Targeting Peripherally Restricted Cannabinoid Receptor 1, Cannabinoid Receptor 2, and Endocannabinoid-Degrading Enzymes for the Treatment of Neuropathic Pain Including Neuropathic Orofacial Pain



Abstract
:1. Introduction
2. Targeting Peripherally Restricted CB1 Receptors for the Treatment of Neuropathic Pain
2.1. Modulation of CB1 Receptor Expression under Neuropathic Pain Conditions
2.2. Peripherally Active CB1 Receptor Agonists for the Treatment of Neuropathic Pain
3. Targeting CB2 Receptors for the Treatment of Neuropathic Pain
3.1. Modulation of CB2 Receptor Expression under Neuropathic Pain Conditions
3.2. Selective CB2 Receptor Agonists for the Treatment of Neuropathic Pain
4. Targeting Endocannabinoids for the Treatment of Neuropathic Pain
4.1. Modulation of Endocannabinoids under Neuropathic Pain Conditions
4.2. Endocannabinoid Degradation Enzyme Inhibitors for the Treatment of Neuropathic Pain
4.2.1. FAAH Inhibitors
Dual FAAH and TRPV1 Inhibitors
Combining FAAH Inhibitors with NSAIDs or COX2 Inhibitors
4.2.2. MAGL Inhibitors
FAAH Inhibitors Combined with MAGL Inhibitors
Dual MAGL and FAAH Inhibitors
MAGL Inhibitors Combined with Opioids
Combining MAGL Inhibitors with COX2 Inhibitors
5. Potential of Endocannabinoid Enzyme Inhibitors and Cannabinoid Receptor Agonists for the Treatment of NOP
5.1. Modulation of Cannabinoid Receptors and Endocannabinoids in the Orofacial Region under Inflammatory and Neuropathic Pain Conditions
5.2. Modulation of Orofacial Neuronal Activity by Cannabinoids and Endocannabinoids
5.3. Evidence of the Anti-Nociceptive Effects of Endocannabinoid Degrading Enzyme Inhibitors and Cannabinoid Receptor Agonists in NOP Models
6. Clinical Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murnion, B.P. Neuropathic pain: Current definition and review of drug treatment. Aust. Prescr. 2018, 41, 60–63. [Google Scholar] [CrossRef]
- Merskey, H. Part III Pain Terms, A Current List with Definitions and Notes on usage. In Classification Chronic Pain-Descriptions Chronic Pain Syndromes and Definitions of Pain Terms; IASP Press: Seattle, WA, USA, 1994; pp. 207–214. [Google Scholar]
- Dueñas, M.; Ojeda, B.; Salazar, A.; Mico, J.A.; Failde, I. A review of chronic pain impact on patients, their social environment and the health care system. J. Pain Res. 2016, 9, 457–467. [Google Scholar] [CrossRef] [Green Version]
- McDermott, A.M.; Toelle, T.R.; Rowbotham, D.J.; Schaefer, C.P.; Dukes, E.M. The burden of neuropathic pain: Results from a cross-sectional survey. Eur. J. Pain 2006, 10, 127. [Google Scholar] [CrossRef]
- Schaefer, C.; Mann, R.; Sadosky, A.; Daniel, S.; Parsons, B.; Nieshoff, E.; Tuchman, M.; Nalamachu, S.; Anschel, A.; Stacey, B.R. Burden of illness associated with peripheral and central neuropathic pain among adults seeking treatment in the united states: A patient-centered evaluation. Pain Med. 2014, 15, 2105–2119. [Google Scholar] [CrossRef]
- Benoliel, R.; Sharav, Y. Chronic orofacial pain. Curr. Pain Headache Rep. 2010, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sessle, B.J. Acute and chronic craniofacial pain: Brainstem mechanisms of nociceptive transmission and neuroplasticity, and their clinical correlates. Crit. Rev. Oral Biol. Med. 2000, 11, 57–91. [Google Scholar] [CrossRef] [PubMed]
- Maarbjerg, S.; Di Stefano, G.; Bendtsen, L.; Cruccu, G. Trigeminal neuralgia—Diagnosis and treatment. Cephalalgia 2017, 37, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, J.M. Differential diagnosis of facial pain and guidelines for management. Br. J. Anaesth. 2013, 111, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, T.V.; Blinkhorn, A.S.; Davies, R.M.; Ryan, P.; Worthington, H.V.; Macfarlane, G.J. Orofacial pain: Just another chronic pain? Results from a population-based survey. Pain 2002, 99, 453–458. [Google Scholar] [CrossRef]
- Mueller, D.; Obermann, M.; Yoon, M.S.; Poitz, F.; Hansen, N.; Slomke, M.A.; Dommes, P.; Gizewski, E.; Diener, H.C.; Katsarava, Z. Prevalence of trigeminal neuralgia and persistent idiopathic facial pain: A population-based study. Cephalalgia 2011, 31, 1542–1548. [Google Scholar] [CrossRef]
- Koopman, J.S.H.A.; Dieleman, J.P.; Huygen, F.J.; de Mos, M.; Martin, C.G.M.; Sturkenboom, M.C.J.M. Incidence of facial pain in the general population. Pain 2009, 147, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Fornasari, D. Pharmacotherapy for Neuropathic Pain: A Review. Pain Ther. 2017, 6, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrance, N.; Ferguson, J.A.; Afolabi, E.; Bennett, M.I.; Serpell, M.G.; Dunn, K.M.; Smith, B.H. Neuropathic pain in the community: More under-treated than refractory? Pain 2013, 154, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Guirguis-Blake, J.; Kelly, C. Are opioids effective in the treatment of neuropathic pain? Am. Fam. Physician 2007, 75, 999–1001. [Google Scholar]
- Thomas, D.A.; Frascella, J.; Hall, T.; Smith, W.; Compton, W.; Koroshetz, W.; Briggs, J.; Grady, P.; Somerman, M.; Volkow, N. Reflections on the role of opioids in the treatment of chronic pain: A shared solution for prescription opioid abuse and pain. J. Intern. Med. 2015, 278, 92–94. [Google Scholar] [CrossRef]
- Russo, E.B. Cannabinoids in the management of difficult to treat pain. Ther. Clin. Risk Manag. 2008, 4, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Lau, B.K.; Vaughan, C.W. Targeting the endogenous cannabinoid system to treat neuropathic pain. Front. Pharmacol. 2014, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The Endogenous Cannabinoid System: A Budding Source of Targets for Treating Inflammatory and Neuropathic Pain. Neuropsychopharmacology 2018, 43, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, M.D.; Richardson, D.; Chapman, V. Endocannabinoid metabolism and uptake: Novel targets for neuropathic and inflammatory pain. Br. J. Pharmacol. 2007, 152, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Jensen, B.; Chen, J.; Furnish, T.; Wallace, M. Medical Marijuana and Chronic Pain: A Review of Basic Science and Clinical Evidence. Curr. Pain Headache Rep. 2015, 19. [Google Scholar] [CrossRef] [PubMed]
- Rahn, E.J.; Hohmann, A.G. Cannabinoids as Pharmacotherapies for Neuropathic Pain: From the Bench to the Bedside. Neurotherapeutics 2009, 6, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish-I. The structure of Cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Radwan, M.M.; ElSohly, M.A.; Slade, D.; Ahmed, S.A.; Khan, I.A.; Ross, S.A. Biologically active cannabinoids from high-potency Cannabis sativa. J. Nat. Prod. 2009, 72, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubart, C.D.; Sommer, I.E.C.; van Gastel, W.A.; Goetgebuer, R.L.; Kahn, R.S.; Boks, M.P.M. Cannabis with high cannabidiol content is associated with fewer psychotic experiences. Schizophr. Res. 2011, 130, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuardi, A.W. History of cannabis as a medicine: A review. Rev. Bras. Psiquiatr. 2006, 28, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Lu, H.C.; MacKie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345–13350. [Google Scholar] [CrossRef] [Green Version]
- Murataeva, N.; Straiker, A.; MacKie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, N.; Tsuboi, K.; Uyama, T.; Ohnishi, T. Biosynthesis and degradation of the endocannabinoid 2-arachidonoylglycerol. BioFactors 2011, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Endocannabinoids: Synthesis and Degradation. In Reviews of Physiology Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–24. [Google Scholar]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An Update on Non-CB 1, Non-CB 2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Fenwick, A.J.; Fowler, D.K.; Wu, S.W.; Shaffer, F.J.; Lindberg, J.E.M.; Kinch, D.C.; Peters, J.H. Direct anandamide activation of TRPV1 produces divergent calcium and current responses. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.P.; Safo, P.K.; Regehr, W.G. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J. Neurosci. 2004, 24, 5623–5631. [Google Scholar] [CrossRef]
- Guo, J.; Ikeda, S.R. Endocannabinoids Modulate N-Type Calcium Channels and G-Protein-Coupled Inwardly Rectifying Potassium Channels via CB1 Cannabinoid Receptors Heterologously Expressed in Mammalian Neurons. Mol. Pharmacol. 2004, 65, 665–674. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar]
- Deadwyler, S.A.; Hampson, R.E.; Mu, J.; Whyte, A.; Childers, S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J. Pharmacol. Exp. Ther. 1995, 273, 734–743. [Google Scholar] [PubMed]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar] [PubMed]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar] [PubMed]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; LE Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.G.; Herkenham, M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: A double-label in situ hybridization study. Neuroscience 1999, 90, 923–931. [Google Scholar] [CrossRef]
- Bridges, D.; Rice, A.S.C.; Egertová, M.; Elphick, M.R.; Winter, J.; Michael, G.J. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience 2003, 119, 803–812. [Google Scholar] [CrossRef]
- Price, T.J.; Helesic, G.; Parghi, D.; Hargreaves, K.M.; Flores, C.M. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience 2003, 120, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Ahluwalia, J.; Urban, L.; Capogna, M.; Bevan, S.; Nagy, I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 2000, 100, 685–688. [Google Scholar] [CrossRef]
- Agarwal, N.; Pacher, P.; Tegeder, I.; Amaya, F.; Constantin, C.E.; Brenner, G.J.; Rubino, T.; Michalski, C.W.; Marsicano, G.; Monory, K.; et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat. Neurosci. 2007, 10, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.P.; Palastro, M.D.; Johnson, B.; Ditre, J.W. Cannabis and Pain: A Clinical Review. Cannabis Cannabinoid Res. 2017, 2, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagerovic, N.; Hernandez-Folgado, L.; Goya, P.; Jagerovic, N.; Hernandez-Folgado, L.; Martin, M.I. Cannabinoids and Neuropathic Pain. Mini Rev. Med. Chem. 2003, 3, 159–165. [Google Scholar]
- Lynch, M.E.; Campbell, F. Cannabinoids for treatment of chronic non-cancer pain; a systematic review of randomized trials. Br. J. Clin. Pharmacol. 2011, 72, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001, 63, 569–611. [Google Scholar] [CrossRef]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R.B. Adverse health effects of marijuana use. N. Engl. J. Med. 2014, 370, 2219–2227. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H. A community survey of adverse effects of cannabis use. Drug Alcohol Depend. 1996, 42, 201–207. [Google Scholar] [CrossRef]
