Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status

 , , , ,
, , , ,
Abstract
:1. Introduction
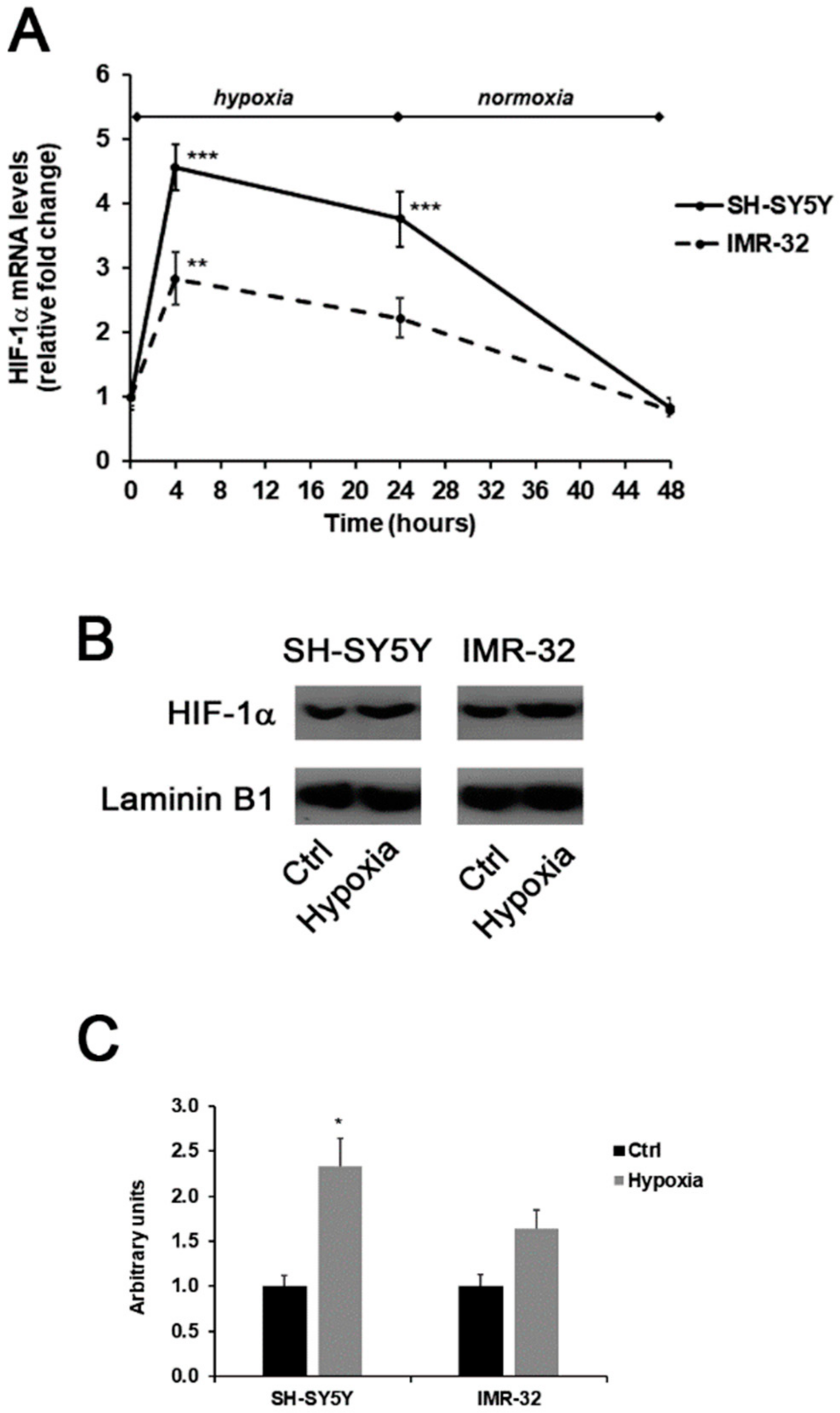
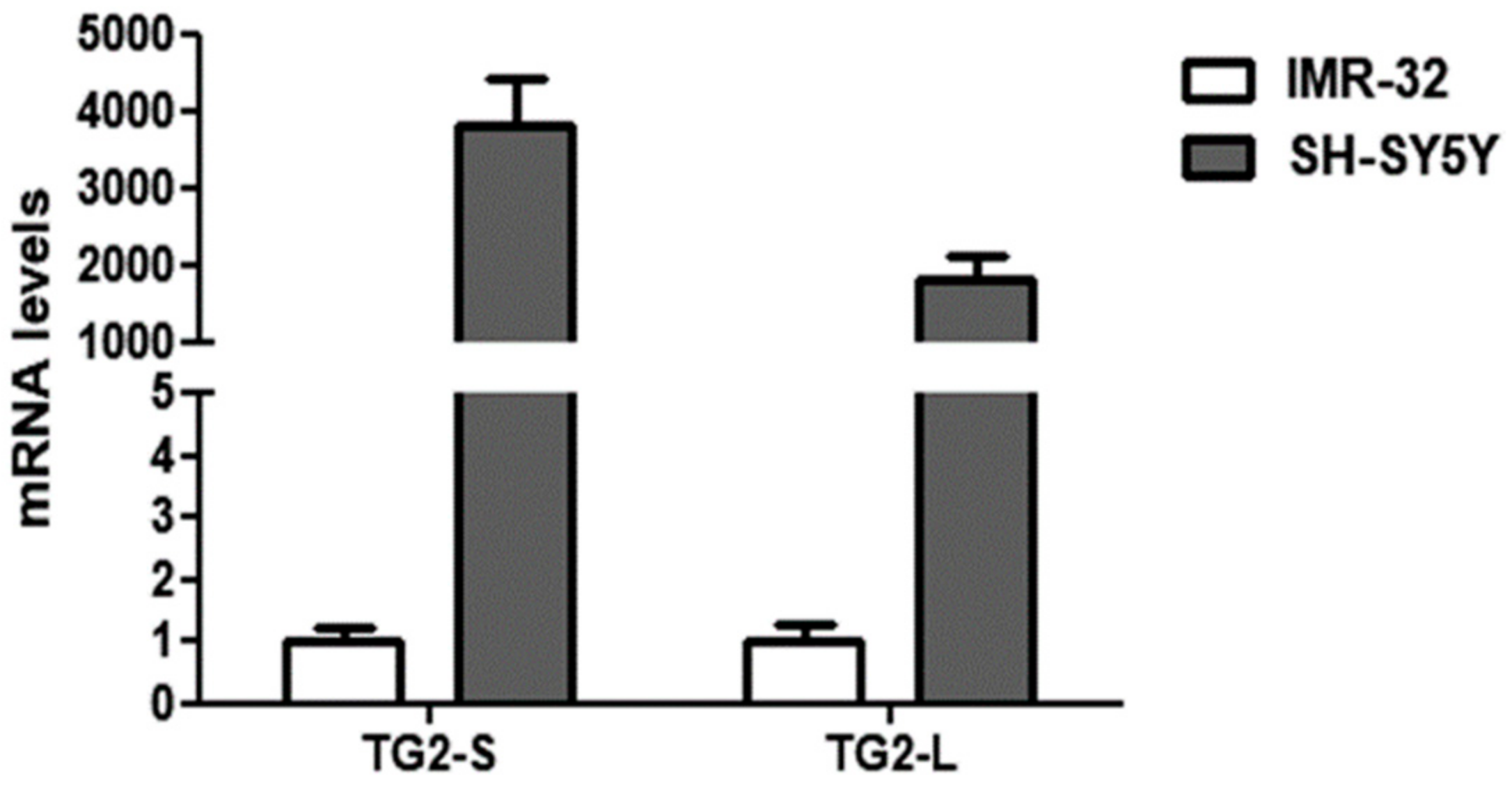
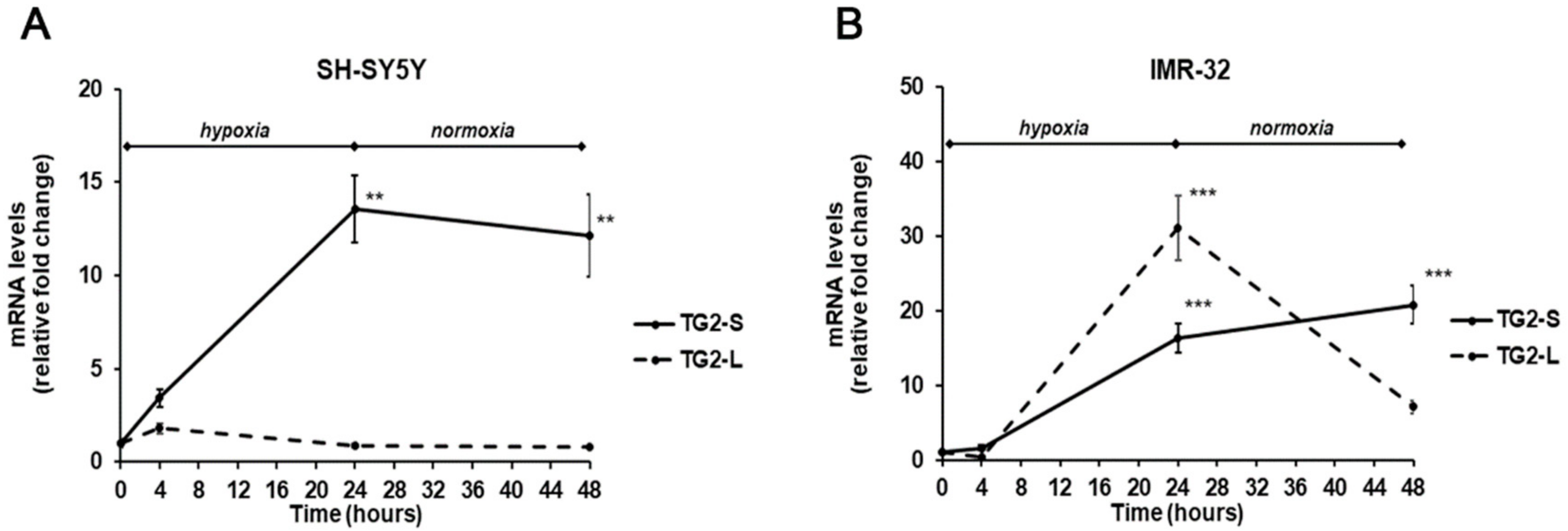
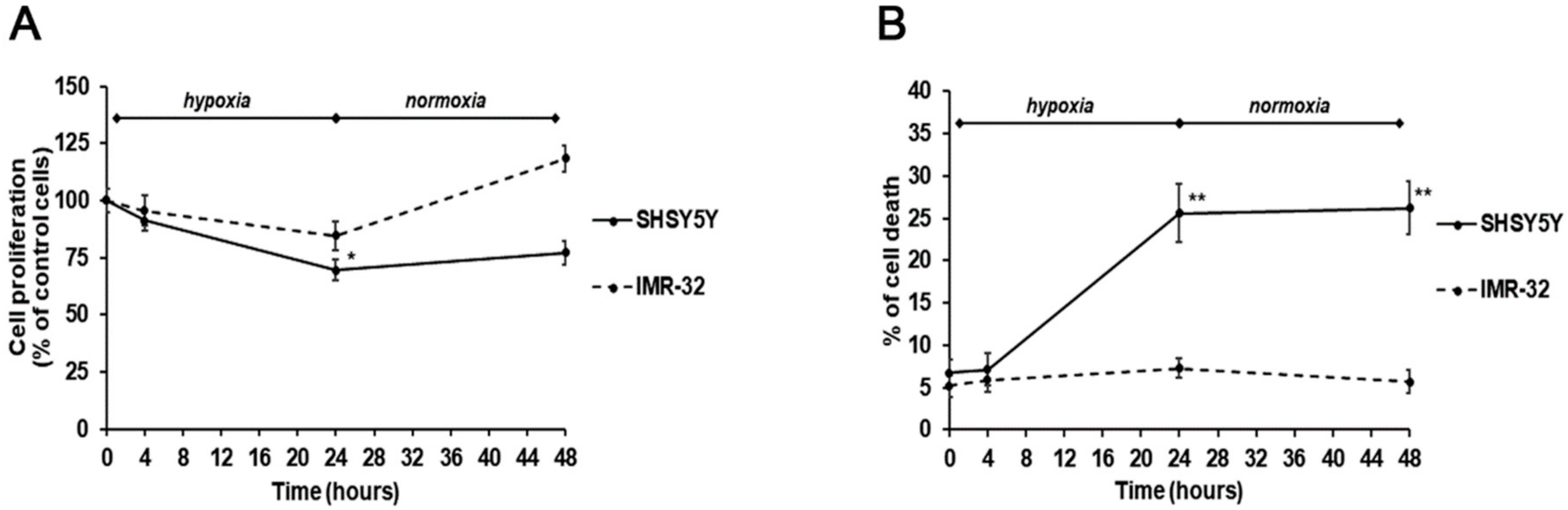
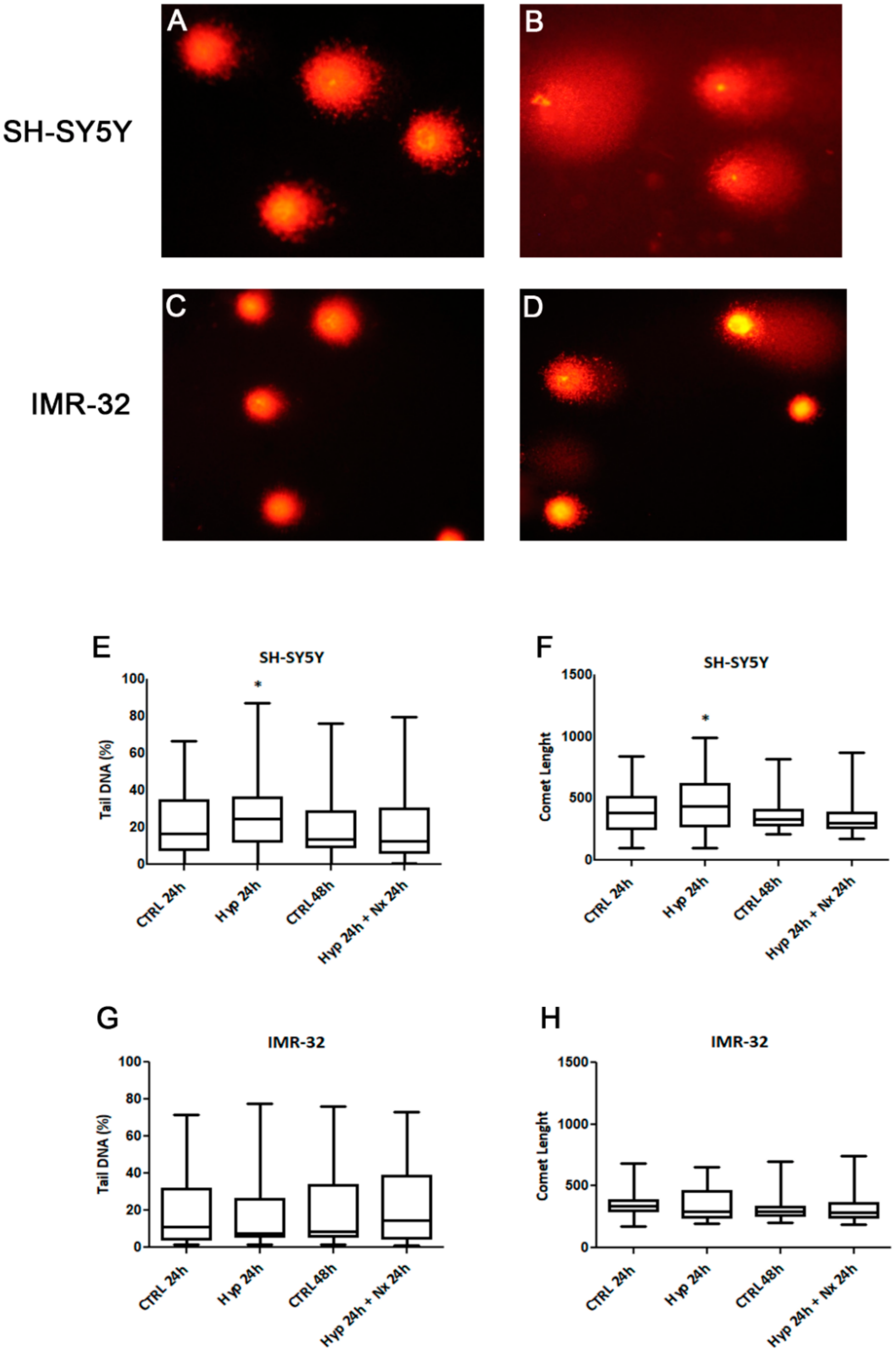
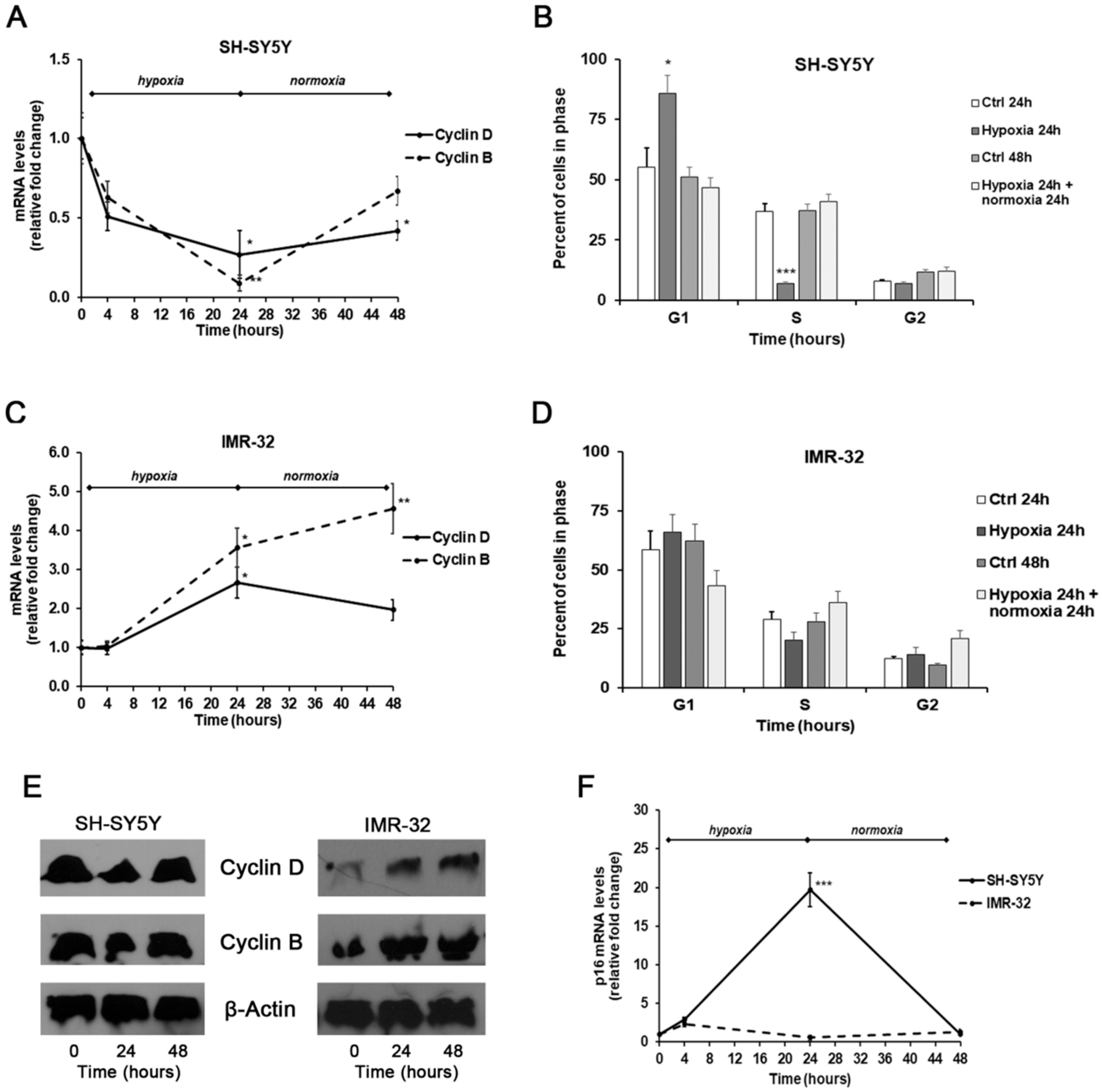
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Cell Culture and Treatment
3.3. Proliferation Assay and Cytotoxicity Study
3.4. Analysis of Gene Expression by Real-Time PCR
3.5. Western Blotting
3.6. Comet Assay
3.7. Cell Cycle Analysis by Flow Cytometry
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, D.; Curro, M.; Ientile, R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, C.; De Martino, A.; Mischiati, C.; Feriotto, G.; Beninati, S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med. Sci. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, J.Y.; Mehta, K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis 2007, 12, 1455–1463. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Tee, A.E.; Marshall, G.M.; Liu, P.Y.; Xu, N.; Haber, M.; Norris, M.D.; Iismaa, S.E.; Liu, T. Opposing effects of two tissue transglutaminase protein isoforms in neuroblastoma cell differentiation. J. Biol. Chem. 2010, 285, 3561–3567. [Google Scholar] [CrossRef] [Green Version]
