The Role Played by Wnt/β-Catenin Signaling Pathway in Acute Lymphoblastic Leukemia

 and
and 
Abstract
:1. Introduction
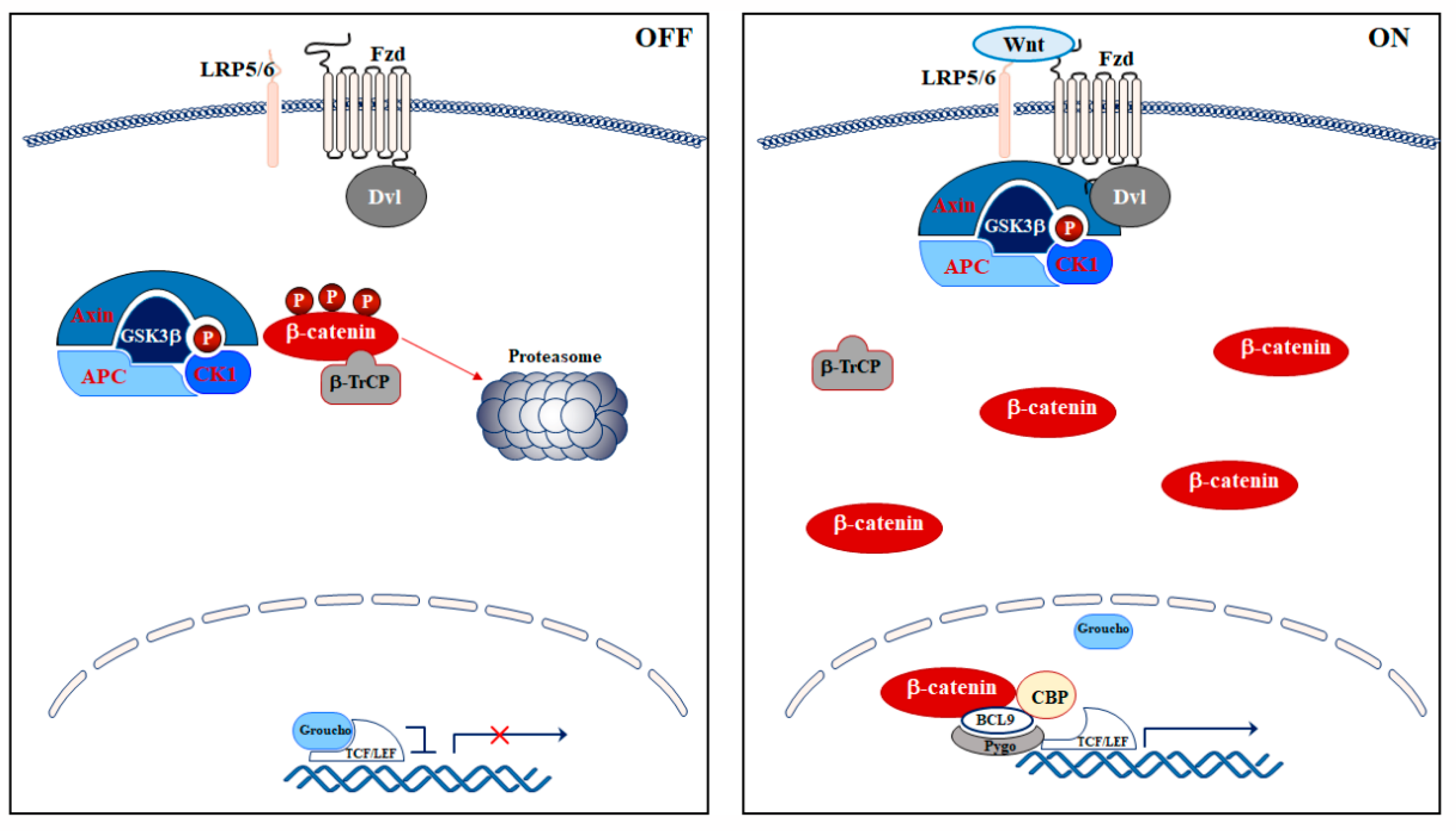
2. Wnt/β-Catenin Signaling Pathway
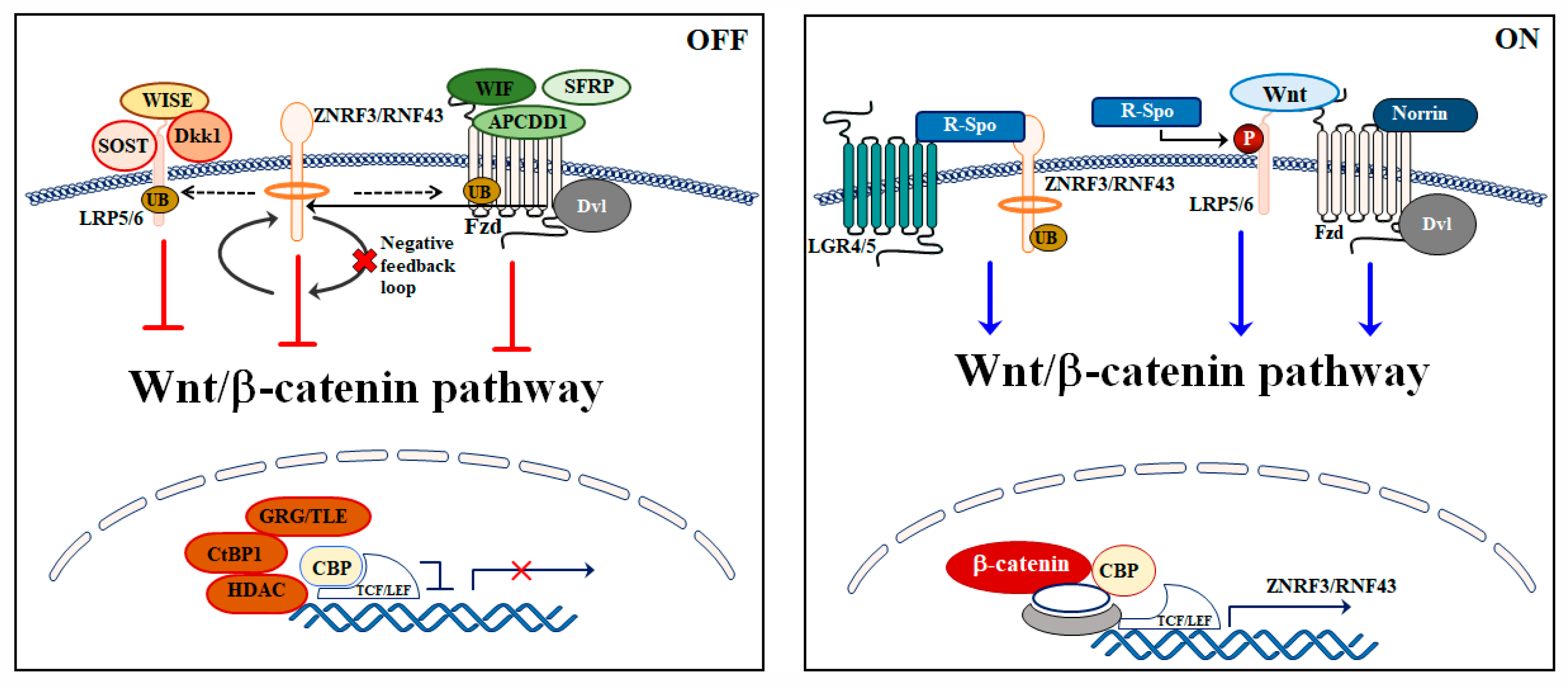
3. Wnt/β-Catenin Signaling Pathway Regulation
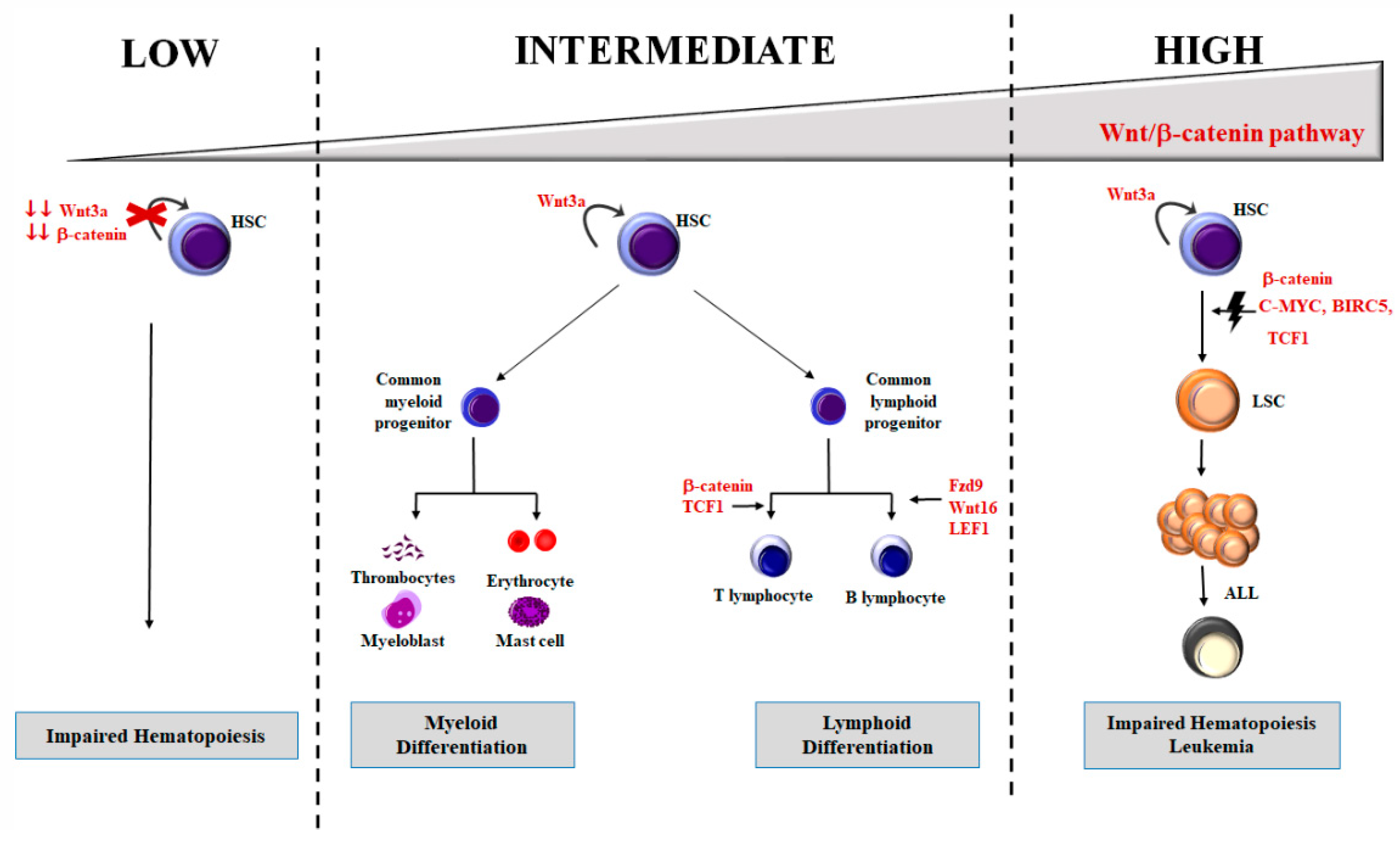
4. Wnt/β-Catenin in Hematopoiesis
5. Wnt/β-Catenin Signaling in Leukemia
5.1. Wnt/β-Catenin in T-ALL
5.2. Wnt/β-Catenin in B-ALL
6. Targeting Wnt/β-Catenin Signaling in ALL
7. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| APC | adenomatous polyposis coli |
| APCDD | adenomatosis polyposis down-regulated |
| B-ALL | B-acute lymphoblastic leukemia |
| β-TrC | β-transducin repeats-containing protein |
| BCL | B-cell lymphoma |
| BET | bromodomains extra terminal proteins |
| BM | bone marrow |
| BRDs | bromodomains |
| BRG1 | ATP-dependent helicase Brahma-related gene |
| CBP | cyclic adenosine mono phosphate response element -binding protein |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CK | casein kinase |
| CREB | cyclic adenosine mono phosphate response element |
| CtBP | C-terminal binding protein |
| Dkk | dickkopf |
| Dvl | disheveled |
| ETP | early T-cell precursor |
| Fzd | frizzled |
| GRG/TLE | groucho/transducin-like enhancer |
| GSK | glycogen synthase kinase |
| HDAC | histone deacetylase |
| Hif | hypoxia-inducible factor |
| HSC | hematopoietic stem cells |
| LEF | lymphoid enhancing factor |
| LGR | leucine-rich repeat-containing G-protein coupled receptor |
| LRP | low-density lipoprotein receptor-related protein |
| LSC | leukemic stem cells |
| MAPK | mitogen-activated protein kinase |
| MEF | myocyte enhancer factor |
| MLL | mixed lineage leukemia |
| mTOR | mechanistic target of rapamycin |
| Ph+ | Philadelphia positive |
| PI3K | phosphatidylinositol 3-kinase |
| PP | protein phosphatase |
| PRL-3 | phosphatase of regenerating liver-3 |
| RNF43 | ring finger 43 |
| SFRP | secreted frizzled-related proteins |
| SOST | sclerostin |
| SOSTDC | sclerostin domain containing |
| T-ALL | T-acute lymphoblastic leukemia |
| TCF | T-cell factor |
| WIF | Wnt inhibitory factor |
| ZNRF3 | cell-surface transmembrane E3 ubiquitin ligase zinc and ring finger 3 |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faderl, S.; O’Brien, S.; Pui, C.H.; Stock, W.; Wetzler, M.; Hoelzer, D.; Kantarjian, H.M. Adult acute lymphoblastic leukemia: Concepts and strategies. Cancer 2010, 116, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef]
- Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. Int. J. Mol. Sci. 2018, 19, 1878. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef]