- Kalant, H. Adverse effects of cannabis on health: An update of the literature since 1996. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2004, 28, 849–863. [Google Scholar] [CrossRef]
- Moreira, F.A.; Grieb, M.; Lutz, B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: Focus on anxiety and depression. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 133–144. [Google Scholar] [CrossRef]
- Spigelman, I. Therapeutic Targeting of Peripheral Cannabinoid Receptors in Inflammatory and Neuropathic Pain States. In Translational Pain Research: From Mouse to Man; CRC Press: Boca Raton, FL, USA, 2010; pp. 99–138. ISBN 9781439812105. [Google Scholar]
- Kunos, G.; Osei-Hyiaman, D.; Bátkai, S.; Sharkey, K.A.; Makriyannis, A. Should peripheral CB1 cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol. Sci. 2009, 30, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Banister, S.D.; Krishna Kumar, K.; Kumar, V.; Kobilka, B.K.; Malhotra, S.V. Selective modulation of the cannabinoid type 1 (CB1) receptor as an emerging platform for the treatment of neuropathic pain. Medchemcomm 2019, 10, 647–659. [Google Scholar] [CrossRef]
- Seltzman, H.H.; Shiner, C.; Hirt, E.E.; Gilliam, A.F.; Thomas, B.F.; Maitra, R.; Snyder, R.; Black, S.L.; Patel, P.R.; Mulpuri, Y.; et al. Peripherally Selective Cannabinoid 1 Receptor (CB1R) Agonists for the Treatment of Neuropathic Pain. J. Med. Chem. 2016, 59, 7525–7543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, G.; Lee, G.; Valenzano, K. The Role of the Cannabinoid CB2 Receptor in Pain Transmission and Therapeutic Potential of Small Molecule CB2 Receptor Agonists. Curr. Med. Chem. 2007, 14, 917–936. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A.G. Cannabinoid CB 2 receptors: A therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, J.; Hohmann, A. The Endocannabinoid System and Pain. CNS Neurol. Disord. Drug Targets 2012, 8, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Woodhams, S.G.; Sagar, D.R.; Burston, J.J.; Chapman, V. The role of the endocannabinoid system in pain. Handb. Exp. Pharmacol. 2015, 227, 119–143. [Google Scholar]
- Pertwee, R.G. Targeting the endocannabinoid system with cannabinoid receptor agonists: Pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3353–3363. [Google Scholar] [CrossRef]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, A.H.; Cook, S.A.; Martin, B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: Evidence supporting periaqueductal gray involvement. J. Pharmacol. Exp. Ther. 1996, 276, 585–593. [Google Scholar]
- Van Sickle, M.D.; Oland, L.D.; Ho, W.; Hillard, C.J.; Mackie, K.; Davison, J.S.; Sharkey, K.A. Cannabinoids inhibit emesis through CB1 receptors in the brainstem of the ferret. Gastroenterology 2001, 121, 767–774. [Google Scholar] [CrossRef]
- Sañudo-Peña, M.C.; Strangman, N.M.; Mackie, K.; Walker, J.M.; Kang, T. CB1 receptor localization in rat spinal cord and roots, dorsal root ganglion, and peripheral nerve. Acta Pharmacol. Sin. 1999, 20, 1115–1120. [Google Scholar]
- Binzen, U.; Greffrath, W.; Hennessy, S.; Bausen, M.; Saaler-Reinhardt, S.; Treede, R.D. Co-expression of the voltage-gated potassium channel Kv1.4 with transient receptor potential channels (TRPV1 and TRPV2) and the cannabinoid receptor CB1 in rat dorsal root ganglion neurons. Neuroscience 2006, 142, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Mostafeezur, R.M.; Zakir, H.M.; Takatsuji, H.; Yamada, Y.; Yamamura, K.; Kitagawa, J. Cannabinoids Facilitate the Swallowing Reflex Elicited by the Superior Laryngeal Nerve Stimulation in Rats. PLoS ONE 2012, 7, e50703. [Google Scholar] [CrossRef] [PubMed]
- Khasabova, I.A.; Simone, D.A.; Seybold, V.S. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience 2002, 115, 613–625. [Google Scholar] [CrossRef]
- Mitrirattanakul, S.; Ramakul, N.; Guerrero, A.V.; Matsuka, Y.; Ono, T.; Iwase, H.; Mackie, K.; Faull, K.F.; Spigelman, I. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 2006, 126, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Ständer, S.; Schmelz, M.; Metze, D.; Luger, T.; Rukwied, R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J. Dermatol. Sci. 2005, 38, 177–188. [Google Scholar] [CrossRef]
- Amaya, F.; Shimosato, G.; Kawasaki, Y.; Hashimoto, S.; Tanaka, Y.; Ji, R.R.; Tanaka, M. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain 2006, 124, 175–183. [Google Scholar] [CrossRef]
- Lim, G.; Sung, B.; Ji, R.R.; Mao, J. Upregulation of spinal cannabinoid-1-receptors following nerve injury enhances the effects of Win 55,212-2 on neuropathic pain behaviors in rats. Pain 2003, 105, 275–283. [Google Scholar] [CrossRef]
- Siegling, A.; Hofmann, H.A.; Denzer, D.; Mauler, F.; De Vry, J. Cannabinoid CB1 receptor upregulation in a rat model of chronic neuropathic pain. Eur. J. Pharmacol. 2001, 415, R5–R7. [Google Scholar] [CrossRef]
- Walczak, J.S.; Pichette, V.; Leblond, F.; Desbiens, K.; Beaulieu, P. Characterization of chronic constriction of the saphenous nerve, a model of neuropathic pain in mice showing rapid molecular and electrophysiological changes. J. Neurosci. Res. 2006, 83, 1310–1322. [Google Scholar] [CrossRef]
- Walczak, J.S.; Pichette, V.; Leblond, F.; Desbiens, K.; Beaulieu, P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: A new model of neuropathic pain. Neuroscience 2005, 132, 1093–1102. [Google Scholar] [CrossRef]
- Liang, Y.C.; Huang, C.C.; Hsu, K. Sen The synthetic cannabinoids attenuate allodynia and hyperalgesia in a rat model of trigeminal neuropathic pain. Neuropharmacology 2007, 53, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Kesingland, A.; Gentry, C.; McNair, K.; Patel, S.; Urban, L.; James, I. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain 2001, 92, 91–100. [Google Scholar] [CrossRef]
- Potenzieri, C.; Brink, T.S.; Pacharinsak, C.; Simone, D.A. Cannabinoid modulation of cutaneous Aδ nociceptors during inflammation. J. Neurophysiol. 2008, 100, 2794–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagar, D.R.; Kelly, S.; Millns, P.J.; O’Shaughnessey, C.T.; Kendall, D.A.; Chapman, V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur. J. Neurosci. 2005, 22, 371–379. [Google Scholar] [CrossRef]
- Vera, G.; Cabezos, P.A.; Martín, M.I.; Abalo, R. Characterization of cannabinoid-induced relief of neuropathic pain in a rat model of cisplatin-induced neuropathy. Pharmacol. Biochem. Behav. 2013, 105, 205–212. [Google Scholar] [CrossRef]
- Dziadulewicz, E.K.; Bevan, S.J.; Brain, C.T.; Coote, P.R.; Culshaw, A.J.; Davis, A.J.; Edwards, L.J.; Fisher, A.J.; Fox, A.J.; Gentry, C.; et al. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone: A potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J. Med. Chem. 2007, 50, 3851–3856. [Google Scholar] [CrossRef]
- Groblewski, T.; Yu, X.H.; Lessard, E.; St-Onge, S.; Yang, H.; Panetta, R.; Cao, C.Q.; Swedberg, M.; Cebers, G.; Nyberg, S.; et al. Pre-clinical pharmacological properties of novel peripherally-acting CB1-CB2 agonists. In Proceedings of the 20th Annual Symposium Cannabinoids, Lund, Sweden, 24–27 July 2010; p. 37. [Google Scholar]
- Yu, X.H.; Cao, C.Q.; Martino, G.; Puma, C.; Morinville, A.; St-Onge, S.; Lessard, É.; Perkins, M.N.; Laird, J.M.A. A peripherally restricted cannabinoid receptor agonist produces robust anti-nociceptive effects in rodent models of inflammatory and neuropathic pain. Pain 2010, 151, 337–344. [Google Scholar] [CrossRef]
- Adam, J.M.; Clark, J.K.; Davies, K.; Everett, K.; Fields, R.; Francis, S.; Jeremiah, F.; Kiyoi, T.; Maidment, M.; Morrison, A.; et al. Low brain penetrant CB1 receptor agonists for the treatment of neuropathic pain. Bioorganic Med. Chem. Lett. 2012, 22, 2932–2937. [Google Scholar] [CrossRef]
- Page, D.; Wei, Z.; Liu, Z.; Tremblay, M.; Desfosses, H.; Milburn, C.; Srivastava, S.; Yang, H.; Brown, W.; Walpole, C.; et al. 5-Sulfonamide Benzimidazoles: A Class of Cannabinoid Receptors Agonists with Potent In Vivo Antinociception Activity. Lett. Drug Des. Discov. 2010, 7, 208–213. [Google Scholar] [CrossRef]
- Kalliomäki, J.; Segerdahl, M.; Webster, L.; Reimfelt, A.; Huizar, K.; Annas, P.; Karlsten, R.; Quiding, H. Evaluation of the analgesic efficacy of AZD1940, a novel cannabinoid agonist, on post-operative pain after lower third molar surgical removal. Scand. J. Pain 2013, 4, 17–22. [Google Scholar] [CrossRef]
- Kalliomäki, J.; Annas, P.; Huizar, K.; Clarke, C.; Zettergren, A.; Karlsten, R.; Segerdahl, M. Evaluation of the analgesic efficacy and psychoactive effects of AZD1940, a novel peripherally acting cannabinoid agonist, in human capsaicin-induced pain and hyperalgesia. Clin. Exp. Pharmacol. Physiol. 2013, 40, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Lynn, A.B.; Herkenham, M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: Implications for receptor-mediated immune modulation by cannabinoids. J. Pharmacol. Exp. Ther. 1994, 268, 1612–1623. [Google Scholar] [PubMed]
- Griffin, G.; Fernando, S.R.; Ross, R.A.; McKay, N.G.; Ashford, M.L.J.; Shire, D.; Huffman, J.W.; Yu, S.; Lainton, J.A.H.; Pertwee, R.G. Evidence for the presence of CB2-1ike cannabinoid receptors on peripheral nerve terminals. Eur. J. Pharmacol. 1997, 339, 53–61. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Neuroscience: Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wotherspoon, G.; Fox, A.; McIntyre, P.; Colley, S.; Bevan, S.; Winter, J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience 2005, 135, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: Immunohistochemical localization in rat brain. Brain Res. 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Deng, H.; Zvonok, A.; Cockayne, D.A.; Kwan, J.; Mata, H.P.; Vanderah, T.W.; Lai, J.; Porreca, F.; Makriyannis, A.; et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: Pain inhibition by receptors not present in the CNS. Proc. Natl. Acad. Sci. USA 2003, 100, 10529–10533. [Google Scholar] [CrossRef] [Green Version]
- Stumpf, A.; Parthier, D.; Sammons, R.P.; Stempel, A.V.; Breustedt, J.; Rost, B.R.; Schmitz, D. Cannabinoid type 2 receptors mediate a cell type-specific self-inhibition in cortical neurons. Neuropharmacology 2018, 139, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Beltramo, M.; Bernardini, N.; Bertorelli, R.; Campanella, M.; Nicolussi, E.; Fredduzzi, S.; Reggiani, A. CB2 receptor-mediated antihyperalgesia: Possible direct involvement of neural mechanisms. Eur. J. Neurosci. 2006, 23, 1530–1538. [Google Scholar] [CrossRef]