- Modak, S.; Cheung, N.K. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer Treat. Rev. 2010, 36, 307–317. [Google Scholar] [CrossRef]
- Ruiz-Perez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Sasada, M.; Iyoda, T.; Asayama, T.; Suenaga, Y.; Sakai, S.; Kase, N.; Kodama, H.; Yokoi, S.; Isohama, Y.; Fukai, F. Inactivation of beta1 integrin induces proteasomal degradation of Myc oncoproteins. Oncotarget 2019, 10, 4960–4972. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. MCR 2014, 12, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Bragelmann, J.; Bohm, S.; Guthrie, M.R.; Mollaoglu, G.; Oliver, T.G.; Sos, M.L. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 2017, 16, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. c-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.E.; Schwarzbauer, J.E. Collaboration of fibronectin matrix with other extracellular signals in morphogenesis and differentiation. Curr. Opin. Cell Biol. 2016, 42, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wolfenson, H.; Lavelin, I.; Geiger, B. Dynamic regulation of the structure and functions of integrin adhesions. Dev. Cell 2013, 24, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Lutz, W.; Schwab, M. In vivo regulation of single copy and amplified N-myc in human neuroblastoma cells. Oncogene 1997, 15, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.X.; Zhao, H.; Kung, B.; Kim, D.Y.; Hicks, S.L.; Cohn, S.L.; Cheung, N.K.; Seeger, R.C.; Evans, A.E.; Ikegaki, N. The MYCN enigma: Significance of MYCN expression in neuroblastoma. Cancer Res. 2006, 66, 2826–2833. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Kaczowka, P.; Wieczorek, A.; Czogala, M.; Ksiazek, T.; Szewczyk, K.; Balwierz, W. The role of N-Myc gene amplification in neuroblastoma childhood tumour—single-centre experience. Contemp. Oncol. 2018, 22, 223–228. [Google Scholar] [CrossRef]
- Montalto, A.S.; Curro, M.; Russo, T.; Visalli, G.; Impellizzeri, P.; Antonuccio, P.; Arena, S.; Borruto, F.A.; Scalfari, G.; Ientile, R.; et al. In vitro CO2-induced ROS production impairs cell cycle in SH-SY5Y neuroblastoma cells. Pediatric Surg. Int. 2013, 29, 51–59. [Google Scholar] [CrossRef]
- Curro, M.; Montalto, A.S.; Impellizzeri, P.; Montalto, E.; Risitano, R.; Russo, T.; Perrone, P.; Chirico, V.; Arrigo, T.; Salpietro, C.; et al. CO(2) pneumoperitoneum induces in vitro hypoxic response culminating in apoptosis of human neuroblastoma cells. J. Biol. Regul. Homeost. Agents 2014, 28, 497–506. [Google Scholar]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS ONE 2012, 7, e30905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabjeesh, N.J.; Amir, S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol. Histopathol. 