- Staal, F.J.; Sen, J.M. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol. 2008, 38, 1788–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Bienz, M. Signalosome assembly by domains undergoing dynamic head-to-tail polymerization. Trends Biochem. Sci. 2014, 39, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Huang, H.; Garcia Abreu, J.; He, X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE 2009, 4, e4926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, J.L.; Chu, M.L.; Enos, M.D.; Shah, N.; Weis, W.I. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. eLife 2014, 3, e01998. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Graeb, M.; Mieszczanek, J.; Rutherford, T.J.; Johnson, C.M.; Bienz, M. An ancient Pygo-dependent Wnt enhanceosome integrated by Chip/LDB-SSDP. eLife 2015, 4. [Google Scholar] [CrossRef]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Kim, K.A.; Kakitani, M.; Zhao, J.; Oshima, T.; Tang, T.; Binnerts, M.; Liu, Y.; Boyle, B.; Park, E.; Emtage, P.; et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science 2005, 309, 1256–1259. [Google Scholar] [CrossRef]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [Green Version]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.X.; Jiang, X.; Cong, F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers 2016, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Braunger, B.M.; Tamm, E.R. The different functions of Norrin. Adv. Exp. Med. Biol. 2012, 723, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Harikumar, K.G.; Erice, C.; Chen, C.; Gu, X.; Wang, L.; Parker, N.; Cheng, Z.; Xu, W.; Williams, B.O.; et al. Structure and function of Norrin in assembly and activation of a Frizzled 4-Lrp5/6 complex. Genes Dev. 2013, 27, 2305–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. Norrin and frizzled: A new vein for the eye. Dev. Cell 2004, 6, 453–454. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, Y.; Agalliu, D.; Vonica, A.; Luria, V.; Wajid, M.; Baumer, A.; Belli, S.; Petukhova, L.; Schinzel, A.; Brivanlou, A.H.; et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature 2010, 464, 1043–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.; Sanderson, B.W.; Klein, O.D.; Krumlauf, R. Inhibition of Wnt signaling by Wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development 2010, 137, 3221–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenov, M.; Tamai, K.; He, X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005, 280, 26770–26775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, T.; Zaret, K.S. Repression by Groucho/TLE/Grg proteins: Genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol. Cell. 2007, 28, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Shooshtarizadeh, P.; Helness, A.; Vadnais, C.; Brouwer, N.; Beauchemin, H.; Chen, R.; Bagci, H.; Staal, F.J.T.; Cote, J.F.; Moroy, T. Gfi1b regulates the level of Wnt/beta-catenin signaling in hematopoietic stem cells and megakaryocytes. Nat. Commun. 2019, 10, 1270. [Google Scholar] [CrossRef] [Green Version]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef]
