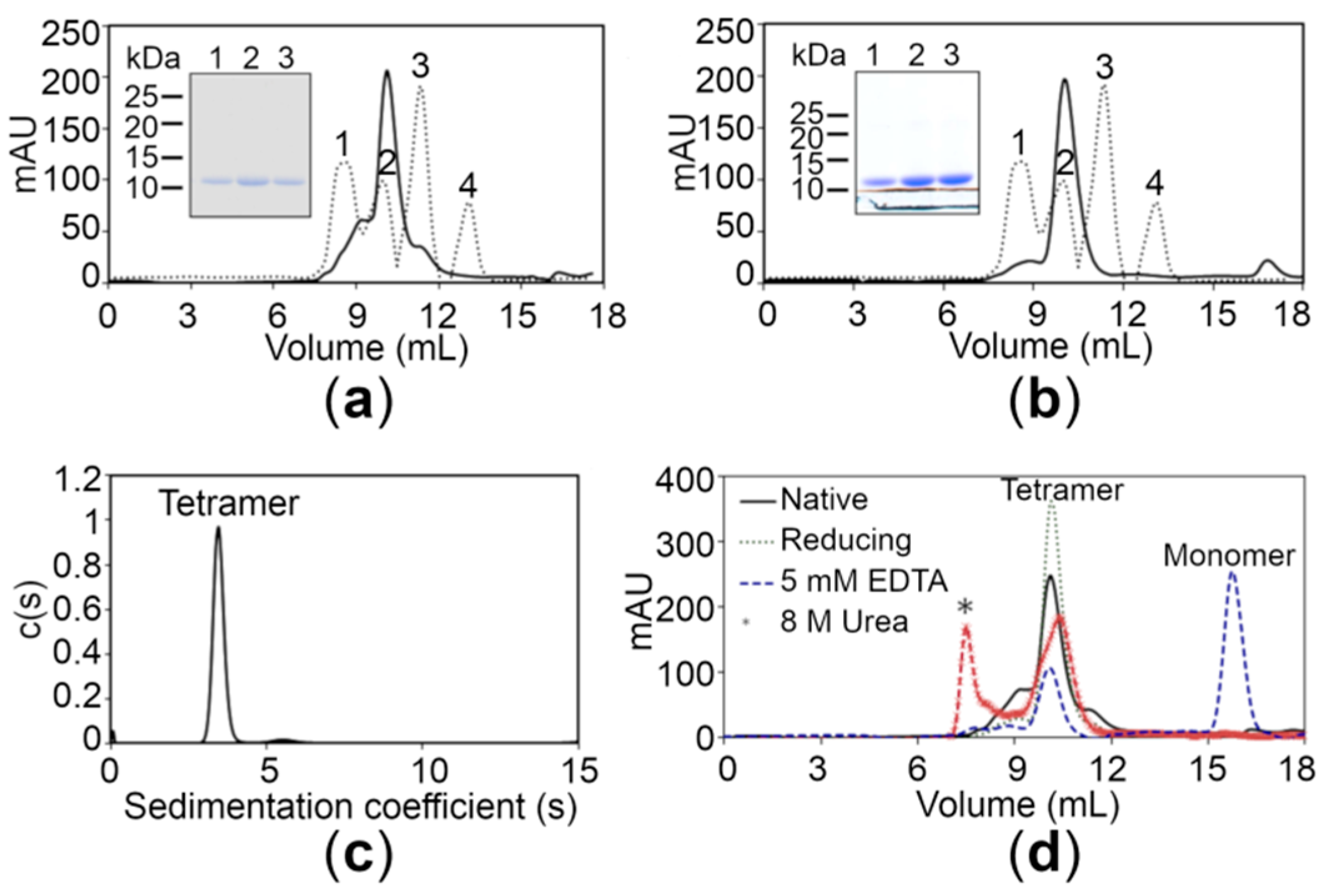
2.2. The CTDs Exist as Tetramers in Solution
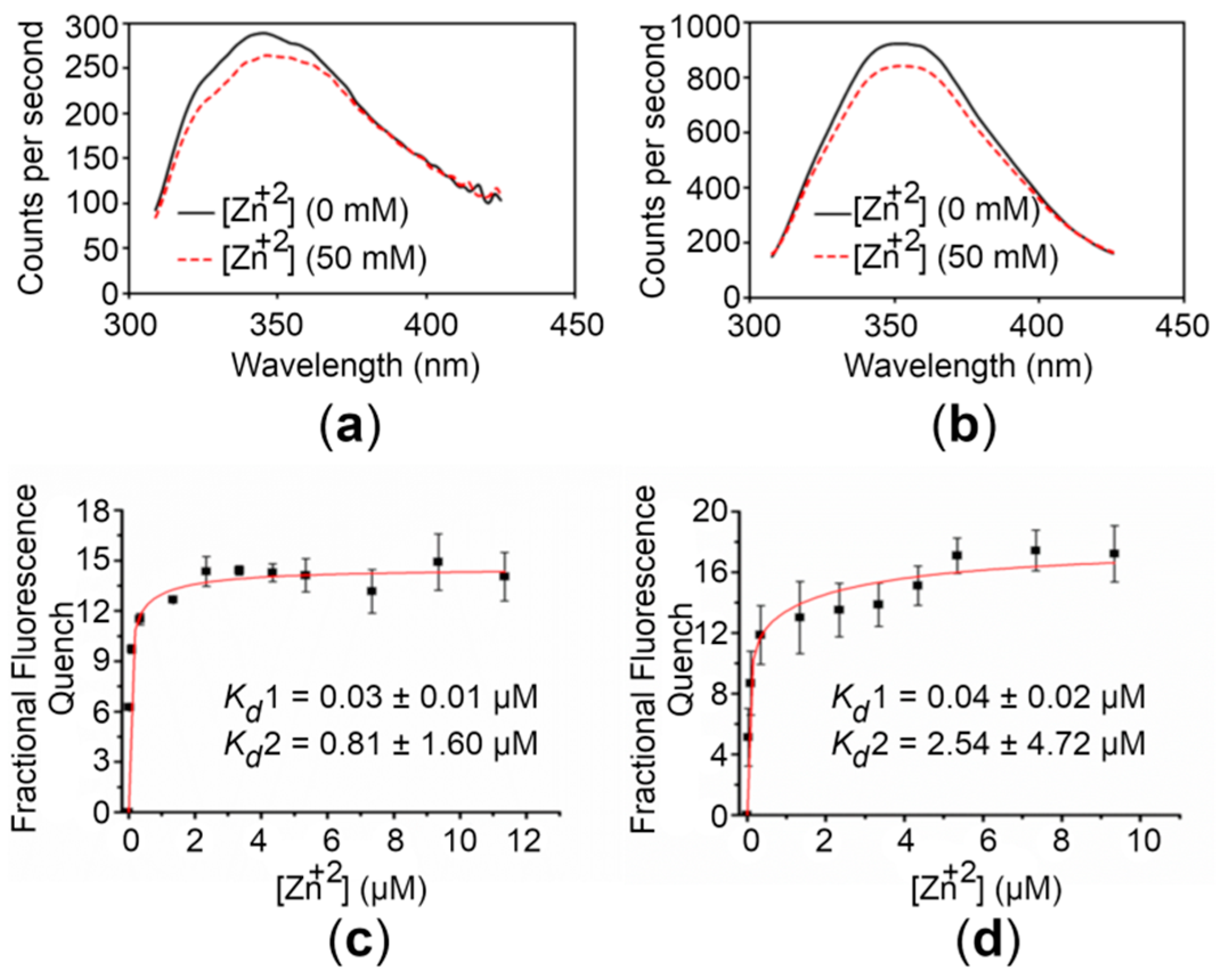
2.3. The CTDs Bind Zn+2 with High Affinity
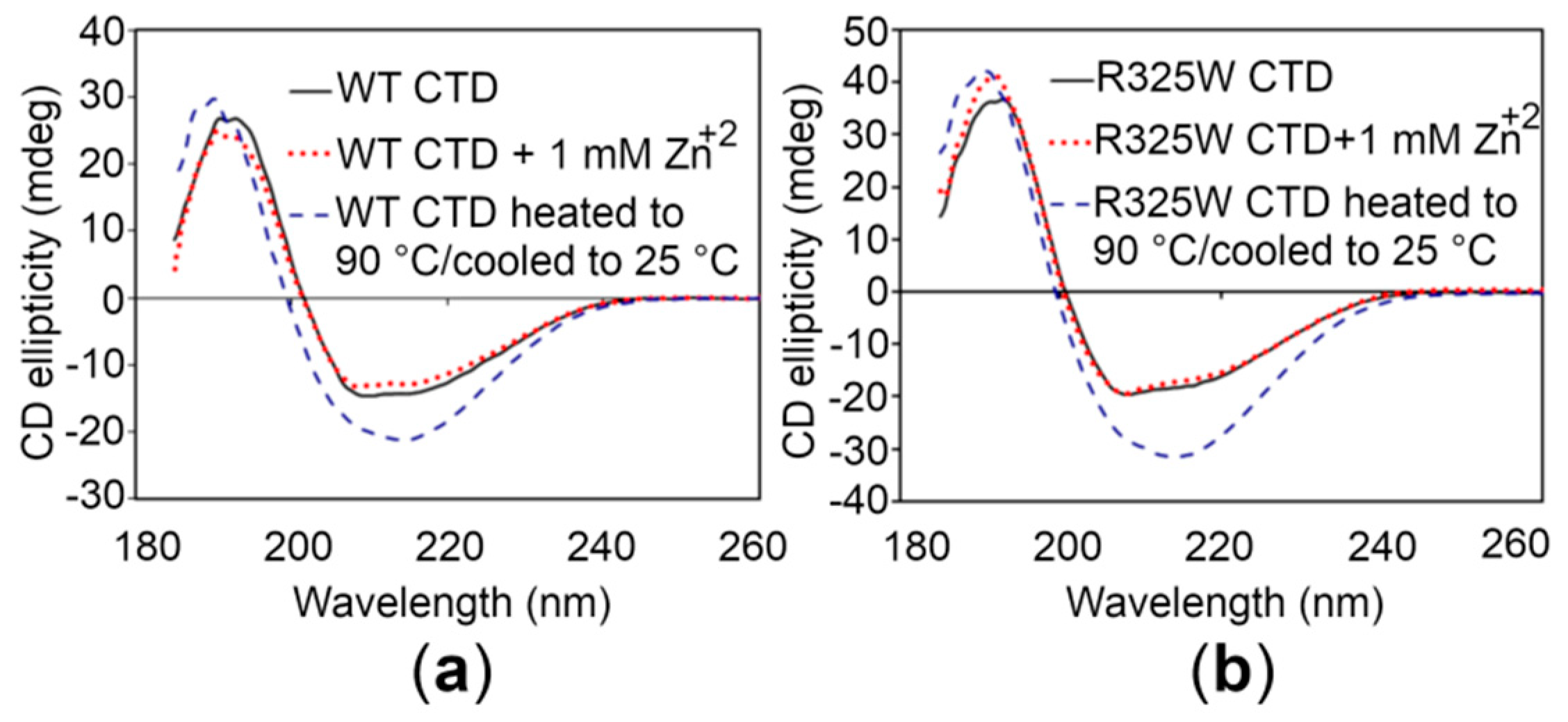
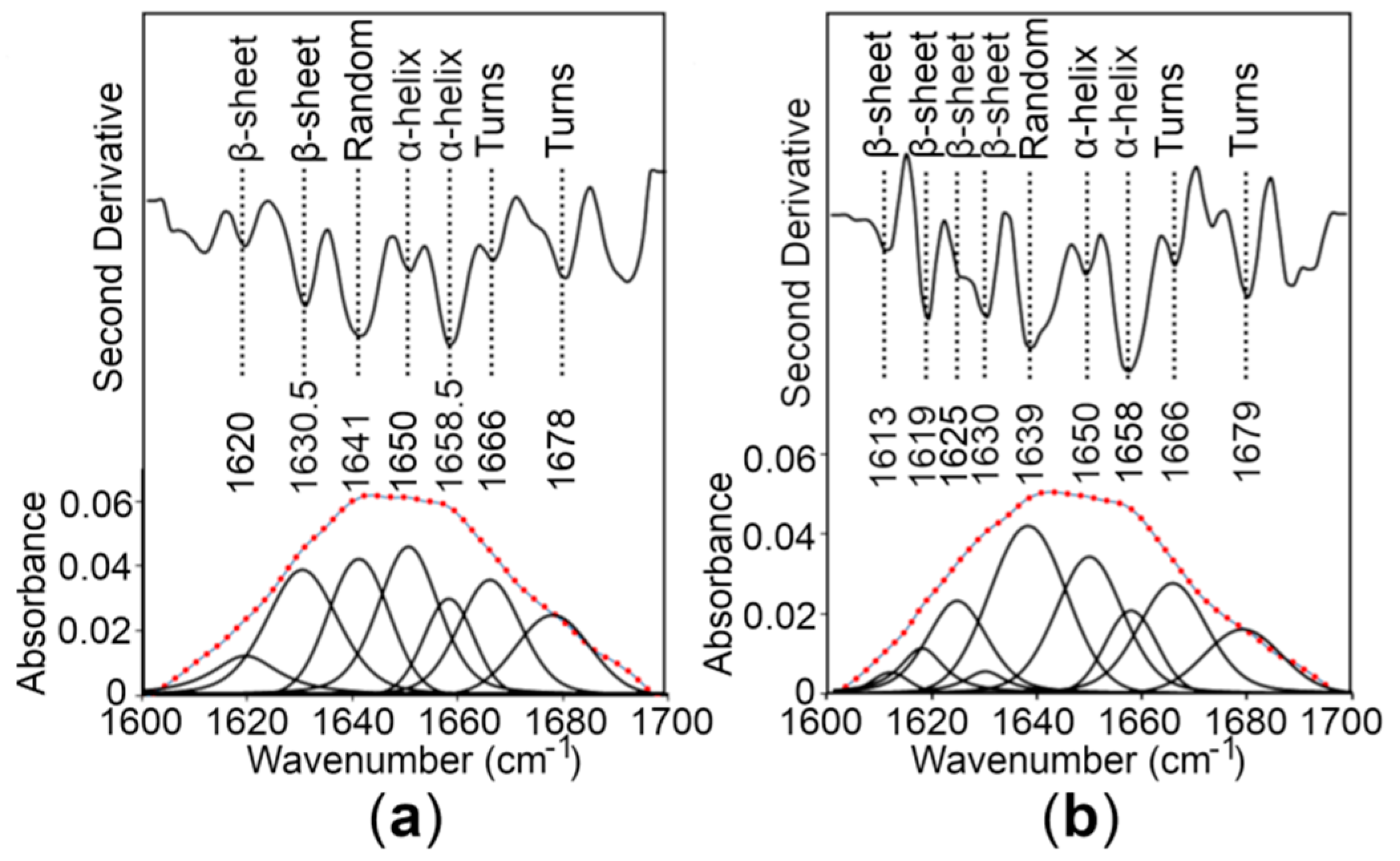
2.4. The CTDs Share Similar Structural Features in the Presence and Absence of Zn+2
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of the CTDs
4.2. Gel Filtration
4.3. Analytical Ultracentrifugation
4.4. Mass Spectrometry
4.5. Negative-Stain EM
4.6. Metal Binding Analysis
4.7. CD Spectroscopy
4.8. FT-IR Spectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Circular dichroism |
| CDF | Cation diffusion facilitator |
| CTD | C-terminal domain |
| EM | Electron microscopy |
| FT-IR | Fourier transform infrared |
| GWAS | Genome-wide association study |
| hZnT8 | Human zinc transporter 8 |
| MBP | Maltose-binding protein |
| MW | Molecular weight |
| SEC | Size-exclusion chromatography |
| SNP | Single nucleotide polymorphism |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TEV site | Tobacco etch virus protease recognition site |
| TMD | Transmembrane domain |
| WT | Wild-type |
| ZIP | Zrt- and Irt-like proteins |
| ZnT | Zinc transporter |
References
- Lichten, L.A.; Cousins, R.J. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef]