2007, 22, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 (Suppl. 5), 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockel, M.; Vaupel, P. Biological consequences of tumor hypoxia. Semin. Oncol. 2001, 28, 36–41. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.Y.; Jeon, J.H.; Cho, S.Y.; Shin, D.M.; Kim, C.W.; Jeong, E.M.; Bae, H.C.; Kim, T.W.; Lee, S.H.; Choi, Y.; et al. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-kappaB activity in hypoxic tumor cells. Oncogene 2010, 29, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Nezir, A.E.; Ulukan, B.; Telci, D. Transglutaminase 2: The Maestro of the Oncogenic Mediators in Renal Cell Carcinoma. Med. Sci. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.; Yao, S.; Luo, G.; Zhang, X. Identification of differentially expressed genes in human breast cancer cells induced by 4-hydroxyltamoxifen and elucidation of their pathophysiological relevance and mechanisms. Oncotarget 2018, 9, 2475–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Hitora, T.; Tei, M.; Sekimoto, M.; Doki, Y.; Mori, M. TGM2 is a novel marker for prognosis and therapeutic target in colorectal cancer. Ann. Surg. Oncol. 2010, 17, 967–972. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, H.K.; Lee, J.H.; Choi, Y.B.; Park, S.Y.; Yang, S.H.; Kim, S.Y.; Hong, K.M. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 493–502. [Google Scholar] [CrossRef]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Compact spheroid formation by ovarian cancer cells is associated with contractile behavior and an invasive phenotype. Int. J. Cancer 2009, 124, 2060–2070. [Google Scholar] [CrossRef]
- Shan, H.; Zhou, X.; Chen, C. MicroRNA214 suppresses the viability, migration and invasion of human colorectal carcinoma cells via targeting transglutaminase 2. Mol. Med. Rep. 2019, 20, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Med. Sci. 2018, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Lentini, A.; Forni, C.; Provenzano, B.; Beninati, S. Enhancement of transglutaminase activity and polyamine depletion in B16-F10 melanoma cells by flavonoids naringenin and hesperitin correlate to reduction of the in vivo metastatic potential. Amino Acids 2007, 32, 95–100. [Google Scholar] [CrossRef]
- Phatak, V.M.; Croft, S.M.; Setty, S.G.R.; Scarpellini, A.; Hughes, D.C.; Rees, R.; McArdle, S.; Verderio, E.A.M. Expression of transglutaminase-2 isoforms in normal human tissues and cancer cell lines: Dysregulation of alternative splicing in cancer. Amino Acids 2013, 44, 33–44. [Google Scholar] [CrossRef]