- Hsieh, G.C.; Pai, M.; Chandran, P.; Hooker, B.A.; Zhu, C.Z.; Salyers, A.K.; Wensink, E.J.; Zhan, C.; Carroll, W.A.; Dart, M.J.; et al. Central and peripheral sites of action for CB 2 receptor mediated analgesic activity in chronic inflammatory and neuropathic pain models in rats. Br. J. Pharmacol. 2011, 162, 428–440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hoffert, C.; Vu, H.K.; Groblewski, T.; Ahmad, S.; O’Donnell, D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur. J. Neurosci. 2003, 17, 2750–2754. [Google Scholar] [CrossRef] [PubMed]
- Klauke, A.L.; Racz, I.; Pradier, B.; Markert, A.; Zimmer, A.M.; Gertsch, J.; Zimmer, A. The cannabinoid CB2 receptor-selective phytocannabinoid beta-caryophyllene exerts analgesic effects in mouse models of inflammatory and neuropathic pain. Eur. Neuropsychopharmacol. 2014, 24, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racz, I.; Nadal, X.; Alferink, J.; Baños, J.E.; Rehnelt, J.; Martín, M.; Pintado, B.; Gutierrez-Adan, A.; Sanguino, E.; Manzanares, J.; et al. Crucial role of CB2 cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J. Neurosci. 2008, 28, 12125–12135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segat, G.C.; Manjavachi, M.N.; Matias, D.O.; Passos, G.F.; Freitas, C.S.; Costa, R.; Calixto, J.B. Antiallodynic effect of β-caryophyllene on paclitaxel-induced peripheral neuropathy in mice. Neuropharmacology 2017, 125, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Kim, W.K.; Chavarría, I.; Hillard, C.J.; Mackie, K.; Tolón, R.M.; Williams, K.; Romero, J. A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunodeficiency virus-induced encephalitis. J. Neurosci. 2005, 25, 2530–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Tang, Y.; Xie, M.; Bie, B.; Wu, J.; Yang, H.; Foss, J.F.; Yang, B.; Rosenquist, R.W.; Naguib, M. Activation of cannabinoid receptor 2 attenuates mechanical allodynia and neuroinflammatory responses in a chronic post-ischemic pain model of complex regional pain syndrome type I in rats. Eur. J. Neurosci. 2016, 44, 3046–3055. [Google Scholar] [CrossRef]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.Z.; Unno, S.; Ando, H.; Masuda, Y.; Kitagawa, J. Neuron-Glia crosstalk and neuropathic pain: Involvement in the modulation of motor activity in the Orofacial region. Int. J. Mol. Sci. 2017, 18, 2051. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Wang, H.; Watanabe, M.; Shimizu, K.; Zou, S.; LaGraize, S.C.; Wei, F.; Dubner, R.; Ren, K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 2007, 27, 6006–6018. [Google Scholar] [CrossRef] [Green Version]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sandoval, A.; Nutile-Mcmenemy, N.; Deleo, J.A. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology 2008, 108, 722–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naguib, M.; Xu, J.J.; Diaz, P.; Brown, D.L.; Cogdell, D.; Bie, B.; Hu, J.; Craig, S.; Hittelman, W.N. Prevention of paclitaxel-induced neuropathy through activation of the central cannabinoid type 2 receptor system. Anesth. Analg. 2012, 114, 1104–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, L.; Su, T.; Cao, F.; Meng, X.; Pei, L.; Shi, J.; Pan, H.L.; Li, M. Electroacupuncture increases CB2 receptor expression on keratinocytes and infiltrating inflammatory cells in inflamed skin tissues of rats. J. Pain 2010, 11, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Alvarado-Vazquez, P.A.; Moracho-Vilrriales, C.; Virga, K.G.; Gumina, G.; Romero-Sandoval, A.; Asbill, S. Effects of JWH015 in cytokine secretion in primary human keratinocytes and fibroblasts and its suitability for topical/transdermal delivery. Mol. Pain 2017, 13, 1744806916688220. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [Green Version]
- Elmes, S.J.R.; Jhaveri, M.D.; Smart, D.; Kendall, D.A.; Chapman, V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naïve rats and in rat models of inflammatory and neuropathic pain. Eur. J. Neurosci. 2004, 20, 2311–2320. [Google Scholar] [CrossRef]
- Clayton, N.; Marshall, F.H.; Bountra, C.; O’Shaughnessy, C.T. CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain 2002, 96, 253–260. [Google Scholar] [CrossRef]
- Hanuš, L.; Breuer, A.; Tchilibon, S.; Shiloah, S.; Goldenberg, D.; Horowitz, M.; Pertwee, R.G.; Ross, R.A.; Mechoulam, R.; Fride, E. HU-308: A specific agonist for CB2, a peripheral cannabinoid receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 14228–14233. [Google Scholar] [CrossRef] [Green Version]
- Malan, T.P.; Ibrahim, M.M.; Deng, H.; Makriyannis, A.; Vanderah, T.W. Anti-inflammatory effects of the CB2 cannabinoid receptor-selective agonist AM1241. Anesthesiology 2001, 95, A894. [Google Scholar]
- Quartilho, A.; Mata, H.P.; Ibrahim, M.M.; Vanderah, T.W.; Porreca, F.; Makriyannis, A.; Malan, T.P. Inhibition of inflammatory hyperalgesia by activation of peripheral CB 2 cannabinoid receptors. Anesthesiology 2003, 99, 955–960. [Google Scholar] [CrossRef]
- Leichsenring, A.; Andriske, M.; Bäcker, I.; Stichel, C.C.; Lübbert, H. Analgesic and antiinflammatory effects of cannabinoid receptor agonists in a rat model of neuropathic pain. Naunyn. Schmiedebergs. Arch. Pharmacol. 2009, 379, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, K.J.; Tafesse, L.; Lee, G.; Harrison, J.E.; Boulet, J.M.; Gottshall, S.L.; Mark, L.; Pearson, M.S.; Miller, W.; Shan, S.; et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 2005, 48, 658–672. [Google Scholar] [CrossRef] [PubMed]
- LaBuda, C.J.; Koblish, M.; Little, P.J. Cannabinoid CB2 receptor agonist activity in the hindpaw incision: Model of postoperative pain. Eur. J. Pharmacol. 2005, 527, 172–174. [Google Scholar] [CrossRef]
- Whiteside, G.T.; Gottshall, S.L.; Boulet, J.M.; Chaffer, S.M.; Harrison, J.E.; Pearson, M.S.; Turchin, P.I.; Mark, L.; Garrison, A.E.; Valenzano, K.J. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur. J. Pharmacol. 2005, 528, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; Mugnaini, C.; Ligresti, A.; Tafi, A.; Brogi, S.; Falciani, C.; Pedani, V.; Pesco, N.; Guida, F.; Luongo, L.; et al. Design, synthesis, and pharmacological characterization of indol-3-ylacetamides, indol-3-yloxoacetamides, and indol-3-ylcarboxamides: Potent and selective CB2 cannabinoid receptor inverse agonists. J. Med. Chem. 2012, 55, 5391–5402. [Google Scholar] [CrossRef]
- Hervera, A.; Negrete, R.; Leánez, S.; Martín-Campos, J.; Pol, O. The role of nitric oxide in the local antiallodynic and antihyperalgesic effects and expression of δ-opioid and cannabinoid-2 receptors during neuropathic pain in mice. J. Pharmacol. Exp. Ther. 2010, 334, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Castany, S.; Carcolé, M.; Leánez, S.; Pol, O. The role of carbon monoxide on the anti-nociceptive effects and expression of cannabinoid 2 receptors during painful diabetic neuropathy in mice. Psychopharmacology 2016, 233, 2209–2219. [Google Scholar] [CrossRef]
- Rahn, E.J.; Makriyannis, A.; Hohmann, A.G. Activation of cannabinoid CB 1 and CB 2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. Br. J. Pharmacol. 2007, 152, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Rahn, E.J.; Zvonok, A.M.; Thakur, G.A.; Khanolkar, A.D.; Makriyannis, A.; Hohmann, A.G. Selective activation of cannabinoid CB2 receptors suppresses neuropathic nociception induced by treatment with the chemotherapeutic agent paclitaxel in rats. J. Pharmacol. Exp. Ther. 2008, 327, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Bujalska, M. Effect of cannabinoid receptor agonists on streptozotocin-induced hyperalgesia in diabetic neuropathy. Pharmacology 2008, 82, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bujalska-Zadrozny, M.; De Cordé, A.; Pawlik, K. Influence of nitric oxide synthase or cyclooxygenase inhibitors on cannabinoids activity in streptozotocin-induced neuropathy. Pharmacol. Reports 2015, 67, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, T.; Crystal, J.D.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Self-medication of a cannabinoid CB 2 agonist in an animal model of neuropathic pain. Pain 2011, 152, 1976–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, J.L.; Gentry, K.R.; Dengler, E.C.; Wallace, J.A.; Kerwin, A.A.; Kuhn, M.N.; Zvonok, A.M.; Thakur, G.A.; Makriyannis, A.; Milligan, E.D. Immunofluorescent spectral analysis reveals the intrathecal cannabinoid agonist, AM1241, produces spinal anti-inflammatory cytokine responses in neuropathic rats exhibiting relief from allodynia. Brain Behav. 2012, 2, 155–177. [Google Scholar] [CrossRef]
- Wilkerson, J.L.; Gentry, K.R.; Dengler, E.C.; Wallace, J.A.; Kerwin, A.A.; Armijo, L.M.; Kuhn, M.N.; Thakur, G.A.; Makriyannis, A.; Milligan, E.D. Intrathecal cannabilactone CB 2R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain 2012, 153, 1091–1106. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Doods, H.; Treede, R.D.; Ceci, A. Depression-like behaviour in rats with mononeuropathy is reduced by the CB2-selective agonist GW405833. Pain 2009, 143, 206–212. [Google Scholar] [CrossRef]
- Naguib, M.; Diaz, P.; Xu, J.J.; Astruc-Diaz, F.; Craig, S.; Vivas-Mejia, P.; Brown, D.L. MDA7: A novel selective agonist for CB 2 receptors that prevents allodynia in rat neuropathic pain models. Br. J. Pharmacol. 2008, 155, 1104–1116. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.J.; Diaz, P.; Astruc-Diaz, F.; Craig, S.; Munoz, E.; Naguib, M. Pharmacological characterization of a novel cannabinoid ligand, MDA19, for treatment of neuropathic pain. Anesth. Analg. 2010, 111, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Luongo, L.; Palazzo, E.; Tambaro, S.; Giordano, C.; Gatta, L.; Scafuro, M.A.; sca Rossi, F.; Lazzari, P.; Pani, L.; de Novellis, V.; et al. 1-(2′,4′-dichlorophenyl)-6-methyl-N-cyclohexylamine-1,4-dihy droindeno[1,2-c]pyrazole-3-carboxamide, a novel CB2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiol. Dis. 2010, 37, 177–185. [Google Scholar] [CrossRef]
- Yamamoto, W.; Mikami, T.; Iwamura, H. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur. J. Pharmacol. 2008, 583, 56–61. [Google Scholar] [CrossRef]