- Wu, W.; Glinka, A.; Delius, H.; Niehrs, C. Mutual antagonism between dickkopf1 and dickkopf2 regulates Wnt/beta-catenin signalling. Curr. Biol. 2000, 10, 1611–1614. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Mao, J.; Sun, L.; Liu, W.; Wu, D. Second cysteine-rich domain of Dickkopf-2 activates canonical Wnt signaling pathway via LRP-6 independently of dishevelled. J. Biol. Chem. 2002, 277, 5977–5981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, B.; Niehrs, C. Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signaling. Gene 2003, 302, 179–183. [Google Scholar] [CrossRef]
- Brott, B.K.; Sokol, S.Y. Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol. Cell Biol. 2002, 22, 6100–6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 2016, 1863, 449–463. [Google Scholar] [CrossRef]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Scheller, M.; Huelsken, J.; Rosenbauer, F.; Taketo, M.M.; Birchmeier, W.; Tenen, D.G.; Leutz, A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat. Immunol. 2006, 7, 1037–1047. [Google Scholar] [CrossRef]
- Kirstetter, P.; Anderson, K.; Porse, B.T.; Jacobsen, S.E.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056. [Google Scholar] [CrossRef]
- Luis, T.C.; Naber, B.A.; Roozen, P.P.; Brugman, M.H.; de Haas, E.F.; Ghazvini, M.; Fibbe, W.E.; van Dongen, J.J.; Fodde, R.; Staal, F.J. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 2011, 9, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Famili, F.; Brugman, M.H.; Taskesen, E.; Naber, B.E.A.; Fodde, R.; Staal, F.J.T. High Levels of Canonical Wnt Signaling Lead to Loss of Stemness and Increased Differentiation in Hematopoietic Stem Cells. Stem Cell Rep. 2016, 6, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobas, M.; Wilson, A.; Ernst, B.; Mancini, S.J.; MacDonald, H.R.; Kemler, R.; Radtke, F. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J. Exp. Med. 2004, 199, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeannet, G.; Scheller, M.; Scarpellino, L.; Duboux, S.; Gardiol, N.; Back, J.; Kuttler, F.; Malanchi, I.; Birchmeier, W.; Leutz, A.; et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood 2008, 111, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Luis, T.C.; Weerkamp, F.; Naber, B.A.; Baert, M.R.; de Haas, E.F.; Nikolic, T.; Heuvelmans, S.; De Krijger, R.R.; van Dongen, J.J.; Staal, F.J. Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood 2009, 113, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cui, Z.; Wang, F.; Yao, Y.; Yu, G.; Cao, D.; Niu, S.; You, M.; Sun, Z.; Lian, D.; et al. Lrp5 and Lrp6 are required for maintaining self-renewal and differentiation of hematopoietic stem cells. FASEB J. 2019, 33, 5615–5625. [Google Scholar] [CrossRef]
- Wang, Y.; Nakayama, N. WNT and BMP signaling are both required for hematopoietic cell development from human ES cells. Stem Cell Res. 2009, 3, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Gertow, K.; Hirst, C.E.; Yu, Q.C.; Ng, E.S.; Pereira, L.A.; Davis, R.P.; Stanley, E.G.; Elefanty, A.G. WNT3A promotes hematopoietic or mesenchymal differentiation from hESCs depending on the time of exposure. Stem Cell Rep. 2013, 1, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Luis, T.C.; Naber, B.A.; Fibbe, W.E.; van Dongen, J.J.; Staal, F.J. Wnt3a nonredundantly controls hematopoietic stem cell function and its deficiency results in complete absence of canonical Wnt signaling. Blood 2010, 116, 496–497. [Google Scholar] [CrossRef]