- Liu, T.; Tee, A.E.; Porro, A.; Smith, S.A.; Dwarte, T.; Liu, P.Y.; Iraci, N.; Sekyere, E.; Haber, M.; Norris, M.D.; et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18682–18687. [Google Scholar] [CrossRef] [Green Version]
- Condello, S.; Caccamo, D.; Curro, M.; Ferlazzo, N.; Parisi, G.; Ientile, R. Transglutaminase 2 and NF-kappaB interplay during NGF-induced differentiation of neuroblastoma cells. Brain Res. 2008, 1207, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tucholski, J.; Lesort, M.; Johnson, G.V.W. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience 2001, 102, 481–491. [Google Scholar] [CrossRef]
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Begg, G.E.; Carrington, L.; Stokes, P.H.; Matthews, J.M.; Wouters, M.A.; Husain, A.; Lorand, L.; Iismaa, S.E.; Graham, R.M. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. USA 2006, 103, 19683–19688. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, L.; Jogi, A.; Pahlman, S. Phenotypic persistence after reoxygenation of hypoxic neuroblastoma cells. Int. J. Cancer 2005, 116, 218–225. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Alterations in DNA repair gene expression under hypoxia: Elucidating the mechanisms of hypoxia-induced genetic instability. Ann. N. Y. Acad. Sci. 2005, 1059, 184–195. [Google Scholar] [CrossRef]