- Deng, L.; Guindon, J.; Vemuri, V.K.; Thakur, G.A.; White, F.A.; Makriyannis, A.; Hohmann, A.G. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB2 receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Mol. Pain 2012, 8, 1744–8069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahn, E.J.; Deng, L.; Thakur, G.A.; Vemuri, K.; Zvonok, A.M.; Lai, Y.Y.; Makriyannis, A.; Hohmann, A.G. Prophylactic cannabinoid administration blocks the development of paclitaxel-induced neuropathic nociception during analgesic treatment and following cessation of drug delivery. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Guindon, J.; Cornett, B.L.; Makriyannis, A.; Mackie, K.; Hohmann, A.G. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol. Psychiatry 2015, 77, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Ikegami, M.; Kai, M.; Ohsawa, M.; Kamei, J. Activation of spinal cannabinoid CB2 receptors inhibits neuropathic pain in streptozotocin-induced diabetic mice. Neuroscience 2013, 250, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Dhopeshwarkar, A.S.; Huibregtse, M.; MacKie, K.; Hohmann, A.G. Slowly signaling G protein-biased CB2 cannabinoid receptor agonist LY2828360 suppresses neuropathic pain with sustained efficacy and attenuates morphine tolerance and dependence. Mol. Pharmacol. 2018, 93, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Malan, T.P.; Ibrahim, M.M.; Deng, H.; Liu, Q.; Mata, H.P.; Vanderah, T.; Porreca, F.; Makriyannis, A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain 2001, 93, 239–245. [Google Scholar] [CrossRef]
- Hohmann, A.G.; Farthing, J.N.; Zvonok, A.M.; Makriyannis, A. Selective Activation of Cannabinoid CB2 Receptors Suppresses Hyperalgesia Evoked by Intradermal Capsaicin. J. Pharmacol. Exp. Ther. 2004, 308, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Rahn, E.J.; Thakur, G.A.; Wood, J.A.T.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Pharmacological characterization of AM1710, a putative cannabinoid CB 2 agonist from the cannabilactone class: Antinociception without central nervous system side-effects. Pharmacol. Biochem. Behav. 2011, 98, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.J.; Diaz, P.; Bie, B.; Astruc-Diaz, F.; Wu, J.; Yang, H.; Brown, D.L.; Naguib, M. Spinal gene expression profiling and pathways analysis of a CB2 agonist (MDA7)-targeted prevention of paclitaxel-induced neuropathy. Neuroscience 2014, 260, 185–194. [Google Scholar] [CrossRef]
- Patel, H.J.; Birrell, M.A.; Crispino, N.; Hele, D.J.; Venkatesan, P.; Barnes, P.J.; Yacoub, M.H.; Belvisi, M.G. Inhibition of guinea-pig and human sensory nerve activity and the cough reflex in guinea-pigs by cannabinoid (CB 2) receptor activation. Br. J. Pharmacol. 2003, 140, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.A.; Coutts, A.A.; McFarlane, S.M.; Anavi-Goffer, S.; Irving, A.J.; Pertwee, R.G.; MacEwan, D.J.; Scott, R.H. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: Implications for antinociception. Neuropharmacology 2001, 40, 221–232. [Google Scholar] [CrossRef]
- Schmid, H.H.O. Pathways and mechanisms of N-acylethanolamine biosynthesis: Can anandamide be generated selectively? Chem. Phys. Lipids 2000, 108, 71–87. [Google Scholar] [CrossRef]
- Pertwee, R.G. Endocannabinoids and their pharmacological actions. In Handbook of Experimental Pharmacology; Springer New York LLC: Cham, Switzerland, 2015; Volume 231, pp. 1–37. [Google Scholar]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.; Plomelli, D. A second endogenous’ cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R. Receptors and Channels Targeted by Synthetic Cannabinoid Receptor Agonists and Antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Regehr, W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.J. The contribution of cyclooxygenase-2 to endocannabinoid metabolism and action. Br. J. Pharmacol. 2007, 152, 594–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strangman, N.M.; Patrick, S.L.; Hohmann, A.G.; Tsou, K.; Walker, J.M. Evidence for a role of endogenous cannabinoids in the modulation of acute and tonic pain sensitivity. Brain Res. 1998, 813, 323–328. [Google Scholar] [CrossRef]
- Smith, P.B.; Compton, D.R.; Welch, S.P.; Razdan, R.K.; Mechoulam, R.; Martin, B.R. The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J. Pharmacol. Exp. Ther. 1994, 270, 219–227. [Google Scholar] [PubMed]
- Smith, S.C.; Wagner, M.S. Clinical endocannabinoid deficiency (CECD) revisited: Can this concept explain the therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuroendocrinol. Lett. 2014, 35, 198–201. [Google Scholar] [PubMed]
- Russo, E.B. Clinical Endocannabinoid Deficiency Reconsidered: Current Research Supports the Theory in Migraine, Fibromyalgia, Irritable Bowel, and Other Treatment-Resistant Syndromes. Cannabis Cannabinoid Res. 2016, 1, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Masocha, W. Targeting the Endocannabinoid System for Prevention or Treatment of Chemotherapy-Induced Neuropathic Pain: Studies in Animal Models. Pain Res. Manag. 2018, 2018. [Google Scholar] [CrossRef]
- Pandey, R.; Mousawy, K.; Nagarkatti, M.; Nagarkatti, P. Endocannabinoids and immune regulation. Pharmacol. Res. 2009, 60, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Pestonjamasp, V.K.; Burstein, S.H. Anandamide synthesis is induced by arachidonate mobilizing agonists in cells of the immune system. Biochim. Biophys. Acta Lipids Lipid Metab. 1998, 1394, 249–260. [Google Scholar] [CrossRef]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar] [CrossRef] [Green Version]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain. Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol. Ther. 2016, 166, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, M.; Kubo, A.; Hayashi, Y.; Iwata, K. Peripheral and Central Mechanisms of Persistent Orofacial Pain. Front. Neurosci. 2019, 13, 1227. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Palazzo, E.; de Novellis, V.; Bisogno, T.; Rossi, F.; Maione, S.; Di Marzo, V. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 2007, 52, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Guasti, L.; Richardson, D.; Jhaveri, M.; Eldeeb, K.; Barrett, D.; Elphick, M.R.; Alexander, S.P.H.; Kendall, D.; Michael, G.J.; Chapman, V. Minocycline Treatment Inhibits Microglial Activation and Alters Spinal Levels of Endocannabinoids in a Rat Model of Neuropathic Pain. Mol. Pain 2009, 5, 1744–8069. [Google Scholar] [CrossRef]
- Giordano, C.; Cristino, L.; Luongo, L.; Siniscalco, D.; Petrosino, S.; Piscitelli, F.; Marabese, I.; Gatta, L.; Rossi, F.; Imperatore, R.; et al. TRPV1-dependent and-independent alterations in the limbic cortex of neuropathic mice: Impact on glial caspases and pain perception. Cereb. Cortex 2012, 22, 2495–2518. [Google Scholar] [CrossRef] [Green Version]
- Guindon, J.; Lai, Y.; Takacs, S.M.; Bradshaw, H.B.; Hohmann, A.G. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: Effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol. Res. 2013, 67, 94–109. [Google Scholar]
- Palazzo, E.; De Novellis, V.; Petrosino, S.; Marabese, I.; Vita, D.; Giordano, C.; Di Marzo, V.; Mangoni, G.S.; Rossi, F.; Maione, S. Neuropathic pain and the endocannabinoid system in the dorsal raphe: Pharmacological treatment and interactions with the serotonergic system. Eur. J. Neurosci. 2006, 24, 2011–2020. [Google Scholar] [CrossRef]
- Walker, J.M.; Huang, S.M.; Strangman, N.M.; Tsou, K.; Sañudo-Peña, M.C. Pain modulation by release of the endogenous cannabinoid anandamide. Proc. Natl. Acad. Sci. USA 1999, 96, 12198–12203. [Google Scholar] [CrossRef] [Green Version]
- Guindon, J.; Desroches, J.; Beaulieu, P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB 2 receptors. Br. J. Pharmacol. 2007, 150, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, M.D.; Richardson, D.; Kendall, D.A.; Barrett, D.A.; Chapman, V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J. Neurosci. 2006, 26, 13318–13327. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Di Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006, 20, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Sokal, D.M.; Elmes, S.J.R.; Kendall, D.A.; Chapman, V. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurones via activation of CB2 receptors in anaesthetised rats. Neuropharmacology 2003, 45, 404–411. [Google Scholar] [CrossRef]
- Jardín, I.; López, J.J.; Diez, R.; Sánchez-Collado, J.; Cantonero, C.; Albarrán, L.; Woodard, G.E.; Redondo, P.C.; Salido, G.M.; Smani, T.; et al. TRPs in pain sensation. Front. Physiol. 2017, 8, 392. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.Z.; Bakri, M.M.; Yahya, F.; Ando, H.; Unno, S.; Kitagawa, J. The role of transient receptor potential (TRP) channels in the transduction of dental pain. Int. J. Mol. Sci. 2019, 20, 526. [Google Scholar] [CrossRef] [Green Version]
- Marrone, M.C.; Morabito, A.; Giustizieri, M.; Chiurchiù, V.; Leuti, A.; Mattioli, M.; Marinelli, S.; Riganti, L.; Lombardi, M.; Murana, E.; et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun. 2017, 8, 1–18. [Google Scholar] [CrossRef]
- Vyklický, L.; Nováková-Toušová, K.; Benedikt, J.; Samad, A.; Touška, F.; Vlachova, V. Calcium-dependent desensitization of vanilloid receptor TRPV1: A mechanism possibly involved in analgesia induced by topical application of capsaicin. Physiol. Res. 2008, 57 (Suppl. 3), S59–S68. [Google Scholar]
- Fride, E.; Mechoulam, R. Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur. J. Pharmacol. 1993, 231, 313–314. [Google Scholar] [CrossRef]
- Jaggar, S.I.; Hasnie, F.S.; Sellaturay, S.; Rice, A.S.C. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 1998, 76, 189–199. [Google Scholar] [CrossRef]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, K.; Smith, S.E.; Liimatta, M.B.; Beidler, D.; Sadagopan, N.; Dudley, D.T.; Young, T.; Wren, P.; Zhang, Y.; Swaney, S.; et al. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, S.G.; Long, J.Z.; O’Neal, S.T.; Abdullah, R.A.; Poklis, J.L.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J. Pharmacol. Exp. Ther. 2009, 330, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprioli, A.; Coccurello, R.; Rapino, C.; Di Serio, S.; Di Tommaso, M.; Vertechy, M.; Vacca, V.; Battista, N.; Pavone, F.; Maccarrone, M.; et al. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J. Pharmacol. Exp. Ther. 2012, 342, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Adamson Barnes, N.S.; Mitchell, V.A.; Kazantzis, N.P.; Vaughan, C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine neuropathic pain model. Br. J. Pharmacol. 2016, 173, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.; LoVerme, J.; La Rana, G.; Compton, T.R.; Parrott, J.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Calignano, A.; et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J. Pharmacol. Exp. Ther. 2007, 322, 236–242. [Google Scholar] [CrossRef]
- Starowicz, K.; Makuch, W.; Osikowicz, M.; Piscitelli, F.; Petrosino, S.; Di Marzo, V.; Przewlocka, B. Spinal anandamide produces analgesia in neuropathic rats: Possible CB 1- and TRPV1-mediated mechanisms. Neuropharmacology 2012, 62, 1746–1755. [Google Scholar] [CrossRef]