- Richter, J.; Stanley, E.G.; Ng, E.S.; Elefanty, A.G.; Traver, D.; Willert, K. WNT9A Is a Conserved Regulator of Hematopoietic Stem and Progenitor Cell Development. Genes 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezer, D.; Vukovic, M.; Soga, T.; Pollard, P.J.; Kranc, K.R. Concise review: Genetic dissection of hypoxia signaling pathways in normal and leukemic stem cells. Stem Cells 2014, 32, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Lento, W.; Congdon, K.; Voermans, C.; Kritzik, M.; Reya, T. Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Biol. 2013, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Ma, Z.; Hsieh, J.C.; Fan, C.W.; Chen, B.; Longgood, J.C.; Williams, N.S.; Amatruda, J.F.; Lum, L.; Chen, C. Structure-activity relationship studies of small-molecule inhibitors of Wnt response. Bioorg. Med. Chem. Lett. 2009, 19, 3825–3827. [Google Scholar] [CrossRef] [Green Version]
- Derksen, P.W.; Tjin, E.; Meijer, H.P.; Klok, M.D.; MacGillavry, H.D.; van Oers, M.H.; Lokhorst, H.M.; Bloem, A.C.; Clevers, H.; Nusse, R.; et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6122–6127. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.I.; Bradstock, K.F.; Bendall, L.J. Activation of Wnt/beta-catenin pathway mediates growth and survival in B-cell progenitor acute lymphoblastic leukaemia. Br. J. Haematol. 2007, 138, 338–348. [Google Scholar] [CrossRef]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, S.; Romanos-Sirakis, E.; Pais, F.; Bhatla, T.; Jones, C.; Bourgeois, W.; Hunger, S.P.; Raetz, E.A.; Hermiston, M.L.; Dasgupta, R.; et al. Wnt inhibition leads to improved chemosensitivity in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 167, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gang, E.J.; Hsieh, Y.T.; Pham, J.; Zhao, Y.; Nguyen, C.; Huantes, S.; Park, E.; Naing, K.; Klemm, L.; Swaminathan, S.; et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33, 2169–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.L.; Zierold, C.; Ranheim, E.A. Dysregulation of Frizzled 6 is a critical component of B-cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 2009, 113, 3031–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Cordeu, L.; Vilas-Zornoza, A.; San Jose-Eneriz, E.; Garate, L.; Castillejo, J.A.; Martin, V.; Prosper, F.; Heiniger, A.; et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur. J. Cancer 2007, 43, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.H.; Erbilgin, Y.; Firtina, S.; Celkan, T.; Karakas, Z.; Aydogan, G.; Turkkan, E.; Yildirmak, Y.; Timur, C.; Zengin, E.; et al. Deregulated WNT signaling in childhood T-cell acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Zhou, X.; Steinke, F.C.; Liu, C.; Chen, S.C.; Zagorodna, O.; Jing, X.; Yokota, Y.; Meyerholz, D.K.; Mullighan, C.G.; et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 2012, 37, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Tiemessen, M.M.; Baert, M.R.; Schonewille, T.; Brugman, M.H.; Famili, F.; Salvatori, D.C.; Meijerink, J.P.; Ozbek, U.; Clevers, H.; van Dongen, J.J.; et al. The nuclear effector of Wnt-signaling, Tcf1, functions as a T-cell-specific tumor suppressor for development of lymphomas. PLoS Biol. 2012, 10, e1001430. [Google Scholar] [CrossRef] [Green Version]
- Nygren, M.K.; Dosen, G.; Hystad, M.E.; Stubberud, H.; Funderud, S.; Rian, E. Wnt3A activates canonical Wnt signalling in acute lymphoblastic leukaemia (ALL) cells and inhibits the proliferation of B-ALL cell lines. Br. J. Haematol. 2007, 136, 400–413. [Google Scholar] [CrossRef]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.C.; Rodrigues Alves, A.P.N.; Machado-Neto, J.A.; Scopim-Ribeiro, R.; Fenerich, B.A.; da Silva, F.B.; Simoes, B.P.; Rego, E.M.; Traina, F. IRS1/beta-Catenin Axis Is Activated and Induces MYC Expression in Acute Lymphoblastic Leukemia Cells. J. Cell Biochem. 2017, 118, 1774–1781. [Google Scholar] [CrossRef]
- Gekas, C.; D’Altri, T.; Aligue, R.; Gonzalez, J.; Espinosa, L.; Bigas, A. beta-Catenin is required for T-cell leukemia initiation and MYC transcription downstream of Notch1. Leukemia 2016, 30, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Dose, M.; Kovalovsky, D.; Chang, R.; O’Neil, J.; Look, A.T.; von Boehmer, H.; Khazaie, K.; Gounari, F. Beta-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood 2007, 109, 5463–5472. [Google Scholar] [CrossRef]
- Guo, W.; Lasky, J.L.; Chang, C.J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M.; et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Giambra, V.; Jenkins, C.E.; Lam, S.H.; Hoofd, C.; Belmonte, M.; Wang, X.; Gusscott, S.; Gracias, D.; Weng, A.P. Leukemia stem cells in T-ALL require active Hif1alpha and Wnt signaling. Blood 2015, 125, 3917–3927. [Google Scholar] [CrossRef] [Green Version]
- Kaveri, D.; Kastner, P.; Dembele, D.; Nerlov, C.; Chan, S.; Kirstetter, P. beta-Catenin activation synergizes with Pten loss and Myc overexpression in Notch-independent T-ALL. Blood 2013, 122, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, R.; Liu, J.; Li, M.; Song, C.; Dovat, S.; Li, J.; Ge, Z. Characterization of LEF1 High Expression and Novel Mutations in Adult Acute Lymphoblastic Leukemia. PLoS ONE 2015, 10, e0125429. [Google Scholar] [CrossRef]
- Gutierrez, A.; Sanda, T.; Ma, W.; Zhang, J.; Grebliunaite, R.; Dahlberg, S.; Neuberg, D.; Protopopov, A.; Winter, S.S.; Larson, R.S.; et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood 2010, 115, 2845–2851. [Google Scholar] [CrossRef] [Green Version]
- McWhirter, J.R.; Neuteboom, S.T.; Wancewicz, E.V.; Monia, B.P.; Downing, J.R.; Murre, C. Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia. Proc. Natl. Acad. Sci. USA 1999, 96, 11464–11469. [Google Scholar] [CrossRef] [Green Version]