- Hui, A.S.; Bauer, A.L.; Striet, J.B.; Schnell, P.O.; Czyzyk-Krzeska, M.F. Calcium signaling stimulates translation of HIF-alpha during hypoxia. FASEB J. 2006, 20, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zheng, W.; Zhu, Q.; Gu, L.; Du, Y.; Han, Z.; Zhang, X.; Carter, D.R.; Cheung, B.B.; Qiu, A.; et al. Increase in DNA Damage by MYCN Knockdown Through Regulating Nucleosome Organization and Chromatin State in Neuroblastoma. Front. Genet. 2019, 10, 684. [Google Scholar] [CrossRef]
- Bedessem, B.; Stephanou, A. A mathematical model of HiF-1alpha-mediated response to hypoxia on the G1/S transition. Math. Biosci. 2014, 248, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Pattyn, F.; Speleman, F.; De Paepe, A.; Vandesompele, J. RTPrimerDB: The Real-Time PCR primer and probe database. Nucleic Acids Res. 2003, 31, 122–123. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, D.; Campisi, A.; Curro, M.; Aguennouz, M.; Volti, G.L.; Avola, R.; Ientile, R. Nuclear factor-kappa B activation is associated with glutamate-evoked tissue transglutaminase up-regulation in primary astrocyte cultures. J. Neurosci. Res. 2005, 82, 858–865. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Cyclin B1 | 5′-ctgctgcctggtgaagag-3′ | 5′-cgcctgccatgttgatct-3′ |
| Cyclin D1 | 5′-tattgcgctgctaccgttga-3′ | 5′-ccaatagcagcaaacaatgtgaaa-3′ |