- Starowicz, K.; Makuch, W.; Korostynski, M.; Malek, N.; Slezak, M.; Zychowska, M.; Petrosino, S.; De Petrocellis, L.; Cristino, L.; Przewlocka, B.; et al. Full Inhibition of Spinal FAAH Leads to TRPV1-Mediated Analgesic Effects in Neuropathic Rats and Possible Lipoxygenase-Mediated Remodeling of Anandamide Metabolism. PLoS ONE 2013, 8, e60040. [Google Scholar] [CrossRef]
- Desroches, J.; Charron, S.; Bouchard, J.F.; Beaulieu, P. Endocannabinoids decrease neuropathic pain-Related behavior in mice through the activation of one or both peripheral CB1 and CB2 receptors. Neuropharmacology 2014, 77, 441–452. [Google Scholar] [CrossRef]
- Desroches, J.; Guindon, J.; Lambert, C.; Beaulieu, P. Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br. J. Pharmacol. 2008, 155, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, N.; Kostrzewa, M.; Makuch, W.; Pajak, A.; Kucharczyk, M.; Piscitelli, F.; Przewlocka, B.; Di Marzo, V.; Starowicz, K. The multiplicity of spinal AA-5-HT anti-nociceptive action in a rat model of neuropathic pain. Pharmacol. Res. 2016, 111, 251–263. [Google Scholar] [CrossRef] [PubMed]
- de Novellis, V.; Vita, D.; Gatta, L.; Luongo, L.; Bellini, G.; De Chiaro, M.; Marabese, I.; Siniscalco, D.; Boccella, S.; Piscitelli, F.; et al. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol. Pain 2011, 7, 1744–8069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slivicki, R.A.; Saberi, S.A.; Iyer, V.; Vemuri, V.K.; Makriyannis, A.; Hohmann, A.G. Brain-permeant and -impermeant inhibitors of fatty acid amide hydrolase synergize with the opioid analgesic morphine to suppress chemotherapy-induced neuropathic nociception without enhancing effects of morphine on gastrointestinal transit. J. Pharmacol. Exp. Ther. 2018, 367, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niphakis, M.J.; Cognetta, A.B.; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Kinsey, S.G.; Liu, Q.S.; Hruba, L.; McMahon, L.R.; Grim, T.W.; Merritt, C.R.; Wise, L.E.; Abdullah, R.A.; Selley, D.E.; et al. Full fatty acid amide hydrolase inhibition combined with partial monoacylglycerol lipase inhibition: Augmented and sustained antinociceptive effects with reduced cannabimimetic side effects in mice. J. Pharmacol. Exp. Ther. 2015, 354, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Grim, T.W.; Ghosh, S.; Hsu, K.L.; Cravatt, B.F.; Kinsey, S.G.; Lichtman, A.H. Combined inhibition of FAAH and COX produces enhanced anti-allodynic effects in mouse neuropathic and inflammatory pain models. Pharmacol. Biochem. Behav. 2014, 124, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, S.G.; Long, J.Z.; Cravatt, B.F.; Lichtman, A.H. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J. Pain 2010, 11, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Clapper, J.R.; Moreno-Sanz, G.; Russo, R.; Guijarro, A.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Sciolino, N.R.; Spradley, J.M.; et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010, 13, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Sasso, O.; Bertorelli, R.; Bandiera, T.; Scarpelli, R.; Colombano, G.; Armirotti, A.; Moreno-Sanz, G.; Reggiani, A.; Piomelli, D. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol. Res. 2012, 65, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Jayamanne, A.; Greenwood, R.; Mitchell, V.A.; Aslan, S.; Piomelli, D.; Vaughan, C.W. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 2006, 147, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.; Johnson, D.S.; Mileni, M.; Beidler, D.; Long, J.Z.; McKinney, M.K.; Weerapana, E.; Sadagopan, N.; Liimatta, M.; Smith, S.E.; et al. Discovery and Characterization of a Highly Selective FAAH Inhibitor that Reduces Inflammatory Pain. Chem. Biol. 2009, 16, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Wise, L.E.; Chen, Y.; Gujjar, R.; Mahadevan, A.; Cravatt, B.F.; Lichtman, A.H. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci. 2013, 92, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maione, S.; Morera, E.; Marabese, I.; Ligresti, A.; Luongo, L.; Ortar, G.; Di Marzo, V. Antinociceptive effects of tetrazole inhibitors of endocannabinoid inactivation: Cannabinoid and non-cannabinoid receptor-mediated mechanisms. Br. J. Pharmacol. 2008, 155, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Naidu, P.S.; Booker, L.; Cravatt, B.F.; Lichtman, A.H. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J. Pharmacol. Exp. Ther. 2009, 329, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, S.G.; Wise, L.E.; Ramesh, D.; Abdullah, R.; Selley, D.E.; Cravatt, B.F.; Lichtman, A.H. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J. Pharmacol. Exp. Ther. 2013, 345, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Ignatowska-Jankowska, B.M.; Ghosh, S.; Crowe, M.S.; Kinsey, S.G.; Niphakis, M.J.; Abdullah, R.A.; Tao, Q.; O’Neal, S.T.; Walentiny, D.M.; Wiley, J.L.; et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2014, 171, 1392–1407. [Google Scholar] [CrossRef] [Green Version]
- Crowe, M.S.; Leishman, E.; Banks, M.L.; Gujjar, R.; Mahadevan, A.; Bradshaw, H.B.; Kinsey, S.G. Combined inhibition of monoacylglycerol lipase and cyclooxygenases synergistically reduces neuropathic pain in mice. Br. J. Pharmacol. 2015, 172, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective monoacylglycerol lipase inhibitors: Antinociceptive versus cannabimimetic effects in Mices. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, Z.A.; Wilkerson, J.L.; Bagdas, D.; Kyte, S.L.; Patel, N.; Donvito, G.; Mustafa, M.A.; Poklis, J.L.; Niphakis, M.J.; Hsu, K.L.; et al. Monoacylglycerol lipase inhibitors reverse paclitaxel-induced nociceptive behavior and proinflammatory markers in a mouse model of chemotherapy-induced neuropathy. J. Pharmacol. Exp. Ther. 2018, 366, 169–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khasabova, I.A.; Yao, X.; Paz, J.; Lewandowski, C.T.; Lindberg, A.E.; Coicou, L.; Burlakova, N.; Simone, D.A.; Seybold, V.S. JZL184 is anti-hyperalgesic in a murine model of cisplatin-induced peripheral neuropathy. Pharmacol. Res. 2014, 90, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makara, J.K.; Mor, M.; Fegley, D.; Szabó, S.I.; Kathuria, S.; Astarita, G.; Duranti, A.; Tontini, A.; Tarzia, G.; Rivara, S.; et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005, 8, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Saario, S.M.; Salo, O.M.H.; Nevalainen, T.; Poso, A.; Laitinen, J.T.; Järvinen, T.; Niemi, R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem. Biol. 2005, 12, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burston, J.J.; Sim-Selley, L.J.; Harloe, J.P.; Mahadevan, A.; Razdan, R.K.; Selley, D.E.; Wiley, J.L. N-arachidonyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonylglycerol through inhibition of monoacylglycerol lipase. J. Pharmacol. Exp. Ther. 2008, 327, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Vandevoorde, S.; Jonsson, K.O.; Labar, G.; Persson, E.; Lambert, D.M.; Fowler, C.J. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br. J. Pharmacol. 2007, 150, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Wiskerke, J.; Irimia, C.; Cravatt, B.F.; De Vries, T.J.; Schoffelmeer, A.N.M.; Pattij, T.; Parsons, L.H. Characterization of the effects of reuptake and hydrolysis inhibition on interstitial endocannabinoid levels in the brain: An in vivo microdialysis study. ACS Chem. Neurosci. 2012, 3, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [Google Scholar] [CrossRef]
- Guindon, J.; Guijarro, A.; Piomelli, D.; Hohmann, A.G. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br. J. Pharmacol. 2011, 163, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- Woodhams, S.G.; Wong, A.; Barrett, D.A.; Bennett, A.J.; Chapman, V.; Alexander, S.P.H. Spinal administration of the monoacylglycerol lipase inhibitor JZL184 produces robust inhibitory effects on nociceptive processing and the development of central sensitization in the rat. Br. J. Pharmacol. 2012, 167, 1609–1619. [Google Scholar] [CrossRef]
- Spradley, J.M.; Guindon, J.; Hohmann, A.G. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol. Res. 2010, 62, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, J.L.; Ghosh, S.; Mustafa, M.; Abdullah, R.A.; Niphakis, M.J.; Cabrera, R.; Maldonado, R.; Cravatt, B.F.; Lichtman, A.H. The endocannabinoid hydrolysis inhibitor SA-57: Intrinsic antinociceptive effects, augmented morphine-induced antinociception, and attenuated heroin seeking behavior in mice. Neuropharmacology 2017, 114, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, W.B.; Gould, M.J.; Torres, R.D.; Mitchell, V.A.; Vaughan, C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine inflammatory pain model. Neuropharmacology 2014, 81, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Sakin, Y.S.; Dogrul, A.; Ilkaya, F.; Seyrek, M.; Ulas, U.H.; Gulsen, M.; Bagci, S. The effect of FAAH, MAGL, and Dual FAAH/MAGL inhibition on inflammatory and colorectal distension-induced visceral pain models in Rodents. Neurogastroenterol. Motil. 2015, 27, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; Burston, J.J.; Sim-Selley, L.J.; Lichtman, A.H.; Wiley, J.L.; et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20270–20275. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, J.L.; Niphakis, M.J.; Grim, T.W.; Mustafa, M.A.; Abdullah, R.A.; Poklis, J.L.; Dewey, W.L.; Akbarali, H.; Banks, M.L.; Wise, L.E.; et al. The selective monoacylglycerol lipase inhibitor MJN110 produces opioid-sparing effects in a mouse neuropathic pain model. J. Pharmacol. Exp. Ther. 2016, 357, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Sessle, B.J. Editorial: Are Cannabinoids Effective for Orofacial Pain States? J. Oral Facial Pain Headache 2015, 29, 5–6. [Google Scholar] [CrossRef] [Green Version]
- McDonough, P.; McKenna, J.P.; McCreary, C.; Downer, E.J. Neuropathic orofacial pain: Cannabinoids as a therapeutic avenue. Int. J. Biochem. Cell Biol. 2014, 55, 72–78. [Google Scholar] [CrossRef]
- Nakajima, Y.; Furuichi, Y.; Biswas, K.K.; Hashiguchi, T.; Kawahara, K.I.; Yamaji, K.; Uchimura, T.; Izumi, Y.; Maruyama, I. Endocannabinoid, anandamide in gingival tissue regulates the periodontal inflammation through NF-κB pathway inhibition. FEBS Lett. 2006, 580, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Borsani, E.; Majorana, A.; Cocchi, M.A.; Conti, G.; Bonadeo, S.; Padovani, A.; Lauria, G.; Bardellini, E.; Rezzani, R.; Rodella, L.F. Epithelial expression of vanilloid and cannabinoid receptors: A potential role in burning mouth syndrome pathogenesis. Histol. Histopathol. 2014, 29, 523–533. [Google Scholar]