- Wijesekara, N.; Chimienti, F.; Wheeler, M.B. Zinc, a regulator of islet function and glucose homeostasis. Diabetes Obes. Metab. 2009, 11, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in cellular regulation: The nature and significance of “zinc signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.M.; Shi, Y. The galvanization of biology: A growing appreciation for the roles of zinc. Science 1996, 271, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Fukada, T.; Kambe, T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics 2011, 3, 662–674. [Google Scholar] [CrossRef]
- Kimura, T.; Kambe, T. The functions of metallothionein and ZIP and ZnT transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [Green Version]
- Kolaj-Robin, O.; Russell, D.; Hayes, K.A.; Pembroke, J.T.; Soulimane, T. Cation diffusion facilitator family: Structure and function. FEBS Lett. 2015, 589, 1283–1295. [Google Scholar] [CrossRef]
- Hara, T.; Takeda, T.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef]
- Chimienti, F.; Devergnas, S.; Favier, A.; Seve, M. Identification and cloning of a β-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes 2004, 53, 2330–2337. [Google Scholar] [CrossRef] [Green Version]
- Chimienti, F.; Favier, A.; Seve, M. ZnT-8, a pancreatic beta-cell-specific zinc transporter. BioMetals 2005, 18, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Chimienti, F.; Schuit, F. Zinc transporters and their role in the pancreatic β-cell. J. Diabetes Investig. 2012, 3, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.M.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]