| HIF-1α | 5′-cgttccttcgatcagttgtc-3′ | 5′-tcagtggtggcagtggtagt-3′ |
| p16 | 5′-accagaggcagtaaccat-3′ | 5′-gtaggaccttcggtgact-3′ |
| TG2-L | 5′-ccttacggagtccaacctca-3′ | 5′-ccgtcttctgctcctcagtc-3′ |
| TG2-S | 5′-accgctgaggagtacgtctg-3′ | 5′-tcaacaaatgctccaggaa-3′ |
| β-Actin | 5′-ttgttacaggaagtcccttgcc-3′ | 5′-atgctatcacctcccctgtgtg-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Currò, M.; Ferlazzo, N.; Giunta, M.L.; Montalto, A.S.; Russo, T.; Arena, S.; Impellizzeri, P.; Caccamo, D.; Romeo, C.; Ientile, R. Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status. Int. J. Mol. Sci. 2020, 21, 1364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041364
Currò M, Ferlazzo N, Giunta ML, Montalto AS, Russo T, Arena S, Impellizzeri P, Caccamo D, Romeo C, Ientile R. Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status. International Journal of Molecular Sciences. 2020; 21(4):1364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041364
Chicago/Turabian StyleCurrò, Monica, Nadia Ferlazzo, Maria Laura Giunta, Angela Simona Montalto, Tiziana Russo, Salvatore Arena, Pietro Impellizzeri, Daniela Caccamo, Carmelo Romeo, and Riccardo Ientile. 2020. "Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status" International Journal of Molecular Sciences 21, no. 4: 1364. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041364