- Cupini, L.M.; Costa, C.; Sarchielli, P.; Bari, M.; Battista, N.; Eusebi, P.; Calabresi, P.; Maccarrone, M. Degradation of endocannabinoids in chronic migraine and medication overuse headache. Neurobiol. Dis. 2008, 30, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, C.; Markert, A.; Zimmer, A. Inhibition of FAAH reduces nitroglycerin-induced migraine-like pain and trigeminal neuronal hyperactivity in mice. Eur. Neuropsychopharmacol. 2015, 25, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Gasperi, V.; Sandrini, G.; Bagetta, G.; Nappi, G.; MacCarrone, M.; Tassorelli, C. Alterations of the endocannabinoid system in an animal model of migraine: Evaluation in cerebral areas of rat. Cephalalgia 2010, 30, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Huang, C.C.; Hsu, K.S.; Takahashi, T. Cannabinoid-induced presynaptic inhibition at the primary afferent trigeminal synapse of juvenile rat brainstem slices. J. Physiol. 2004, 555, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papanastassiou, A.M.; Fields, H.L.; Meng, I.D. Local application of the cannabinoid receptor agonist, WIN 55,212-2, to spinal trigeminal nucleus caudalis differentially affects nociceptive and non-nociceptive neurons. Pain 2004, 107, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Zhang, J.; Ouyang, C.H.; Li, C.Y.; Zhao, F.B.; Liu, Y.W.; Ai, Y.X.; Hu, W.P. Potentiation by WIN 55,212-2 of GABA-activated currents in rat trigeminal ganglion neurones. Br. J. Pharmacol. 2009, 158, 1904–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Yang, R.; Wang, X.; Liu, H.; Zou, L.; Hu, X.; Wu, J.; Zou, A.; Liu, L. Inhibition of 5-HT3 receptors-activated currents by cannabinoids in rat trigeminal ganglion neurons. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 265–271. [Google Scholar] [CrossRef]
- Wang, W.; Cao, X.; Liu, C.; Liu, L. Cannabinoid WIN 55,212-2 inhibits TRPV1 in trigeminal ganglion neurons via PKA and PKC pathways. Neurol. Sci. 2012, 33, 79–85. [Google Scholar] [CrossRef]
- Price, T.J.; Patwardhan, A.; Akopian, A.N.; Hargreaves, K.M.; Flores, C.M. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br. J. Pharmacol. 2004, 141, 1118–1130. [Google Scholar] [CrossRef]
- Greco, R.; Mangione, A.S.; Sandrini, G.; Nappi, G.; Tassorelli, C. Activation of CB2 receptors as a potential therapeutic target for migraine: Evaluation in an animal model. J. Headache Pain 2014, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, R.; Bandiera, T.; Mangione, A.S.; Demartini, C.; Siani, F.; Nappi, G.; Sandrini, G.; Guijarro, A.; Armirotti, A.; Piomelli, D.; et al. Effects of peripheral FAAH blockade on NTG-induced hyperalgesia—Evaluation of URB937 in an animal model of migraine. Cephalalgia 2015, 35, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, R.; Hossain, M.Z.; Unno, S.; Ando, H.; Masuda, Y.; Takahashi, K.; Otake, M.; Saito, I.; Kitagawa, J. Inhibition of 2-arachydonoylgycerol degradation attenuates orofacial neuropathic pain in trigeminal nerve-injured mice. J. Oral Sci. 2018, 60, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leimuranta, P.; Khiroug, L.; Giniatullin, R. Emerging role of (endo)cannabinoids in migraine. Front. Pharmacol. 2018, 9, 420. [Google Scholar] [CrossRef]
- Akerman, S.; Holland, P.R.; Goadsby, P.J. Cannabinoid (CB1) receptor activation inhibits trigeminovascular neurons. J. Pharmacol. Exp. Ther. 2007, 320, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Akerman, S.; Holland, P.R.; Lasalandra, M.P.; Goadsby, P.J. Endocannabinoids in the brainstem modulate dural trigeminovascular nociceptive traffic via CB1 and “Triptan” receptors: Implications in migraine. J. Neurosci. 2013, 33, 14869–14877. [Google Scholar] [CrossRef]
- Burgos, E.; Pascual, D.; Isabel Martín, M.; Goicoechea, C. Antinociceptive effect of the cannabinoid agonist, WIN 55,212-2, in the orofacial and temporomandibular formalin tests. Eur. J. Pain 2010, 14, 40–48. [Google Scholar] [CrossRef]
- Mostafeezur, R.M.; Shinoda, M.; Unno, S.; Zakir, H.M.; Takatsuji, H.; Takahashi, K.; Yamada, Y.; Yamamura, K.; Iwata, K.; Kitagawa, J. Involvement of astroglial glutamate-glutamine shuttle in modulation of the jaw-opening reflex following infraorbital nerve injury. Eur. J. Neurosci. 2014, 39, 2050–2059. [Google Scholar] [CrossRef]
- Mostafeezur, R.M.; Zakir, H.M.; Yamada, Y.; Yamamura, K.; Iwata, K.; Sessle, B.J.; Kitagawa, J. The effect of minocycline on the masticatory movements following the inferior alveolar nerve transection in freely moving rats. Mol. Pain 2012, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Zakir, H.M.; Mostafeezur, R.M.; Suzuki, A.; Hitomi, S.; Suzuki, I.; Maeda, T.; Seo, K.; Yamada, Y.; Yamamura, K.; Lev, S.; et al. Expression of TRPV1 Channels after Nerve Injury Provides an Essential Delivery Tool for Neuropathic Pain Attenuation. PLoS ONE 2012, 7, e44023. [Google Scholar] [CrossRef]
- Mátyás, F.; Urbán, G.M.; Watanabe, M.; Mackie, K.; Zimmer, A.; Freund, T.F.; Katona, I. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 2008, 54, 95–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain. Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Labra, V.C.; Santibáñez, C.A.; Gajardo-Gómez, R.; Díaz, E.F.; Gómez, G.I.; Orellana, J.A. The neuroglial dialog between cannabinoids and hemichannels. Front. Mol. Neurosci. 2018, 11, 79. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; Rodríguez, M.L.L.; Bajjalieh, S.; Stella, N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Barrie, N.; Manolios, N. The endocannabinoid system in pain and inflammation: Its relevance to rheumatic disease. Eur. J. Rheumatol. 2017, 4, 210–218. [Google Scholar] [CrossRef]
- Bruni, N.; Della Pepa, C.; Oliaro-Bosso, S.; Pessione, E.; Gastaldi, D.; Dosio, F. Cannabinoid delivery systems for pain and inflammation treatment. Molecules 2018, 23, 2478. [Google Scholar] [CrossRef] [Green Version]
- Nagarkatti, P.; Pandey, R.; Rieder, S.A.; Hegde, V.L.; Nagarkatti, M. Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem. 2009, 1, 1333–1349. [Google Scholar] [CrossRef] [Green Version]
- Huggins, J.P.; Smart, T.S.; Langman, S.; Taylor, L.; Young, T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of th. Pain 2012, 153, 1837–1846. [Google Scholar] [CrossRef]
- Wagenlehner, F.M.E.; van Till, J.W.O.; Houbiers, J.G.A.; Martina, R.V.; Cerneus, D.P.; Melis, J.H.J.M.; Majek, A.; Vjaters, E.; Urban, M.; Ramonas, H.; et al. Fatty Acid Amide Hydrolase Inhibitor Treatment in Men With Chronic Prostatitis/Chronic Pelvic Pain Syndrome: An Adaptive Double-blind, Randomized Controlled Trial. Urology 2017, 103, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, T.; Price, J.; Albanese, M.; Bullman, J.; Guillard, F.; Meyer, I.; Leeson, R.; Costantin, C.; Ziviani, L.; Nocini, P.F.; et al. A randomized, controlled study to investigate the analgesic efficacy of single doses of the cannabinoid receptor-2 agonist GW842166, ibuprofen or placebo in patients with acute pain following third molar tooth extraction. Clin. J. Pain 2011, 27, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Bradford, D.; Stirling, A.; Ernault, E.; Liosatos, M.; Tracy, K.; Moseley, J.; Blahunka, P.; Smith, M.D. The MOBILE Study-A Phase IIa enriched enrollment randomized withdrawal trial to assess the analgesic efficacy and safety of ASP8477, a fatty acid amide hydrolase inhibitor, in patients with peripheral neuropathic pain. Pain Med. 2017, 18, 2388–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerbrat, A.; Ferré, J.C.; Fillatre, P.; Ronzière, T.; Vannier, S.; Carsin-Nicol, B.; Lavoué, S.; Vérin, M.; Gauvrit, J.Y.; Le Tulzo, Y.; et al. Acute neurologic disorder from an inhibitor of fatty acid amide hydrolase. N. Engl. J. Med. 2016, 375, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, N. Lessons from the fatal french study BIA-10-2474. BMJ 2016, 353, i2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Esbroeck, A.C.M.; Janssen, A.P.A.; Cognetta, A.B.; Ogasawara, D.; Shpak, G.; Van Der Kroeg, M.; Kantae, V.; Baggelaar, M.P.; De Vrij, F.M.S.; Deng, H.; et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchicchi, A.; Lecca, S.; Carta, S.; Pillolla, G.; Muntoni, A.L.; Yasar, S.; Goldberg, S.R.; Pistis, M. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: Involvement of PPAR-α nuclear receptors. Addict. Biol. 2010, 15, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, H.; Drew, G.M.; Christie, M.J.; Vaughan, C.W. Inhibition of fatty acid amide hydrolase unmasks CB 1 receptor and TRPV1 channel-mediated modulation of glutamatergic synaptic transmission in midbrain periaqueductal grey. Br. J. Pharmacol. 2011, 163, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Maione, S.; Bisogno, T.; De Novellis, V.; Palazzo, E.; Cristino, L.; Valenti, M.; Petrosino, S.; Guglielmotti, V.; Rossi, F.; Di Marzo, V. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 re. J. Pharmacol. Exp. Ther. 2006, 316, 969–982. [Google Scholar] [CrossRef]
- Sagar, D.R.; Kendall, D.A.; Chapman, V. Inhibition of fatty acid amide hydrolase produces PPAR-α-mediated analgesia in a rat model of inflammatory pain. Br. J. Pharmacol. 2008, 155, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Saghatelian, A.; McKinney, M.K.; Bandell, M.; Patapoutian, A.; Cravatt, B.F. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry 2006, 45, 9007–9015. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Oleamide: A member of the endocannabinoid family? Br. J. Pharmacol. 2004, 141, 195–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.S.; Rosenkilde, M.M.; Holst, J.J.; Schwartz, T.W. GPR119 as a fat sensor. Trends Pharmacol. Sci. 2012, 33, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Vandevoorde, S.; Saha, B.; Mahadevan, A.; Razdan, R.K.; Pertwee, R.G.; Martin, B.R.; Fowler, C.J. Influence of the degree of unsaturation of the acyl side chain upon the interaction of analogues of 1-arachidonoylglycerol with monoacylglycerol lipase and fatty acid amide hydrolase. Biochem. Biophys. Res. Commun. 2005, 337, 104–109. [Google Scholar] [CrossRef]
- Maldonado, R.; Baños, J.E.; Cabañero, D. The endocannabinoid system and neuropathic pain. Pain 2016, 157, S23–S32. [Google Scholar] [CrossRef] [Green Version]
- Backonja, M.M.; Stacey, B. Neuropathic pain symptoms relative to overall pain rating. J. Pain 2004, 5, 491–497. [Google Scholar] [CrossRef]
- Butler, S.; Jonzon, B.; Branting-Ekenbäck, C.; Wadell, C.; Farahmand, B. Predictors of severe pain in a cohort of 5271 individuals with self-reported neuropathic pain. Pain 2013, 154, 141–146. [Google Scholar] [CrossRef]