- Mazieres, J.; You, L.; He, B.; Xu, Z.; Lee, A.Y.; Mikami, I.; McCormick, F.; Jablons, D.M. Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene 2005, 24, 5396–5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mallampati, S.; Sun, B.; Zhang, J.; Kim, S.B.; Lee, J.S.; Gong, Y.; Cai, Z.; Sun, X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013, 333, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygren, M.K.; Dosen-Dahl, G.; Stubberud, H.; Walchli, S.; Munthe, E.; Rian, E. beta-catenin is involved in N-cadherin-dependent adhesion, but not in canonical Wnt signaling in E2A-PBX1-positive B acute lymphoblastic leukemia cells. Exp. Hematol. 2009, 37, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J. Cell. Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Li, H.; Chen, Y.W.; Srivastava, G.; Gao, Z.; Tao, Q. WNT5A is epigenetically silenced in hematologic malignancies and inhibits leukemia cell growth as a tumor suppressor. Blood 2007, 110, 4130–4132. [Google Scholar] [CrossRef]
- Petropoulos, K.; Arseni, N.; Schessl, C.; Stadler, C.R.; Rawat, V.P.; Deshpande, A.J.; Heilmeier, B.; Hiddemann, W.; Quintanilla-Martinez, L.; Bohlander, S.K.; et al. A novel role for Lef-1, a central transcription mediator of Wnt signaling, in leukemogenesis. J. Exp. Med. 2008, 205, 515–522. [Google Scholar] [CrossRef] [Green Version]
- Kuhnl, A.; Gokbuget, N.; Kaiser, M.; Schlee, C.; Stroux, A.; Burmeister, T.; Mochmann, L.H.; Hoelzer, D.; Hofmann, W.K.; Thiel, E.; et al. Overexpression of LEF1 predicts unfavorable outcome in adult patients with B-precursor acute lymphoblastic leukemia. Blood 2011, 118, 6362–6367. [Google Scholar] [CrossRef]
- Owattanapanich, W.; Rujirachun, P.; Ungprasert, P.; Buaboonnam, J.; Techavichit, P. Prevalence and Clinical Outcome of Philadelphia-Like Acute Lymphoblastic Leukemia: Systematic Review and Meta-analysis. Clin. Lymphoma Myeloma Leuk. 2019, 20, S2152–S2650. [Google Scholar] [CrossRef] [Green Version]
- Martin, V.; Agirre, X.; Jimenez-Velasco, A.; Jose-Eneriz, E.S.; Cordeu, L.; Garate, L.; Vilas-Zornoza, A.; Castillejo, J.A.; Heiniger, A.; Prosper, F.; et al. Methylation status of Wnt signaling pathway genes affects the clinical outcome of Philadelphia-positive acute lymphoblastic leukemia. Cancer Sci. 2008, 99, 1865–1868. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Yang, S.J.; Chen, X.H. Growth of tyrosine kinase inhibitor-resistant Philadelphia-positive acute lymphoblastic leukemia: Role of bone marrow stromal cells. Oncol. Lett. 2017, 13, 2059–2070. [Google Scholar] [CrossRef] [Green Version]