- Huang, Q.; Du, J.; Merriman, C.; Gong, Z. Genetic, functional, and immunological study of ZnT8 in diabetes. Int. J. Endocrinol. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Chauhan, G.; Spurgeon, C.J.; Tabassum, R.; Bhaskar, S.; Kulkarni, S.R.; Mahajan, A.; Chavali, S.; Kumar, M.V.K.; Prakash, S.; Dwivedi, O.P.; et al. Impact of common variants of PPARG, KCNJ11, TCF7L2, SLC30A8, HHEX, CDKN2A, IGF2BP2, and CDKAL1 on the risk of type 2 diabetes in 5164 Indians. Diabetes 2010, 59, 2068–2074. [Google Scholar] [CrossRef] [Green Version]
- Kommoju, U.J.; Maruda, J.; Kadarkarai, S.; Irgam, K.; Kotla, J.P.; Velaga, L.; Mohan Reddy, B. No detectable association of IGF2BP2 and SLC30A8 genes with type 2 diabetes in the population of Hyderabad, India. Meta Gene 2013, 1, 15–23. [Google Scholar] [CrossRef]
- Kulkarni, H.; Mamtani, M.; Peralta, J.M.; Diego, V.; Dyer, T.D.; Goring, H.; Almasy, L.; Mahaney, M.C.; Williams-Blangero, S.; Duggirala, R.; et al. Lack of association between SLC30A8 variants and type 2 diabetes in Mexican American families. J. Diabetes Res. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.P.; Palmer, N.D.; Hicks, P.J.; Sale, M.M.; Langefeld, C.D.; Freedman, B.I.; Divers, J.; Bowden, D.W. Association analysis in African Americans of European-derived type 2 diabetes single nucleotide polymorphisms from whole-genome association studies. Diabetes 2008, 57, 2220–2225. [Google Scholar] [CrossRef] [Green Version]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.R.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef]