- Attal, N.; Lanteri-Minet, M.; Laurent, B.; Fermanian, J.; Bouhassira, D. The specific disease burden of neuropathic pain: Results of a French nationwide survey. Pain 2011, 152, 2836–2843. [Google Scholar] [CrossRef]
- Serpell, M.; Ratcliffe, S.; Hovorka, J.; Schofield, M.; Taylor, L.; Lauder, H.; Ehler, E. A double-blind, randomized, placebo-controlled, parallel group study of THC/CBD spray in peripheral neuropathic pain treatment. Eur. J. Pain 2014, 18, 999–1012. [Google Scholar] [CrossRef]
- Langford, R.M.; Mares, J.; Novotna, A.; Vachova, M.; Novakova, I.; Notcutt, W.; Ratcliffe, S. A double-blind, randomized, placebo-controlled, parallel-group study of THC/CBD oromucosal spray in combination with the existing treatment regimen, in the relief of central neuropathic pain in patients with multiple sclerosis. J. Neurol. 2013, 260, 984–997. [Google Scholar] [CrossRef]
- Lynch, M.E.; Cesar-Rittenberg, P.; Hohmann, A.G. A double-blind, placebo-controlled, crossover pilot trial with extension using an oral mucosal cannabinoid extract for treatment of chemotherapy-induced neuropathic pain. J. Pain Symptom Manage. 2014, 47, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Englund, A.; Morrison, P.D.; Nottage, J.; Hague, D.; Kane, F.; Bonaccorso, S.; Stone, J.M.; Reichenberg, A.; Brenneisen, R.; Holt, D.; et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J. Psychopharmacol. 2013, 27, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.V.; Del Bel, E.A.; Guimarães, F.S. Cannabidiol attenuates catalepsy induced by distinct pharmacological mechanisms via 5-HT1A receptor activation in mice. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 46, 43–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilliard, A.; Stott, C.; Wright, S.; Guy, G.; Pryce, G.; Al-Izki, S.; Bolton, C.; Giovannoni, G. Evaluation of the Effects of Sativex (THC BDS: CBD BDS) on Inhibition of Spasticity in a Chronic Relapsing Experimental Allergic Autoimmune Encephalomyelitis: A Model of Multiple Sclerosis. ISRN Neurol. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Amerongen, G.; Kanhai, K.; Baakman, A.C.; Heuberger, J.; Klaassen, E.; Beumer, T.L.; Strijers, R.L.M.; Killestein, J.; van Gerven, J.; Cohen, A.; et al. Effects on Spasticity and Neuropathic Pain of an Oral Formulation of Δ9-tetrahydrocannabinol in Patients With Progressive Multiple Sclerosis. Clin. Ther. 2018, 40, 1467–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, C.; Mawani, S.; Brady, S.; Chan, C.; Liu, C.; Mehina, E.; Garven, A.; Bestard, J.; Korngut, L. An enriched-enrolment, randomized withdrawal, flexible-dose, double-blind, placebo-controlled, parallel assignment efficacy study of nabilone as adjuvant in the treatment of diabetic peripheral neuropathic pain. Pain 2012, 153, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Ware, M.A.; Fitzcharles, M.-A.; Joseph, L.; Shir, Y. The Effects of Nabilone on Sleep in Fibromyalgia: Results of a Randomized Controlled Trial. Anesth. Analg. 2010, 110, 604–610. [Google Scholar] [CrossRef]
- Pini, L.A.; Guerzoni, S.; Cainazzo, M.M.; Ferrari, A.; Sarchielli, P.; Tiraferri, I.; Ciccarese, M.; Zappaterra, M. Nabilone for the treatment of medication overuse headache: Results of a preliminary double-blind, active-controlled, randomized trial. J. Headache Pain 2012, 13, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, D.; Doupe, M.; Torabi, M.; Gomori, A.; Ethans, K.; Esfahani, F.; Galloway, K.; Namaka, M. Nabilone as an adjunctive to gabapentin for multiple sclerosis-induced neuropathic pain: A randomized controlled trial. Pain Med. 2015, 16, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Narang, S.; Gibson, D.; Wasan, A.D.; Ross, E.L.; Michna, E.; Nedeljkovic, S.S.; Jamison, R.N. Efficacy of Dronabinol as an Adjuvant Treatment for Chronic Pain Patients on Opioid Therapy. J. Pain 2008, 9, 254–264. [Google Scholar] [CrossRef]
- Wilsey, B.; Marcotte, T.; Deutsch, R.; Gouaux, B.; Sakai, S.; Donaghe, H. Low-dose vaporized cannabis significantly improves neuropathic pain. J. Pain 2013, 14, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey-Bloom, J.; Wolfson, T.; Gamst, A.; Jin, S.; Marcotte, T.D.; Bentley, H.; Gouaux, B. Smoked cannabis for spasticity in multiple sclerosis: A randomized, placebo-controlled trial. CMAJ 2012, 184, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.S.; Marcotte, T.D.; Umlauf, A.; Gouaux, B.; Atkinson, J.H. Efficacy of Inhaled Cannabis on Painful Diabetic Neuropathy. J. Pain 2015, 16, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiting, P.F.; Wolff, R.F.; Deshpande, S.; Di Nisio, M.; Duffy, S.; Hernandez, A.V.; Keurentjes, J.C.; Lang, S.; Misso, K.; Ryder, S.; et al. Cannabinoids for medical use: A systematic review and meta-analysis. JAMA J. Am. Med. Assoc. 2015, 313, 2456–2473. [Google Scholar] [CrossRef]
- Martín-Sánchez, E.; Furukawa, T.A.; Taylor, J.; Martin, J.L.R. Systematic review and meta-analysis of cannabis treatment for chronic pain. Pain Med. 2009, 10, 1353–1368. [Google Scholar] [CrossRef] [Green Version]


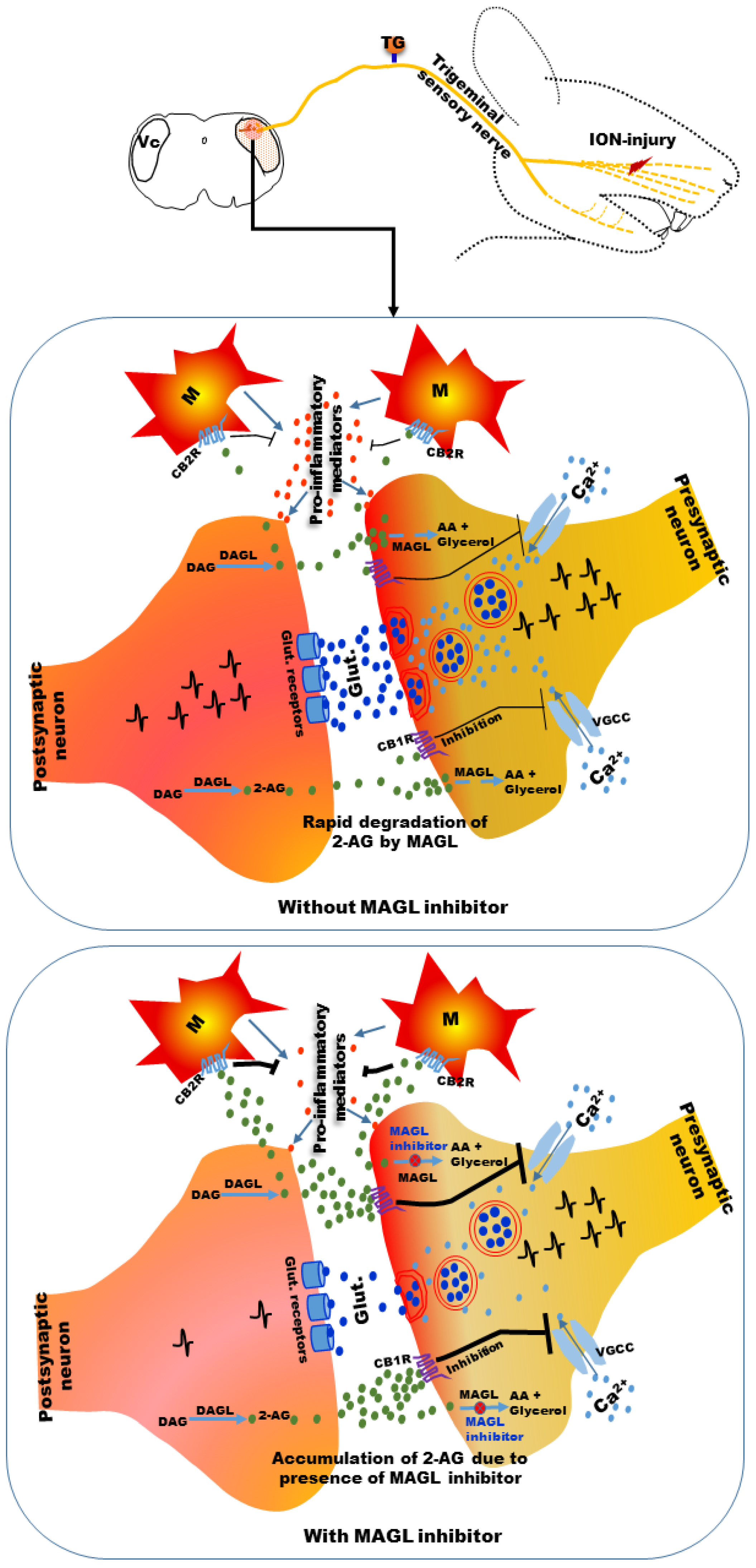{kind=link}
| Compounds (Chemical Name) | Neuropathic Pain Models | Route of Administration | Anti-Nociceptive Effects | Antagonized by /Effectiveness in CB1/CB2 Knockout Mice | Cannabimimetic Side Effects | Ref. |
|---|---|---|---|---|---|---|
| ACEA [(5Z,8Z,11Z,14Z)-N-(2-Chloroethyl)-5,8,11,14-icosatetraen amide] | Spinal nerve ligation injury in rats | Intra-spinal | Attenuation of mechanically-evoked responses of spinal neurons | CB1-selective antagonist | Not tested | [87] |
| Cisplatin-induced neuropathy in rats | Systemic/intra-plantar | Attenuation of mechanical allodynia | CB1-selective antagonist | Did not elicit psychoactive effect at the dose used | [88] | |
| CRA13 [Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl) methanone] | Partial sciatic nerve ligation injury in rats | Oral | Attenuation of mechanical hyperalgesia | CB1-selective antagonist | Did not elicit cardiovascular and central nervous system (CNS) side effects at low but anti-nociceptive dose | [89] |
| AZD1940 [N-{1-[(4,4-Difluorocyclohexyl)methyl]-2-(2-methyl-2-propanyl)-1H-benzimidazol-5-yl} ethanesulfonamide] | Spinal nerve ligation injury in rats | Oral | Attenuation of mechanical allodynia | CB1-selective antagonist | Did not develop tolerance | [90] |
| AZ11713908 [N-{1-(Cyclohexylmethyl)-2-[(5-ethoxy-2-pyridinyl)methyl]-1H-benzimidazol-5-yl}-N-methyl-2-thiophenesulfonamide] | Spinal nerve ligation injury in rats | Systemic | Attenuation of mechanical allodynia | Not tested | Minimal CNS side effects | [91] |
| LBP1 [2-[4-({3-[7-Chloro-1-(tetrahydro-2H-pyran-4-ylmethyl)-1H-indol-3-yl]-1,2,4-oxadiazol-5-yl}methyl)-1-piperazinyl] acetamide] | Spinal nerve ligation injury in rats | Oral | Attenuation of thermal hyperalgesia and mechanical allodynia | CB1-selective antagonist | Did not elicit motor impairment | [92] |
| PrNMI [4-{2-[-(1E)-1[(4-propylnaphthalen-1-yl)methylidene]-1H-inden-3-yl]ethyl} morpholine] | Sciatic nerve entrapment injury in rats | Systemic/oral | Attenuation of mechanical allodynia | CB1-selective antagonist | Minimal CNS side effects | [64] |
| Compounds [Chemical Name] | Neuropathic Pain Models | Route of Administration | Anti-Nociceptive Effects | Antagonized by/Effectiveness in CB1/CB2 Knockout Mice | Cannabimimetic Side Effects | Ref. |
|---|---|---|---|---|---|---|
| JWH015 [(2-Methyl-1-propyl-1H-indol-3-yl)(1-naphthyl) methanone] | Sciatic nerve chronic constriction injury (CCI) in mice | Intra-plantar | Attenuation of mechanical allodynia and thermal hyperalgesia | CB2-selective antagonist | Not tested | [131] |
| Streptozotocin-induced diabetic neuropathy in mice | Intra-plantar | Attenuation of mechanical and thermal allodynia and thermal hyperalgesia | CB2-selective antagonist | Not tested | [132] | |
| AM1241 [(2-Iodo-5-nitrophenyl) {1-[(1-methyl-2-piperidinyl)methyl]-1H-indol-3-yl} methanone] | Spinal nerve ligation injury in rats and mice | Systemic | Attenuation of mechanical allodynia and thermal hyperalgesia | CB2-selective antagonist, effective in CB1 knockout mice | Not tested | [101] |
| Spinal nerve ligation injury in rats | Systemic/intra-spinal/intra-dorsal root ganglia (DRG) | Attenuation of mechanical allodynia | CB2-selective antagonist | Not tested | [103,108] | |
| Vincristine- induced neuropathy in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Did not elicit catalepsy | [133] | |
| Paclitaxel- induced neuropathy in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Not tested | [134] | |
| Streptozotocin- induced diabetic neuropathy in rats | Systemic | Attenuation of mechanical allodynia | Not tested | Not tested | [135,136] | |
| Spared nerve injury of sciatic nerve in rats | Systemic self-administration by animal | Attenuation of mechanical allodynia | Not tested | Did not elicit motor ataxia | [137] | |
| Sciatic nerve CCI in rats | Intra-spinal | Attenuation of mechanical allodynia | Not tested | Not tested | [138,139] | |
| GW405833/L768242 [(2,3-Dichlorophenyl){5-methoxy-2-methyl-3-[2-(4-morpholinyl)ethyl]-1H-indol-1-yl} methanone] | Partial sciatic nerve ligation injury in rats | Systemic | Attenuation of mechanical allodynia | Not tested | Did not elicit sedation and catalepsy at low but effective dose | [127] |
| Partial sciatic nerve ligation injury in mice | Systemic | Attenuation of mechanical allodynia | Not tested | High dose produced motor deficits | [129] | |
| Spinal nerve ligation injury in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Not tested | [103] | |
| Modified spinal nerve ligation injury | Systemic (chronic) | Attenuation of mechanical allodynia | Not tested | Did not observe any motor deficits and catalepsy, although not formally measured | [126] | |
| Sciatic nerve CCI in rats | Systemic | Attenuation of mechanical allodynia and depression-like behavior | Not tested | Did not elicit motor impairment | [140] | |
| MDA7 [1-[(3-benzyl-3-methyl-2,3-dihydro-1-benzofuran-6-yl)carbonyl] piperidine] | Paclitaxel- induced neuropathy in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Did not affect locomotor activity | [141] |
| Spinal nerve ligation injury in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Did not affect locomotor activity | [141] | |
| Paclitaxel- induced neuropathy in rats and mice | Systemic (chronic) | Prevented the development of mechanical allodynia | CB2-selective antagonist, absent in CB2 knockout mice | Not tested | [117] | |