- Linsdell, P. Cystic fibrosis transmembrane conductance regulator (CFTR): Making an ion channel out of an active transporter structure. Channels 2018, 12, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yan, T.; Gong, Y.; Liu, X.; Sun, H.; Xu, W.; Wang, C.; Naren, D.; Zheng, Y. High CFTR expression in Philadelphia chromosome-positive acute leukemia protects and maintains continuous activation of BCR-ABL and related signaling pathways in combination with PP2A. Oncotarget 2017, 8, 24437–24448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Gang, E.J.; Hsieh, Y.T.; Schaefer, P.; Chae, S.; Klemm, L.; Huantes, S.; Loh, M.; Conway, E.M.; Kang, E.S.; et al. Targeting survivin overcomes drug resistance in acute lymphoblastic leukemia. Blood 2011, 118, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Hogan, L.E.; Condos, G.; Bhatla, T.; Germino, N.; Moskowitz, N.P.; Lee, L.; Bhojwani, D.; Horton, T.M.; Belitskaya-Levy, I.; et al. Endogenous knockdown of survivin improves chemotherapeutic response in ALL models. Leukemia 2012, 26, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Raetz, E.A.; Morrison, D.; Romanos-Sirakis, E.; Gaynon, P.; Sposto, R.; Bhojwani, D.; Bostrom, B.C.; Brown, P.; Eckroth, E.; Cassar, J.; et al. A phase I study of EZN-3042, a novel survivin messenger ribonucleic acid (mRNA) antagonist, administered in combination with chemotherapy in children with relapsed acute lymphoblastic leukemia (ALL): A report from the therapeutic advances in childhood leukemia and lymphoma (TACL) consortium. J. Pediatr. Hematol. Oncol. 2014, 36, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Curtin, J.C.; Lorenzi, M.V. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010, 1, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Harb, J.; Lin, P.J.; Hao, J. Recent Development of Wnt Signaling Pathway Inhibitors for Cancer Therapeutics. Curr. Oncol. Rep. 2019, 21, 12. [Google Scholar] [CrossRef]
- Arques, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argiles, G.; Dienstmann, R.; Fernandez, N.; Caratu, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase Inhibition Blocks Wnt/beta-Catenin Pathway and Reverts Resistance to PI3K and AKT Inhibitors in the Treatment of Colorectal Cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.L.; Kim, H.P.; Cho, Y.W.; Min, D.W.; Cheon, S.K.; Lim, Y.J.; Song, S.H.; Kim, S.J.; Han, S.W.; Park, K.J.; et al. Activation of WNT/beta-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int. J. Cancer 2019, 144, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Li, Z.; Wang, Y.; Zhang, L.; Wu, H. Secreted pyruvate kinase M2 facilitates cell migration via PI3K/Akt and Wnt/beta-catenin pathway in colon cancer cells. Biochem. Biophys. Res. Commun. 2015, 459, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Zhao, L.; Yan, X.L.; Huang, Y.H. Loss of G3BP1 suppresses proliferation, migration, and invasion of esophageal cancer cells via Wnt/beta-catenin and PI3K/AKT signaling pathways. J. Cell Physiol. 2019, 234, 20469–20484. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, M.T.; Cox, A.C.; Shorning, B.Y.; Meniel, V.; Griffiths, D.; Kynaston, H.G.; Smalley, M.J.; Clarke, A.R. PTEN loss and activation of K-RAS and beta-catenin cooperate to accelerate prostate tumourigenesis. J. Pathol. 2017, 243, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Toh, S.H.; Chan, Z.L.; Quah, J.Y.; Chooi, J.Y.; Tan, T.Z.; Chong, P.S.Y.; Zeng, Q.; Chng, W.J. A loss-of-function genetic screening reveals synergistic targeting of AKT/mTOR and WTN/beta-catenin pathways for treatment of AML with high PRL-3 phosphatase. J. Hematol. Oncol. 2018, 11, 36. [Google Scholar] [CrossRef]