- Lu, M.; Fu, D. Structure of the zinc transporter YiiP. Science 2007, 317, 1746–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Chai, J.; Fu, D. Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol. 2009, 16, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Rosenkranz, E.; Overbeck, S.; Warmuth, S.; Mocchegiani, E.; Giacconi, R.; Weiskirchen, R.; Karges, W.; Rink, L. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J. Nutr. Biochem. 2012, 23, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.S.; Hogstrand, C.; Maret, W. The C-terminal cytosolic domain of the human zinc transporter ZnT8 and its diabetes risk variant. FEBS J. 2018, 285, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weijers, R.N. Three-dimensional structure of β-cell-specific zinc transporter, ZnT-8, predicted from the type 2 diabetes-associated gene variant SLC30A8 R325W. Diabetol. Metab. Syndr. 2010, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Merriman, C.; Atkinson, M.A.; Wasserfall, C.H.; Mcgrail, K.M.; Liang, Y.; Fu, D.; Dai, H. Proteoliposome-based full-length ZnT8 self-antigen for type 1 diabetes diagnosis on a plasmonic platform. Proc. Natl. Acad. Sci. USA 2017, 114, 10196–10201. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J.M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. I. Assignments and model compounds. Subcell. Biochem. 1994, 23, 329–362. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J.M. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell. Biochem. 1994, 23, 405–450. [Google Scholar] [CrossRef]
- Raussens, V.; Ruysschaert, J.-M.; Goormaghtigh, E. Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: A new scaling method. Anal. Biochem. 2003, 319, 114–121. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W.; Glöckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Murgia, C.; Devirgiliis, C.; Mancini, E.; Donadel, G.; Zalewski, P.; Perozzi, G. Diabetes-linked zinc transporter ZnT8 is a homodimeric protein expressed by distinct rodent endocrine cell types in the pancreas and other glands. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Don, R.H.; Cox, P.T.; Wainwright, B.J.; Baker, K.; Mattick, J.S. “Touchdown” PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991, 19, 4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Iram, N.; Shah, M.S.; Ismat, F.; Habib, M.; Iqbal, M.; Hasnain, S.S.; Rahman, M. Heterologous expression, characterization and evaluation of the matrix protein from Newcastle disease virus as a target for antiviral therapies. Appl. Microbiol. Biotechnol. 2014, 98, 1691–1701. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | α-Helix (%) | β-Sheet (%) | β-Turn (%) | Random Coil (%) | Technique |
|---|---|---|---|---|---|