- Evangelisti, C.; Chiarini, F.; Cappellini, A.; Paganelli, F.; Fini, M.; Santi, S.; Martelli, A.M.; Neri, L.M. Targeting Wnt/beta-catenin and PI3K/Akt/mTOR pathways in T-cell acute lymphoblastic leukemia. J. Cell Physiol. 2020. [Google Scholar] [CrossRef]
- Bigas, A.; Guiu, J.; Gama-Norton, L. Notch and Wnt signaling in the emergence of hematopoietic stem cells. Blood Cells Mol. Dis. 2013, 51, 264–270. [Google Scholar] [CrossRef]
- Hogan, L.E.; Meyer, J.A.; Yang, J.; Wang, J.; Wong, N.; Yang, W.; Condos, G.; Hunger, S.P.; Raetz, E.; Saffery, R.; et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011, 118, 5218–5226. [Google Scholar] [CrossRef] [Green Version]
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 2014, 123, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Loosveld, M.; Castellano, R.; Gon, S.; Goubard, A.; Crouzet, T.; Pouyet, L.; Prebet, T.; Vey, N.; Nadel, B.; Collette, Y.; et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget 2014, 5, 3168–3172. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2019, 33, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, Z.; Numata, A.; Kawasaki, A.; Tenen, D.G.; Virshup, D.M. Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood 2015, 126, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feder, K.; Edmaier-Schroger, K.; Rawat, V.P.S.; Kirsten, N.; Metzeler, K.; Kraus, J.M.; Dohner, K.; Dohner, H.; Kestler, H.A.; Feuring-Buske, M.; et al. Differences in expression and function of LEF1 isoforms in normal versus leukemic hematopoiesis. Leukemia 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Clinical Trials | Reference |
|---|---|---|---|
| XAV939 | Tankyrase | [92] | |
| ICG-001 | CBP/β-catenin | [72] | |
| iCART14 | β-catenin-dep gene expression | [71] | |
| PKF115-584 | β-catenin/LEF1 interaction | [81] | |
| CWP23229 | β-catenin-dep gene expression | NCT01398462 | [107] |
| PRI-724 | CBP/β-catenin | NCT01606579 NCT02195440 | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarini, F.; Paganelli, F.; Martelli, A.M.; Evangelisti, C. The Role Played by Wnt/β-Catenin Signaling Pathway in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031098
Chiarini F, Paganelli F, Martelli AM, Evangelisti C. The Role Played by Wnt/β-Catenin Signaling Pathway in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2020; 21(3):1098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031098
Chicago/Turabian StyleChiarini, Francesca, Francesca Paganelli, Alberto M. Martelli, and Camilla Evangelisti. 2020. "The Role Played by Wnt/β-Catenin Signaling Pathway in Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 21, no. 3: 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031098