| WT CTD | 41 | 15 | 13 | 31 | CD spectroscopy 1 |
| 46 | 10 | 17 | 27 | CD spectroscopy 2 | |
| R325W CTD | 46 | 12 | 13 | 29 | CD spectroscopy 1 |
| 53 | 10 | 17 | 20 | CD spectroscopy 2 | |
| WT CTD | 29.6 | 26.1 | 27 | 17.3 | FT-IR spectroscopy |
| R325W CTD | 27 | 24 | 25 | 24 | FT-IR spectroscopy |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, R.; Shehzad, A.; Shah, M.A.; March, M.D.; Ismat, F.; Iqbal, M.; Onesti, S.; Rahman, M.; McPherson, M.J. C-Terminal Domain of the Human Zinc Transporter hZnT8 Is Structurally Indistinguishable from Its Disease Risk Variant (R325W). Int. J. Mol. Sci. 2020, 21, 926. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030926
Ullah R, Shehzad A, Shah MA, March MD, Ismat F, Iqbal M, Onesti S, Rahman M, McPherson MJ. C-Terminal Domain of the Human Zinc Transporter hZnT8 Is Structurally Indistinguishable from Its Disease Risk Variant (R325W). International Journal of Molecular Sciences. 2020; 21(3):926. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030926
Chicago/Turabian StyleUllah, Raheem, Aamir Shehzad, Majid Ali Shah, Matteo De March, Fouzia Ismat, Mazhar Iqbal, Silvia Onesti, Moazur Rahman, and Michael J. McPherson. 2020. "C-Terminal Domain of the Human Zinc Transporter hZnT8 Is Structurally Indistinguishable from Its Disease Risk Variant (R325W)" International Journal of Molecular Sciences 21, no. 3: 926. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030926