| MDA19 [N′-[(3Z)-1-(1-hexyl)-2-oxo-1,2-dihydro -3H-indol-3-ylidene]benzo hydrazide] | Spinal nerve ligation injury in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Did not affect locomotor activity | [142] |
| Paclitaxel- induced neuropathy in rats and mice | Systemic | Attenuation of mechanical allodynia | Ineffective in CB2 knockout mice | Did not affect locomotor activity | [142] | |
| NES400 [1-(2,4-dichlorophenyl)-6-methyl-N-cyclohexylamine-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide] | Spared nerve injury in mice | Systemic (chronic) | Attenuation of mechanical allodynia and thermal hyperalgesia | Not tested | Not tested | [143] |
| JWH133 [(6aR,10aR)-6,6,9-Trimethyl-3-(2-methyl-2-pentanyl)-6a,7,10,10a-tetrahydro-6H -benzo[c] chromene] | Spinal nerve ligation injury in rats | Intra-spinal | Attenuation of mechanically evoked responses of spinal neurons | CB2-selective antagonist | Not tested | [87] |
| Intra-plantar | Attenuation of mechanical stimuli-induced responses of WDR neurons in spinal cord | CB2-selective antagonist | Not tested | [121] | ||
| Partial sciatic nerve ligation injury in mice | Intra-spinal | Attenuation of mechanical allodynia | Ineffective in CB2 receptor knockout mice | Not tested | [144] | |
| Subcutaneous | Attenuation of mechanical allodynia | CB2-selective antagonist | Not tested | [106] | ||
| Cisplatin- induced neuropathy in rats | Systemic/intra-plantar | Attenuation of mechanical allodynia | CB2-selective antagonist | Did not elicit psychoactive effect at the dose used | [88] | |
| AM1710 [1-Hydroxy-9-methoxy-3-(2-methyl-2-octanyl) -6H-benzo[c] chromen-6-one] | Cisplatin and paclitaxel-induced neuropathy in rats | Systemic | Attenuation of mechanical and cold allodynia | CB2-selective antagonist | Not tested | [145] |
| Paclitaxel- induced neuropathy in rats | Subcutaneous (chronic) | Prevented the development of mechanical and cold allodynia | CB2-selective antagonist | Not tested | [146] | |
| Paclitaxel- induced neuropathy in mice | Systemic (acute and chronic)/intra-spinal | Attenuation of mechanical and cold allodynia | CB2-selective antagonist/ineffective in CB2 knockout mice | Did not elicit motor dysfunction and hypothermia | [147] | |
| AM1714 [1,9-Dihydroxy-3-(2-methyl -2-octanyl)-6H-benzo[c] chromen-6-one] | Paclitaxel- induced neuropathy in rats | Systemic | Attenuation of mechanical allodynia | CB2-selective antagonist | Not tested | [134] |
| L759,656 [(6aR,10aR)-1-Methoxy-6,6-dimethyl-9-methylene-3-(2-methyl-2-octanyl)-6a,7,8,9,10,10a-hexahydro-6H-benzo[c] chromene] | Streptozotocin-induced diabetic neuropathy in mice | Intra-spinal | Attenuation of heat hyperalgesia | CB2-selective antagonist | Not tested | [148] |
| BCP [(1R,9S)-4,11,11-Trimethyl-8-methylenebicyclo [7.2.0] undec-4-ene] | Partial sciatic nerve ligation injury in mice | Oral (chronic) | Attenuation of mechanical allodynia and thermal hyperalgesia | Ineffective in CB2 knockout mice | Did not elicit motor disturbance, hypothermia or catalepsy | [106] |
| Paclitaxel-induced neuropathy in mice | Oral (during and after paclitaxel treatment) | Attenuation of mechanical allodynia and prevention of development of mechanical allodynia | CB2 receptor antagonist | Not tested | [108] | |
| LY2828360 [8-(2-Chlorophenyl)-2-methyl-6-(4-methyl-1-piperazinyl)-9-(tetrahydro-2H-pyran-4-yl)-9H -purine] | Paclitaxel- induced neuropathy in mice | Systemic (acute and chronic) | Attenuation of mechanical and cold allodynia | Not tested | Did not produce tolerance on chronic administration | [149] |
| Compounds [Chemical Name] | Neuropathic Pain Models | Route of Administration | Anti-Nociceptive Effects | Antagonized by/Effectiveness in CB1/CB2/FAAH Knockout Mice | Cannabimimetic Side Effects | Ref. |
|---|---|---|---|---|---|---|
| OL135 [1-oxo-1-[5-(2-pyridyl)-2-yl]-7-phenyl heptane] | Sciatic nerve CCI in mice | Systemic | Attenuation of mechanical allodynia and acetone-induced cold allodynia | CB1 and CB2 receptor antagonists, not affected by the TRPV1 receptor antagonist | Not tested | [199] |
| Oral | Attenuation of mechanical allodynia | Not tested | Not tested | [200] | ||
| URB597 [cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester] | Sciatic nerve CCI in mice | Systemic | Attenuation of mechanical allodynia and acetone-induced cold allodynia | CB1 and CB2 receptor antagonists, ineffective in fatty acid amide hydrolase (FAAH) knockout mice | Not tested | [199] |
| Not tested | Did not elicit catalepsy and motor impairment but slight sedation observed | [201] | ||||
| Oral (chronic) | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 receptor antagonists | Not tested | [202] | ||
| Oral | Attenuation of mechanical allodynia | Not tested | Not tested | [200] | ||
| Sciatic nerve CCI in rats | Intra-spinal | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 or CB1/transient receptor potential vanilloid 1 (TRPV1) antagonists depending on the doses | Not tested | [203] | |
| Attenuation of mechanical allodynia, thermal hyperalgesia and cold allodynia | High dose antagonized by a TRPV1 antagonist | Not tested | [204] | |||
| Partial sciatic nerve ligation injury in mice | Intra-plantar injection | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 receptor antagonists, ineffective in CB1 but not CB2 knockout mice | Not tested | [205] | |
| Partial sciatic nerve ligation injury in rats | Intra-plantar injection | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 and CB2 receptor antagonists | Not tested | [206] | |
| Intra- pre-limbic/infra-limbic cortex microinjection | Attenuation of mechanical allodynia | Not tested | Not tested | [207,208] | ||
| Cisplatin- induced neuropathy in rats | Systemic | Attenuation of mechanical and cold allodynia | CB1, CB2, and TRPV1 receptor antagonists | Not tested | [183] | |
| Paclitaxel- induced neuropathy in mice | Systemic | Attenuation of mechanical and cold allodynia | Not tested | Not tested | [209] | |
| Paclitaxel- induced neuropathy in mice | Systemic (chronic) | Prevented the development of mechanical and cold allodynia and attenuated established allodynia | Not tested | No physical dependence or tolerance | [210] | |
| Streptozotocin-induced diabetic neuropathy in rats | Systemic | Attenuation of mechanical allodynia | Not tested | Not tested | [211] | |
| ST4070 [1-biphenyl-4-ylethenyl piperidine-1-carboxylate] | Sciatic nerve CCI in mice | Oral | Attenuation of mechanical allodynia | CB1, CB2, and PPARα antagonists | Not tested | [200] |
| Vincristine- induced neuropathy in mice | Oral | Attenuation of mechanical allodynia | Not tested | Not tested | [200] | |
| Streptozotocin-induced diabetic neuropathy in mice | Oral | Attenuation of mechanical allodynia | Not tested | Not tested | [200] | |
| PF3845 [N-(3-Pyridinyl)-4-(3-{[5-(trifluoromethyl)-2-pyridinyl]oxy}benzyl)-1-piperidine carboxamide] | Sciatic nerve CCI in mice | Systemic | Attenuation of mechanical allodynia | Not tested | Did not elicit motor impairment, hypothermia and catalepsy | [212] |
| Not tested | Not tested | [213] | ||||
| Attenuation of mechanical and cold allodynia | Not tested | Chronic administration did not induce development of tolerance | [214] | |||
| Ineffective in CB1 and CB2 knockout mice | Not tested | [215] | ||||
| Sciatic nerve CCI in rat | Intra-spinal | Attenuation of mechanical allodynia and thermal hyperalgesia | Not tested | Not tested | [207] | |
| URB937 [3′-Carbamoyl-6-hydroxy-3-biphenylyl cyclohexyl carbamate] | Sciatic nerve CCI in mice | Systemic | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 receptor antagonists | Did not alter spontaneous locomotor activity and daily food intake or feeding pattern | [216] |
| Oral | Attenuation of mechanical allodynia, hyperalgesia and thermal hyperalgesia | Not tested | No gastric ulcerogenic effect | [217] | ||
| Cisplatin- induced neuropathy in rats | Systemic | Attenuation of mechanical and cold allodynia | CB1, CB2, and TRPV1 receptor antagonists | Not tested | [183] | |
| Paclitaxel- induced neuropathy in mice | Systemic (chronic) | Prevented the development of mechanical and cold allodynia and attenuated established allodynia | Not tested | No physical dependence or tolerance | [210] |
| Compounds [Chemical Name] | Neuropathic Pain Models | Route of Administration | Anti-Nociceptive Effects | Antagonized by/Effectiveness in CB1/CB2 Knockout Mice | Cannabimimetic Side Effects | Ref. |
|---|---|---|---|---|---|---|
| URB602 [1,1′-biphenyl]-3-yl- carbamic acid, cyclohexyl ester] | Partial sciatic nerve ligation in rats | Intra-plantar | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 and CB2 receptor antagonists | Not tested | [206] |
| Partial sciatic nerve ligation in mice | Intra-plantar | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 and CB2 receptor antagonists, the effect was altered in both CB1 and CB2 knockout mice | Not tested | [205] | |
| JZl184 [4-nitrophenyl4-(dibenzo[d][1,3]dioxol-5-yl (hydroxy) methyl) piperidine-1- carboxylate] | Sciatic nerve CCI injury in mice | Systemic | Attenuation of mechanical and cold allodynia | CB1 antagonist | Produced hypo-motility | [199] |
| Ineffective in CB1 but not CB2 knockout mice | Not tested | [215] | ||||
| Not tested | Chronic administration induced tolerance, impairment of synaptic plasticity and downregulation and desensitization of CB1 receptors | [214] | ||||
| Not tested | Not tested | [223] | ||||
| Not tested | Not tested | [224] | ||||
| Not tested | Not tested | [225] | ||||
| Not tested | Catalepsy, motor impairment and sedation | [201] | ||||
| Attenuation of mechanical and thermal hyperalgesia | Mechanical allodynia antagonized by both CB1 and CB2 receptor antagonists and thermal hyperalgesia antagonized by CB1 receptor antagonist | Produced hypo-motility | [226] | |||
| Attenuation of mechanical allodynia | Not tested | High dose but not low dose produced impairment of locomotor activity, hypothermia and catalepsy | [212] | |||
| Paclitaxel- induced neuropathy in mice | Systemic | Attenuation of mechanical allodynia | CB1 and CB2 receptor antagonists, ineffective in both CB1 and CB2 knockout mice | Not tested | [227] | |
| Attenuation of mechanical and cold allodynia | Not tested | Not tested | [209] | |||
| Cisplatin- induced neuropathy in rats | Systemic | Attenuation of mechanical and cold allodynia | CB1 and CB2 receptor antagonists | Not tested | [183] | |
| Cisplatin- induced neuropathy in mice | Intra-planter (chronic) | Attenuation of mechanical hyperalgesia | CB1 but not CB2 receptor antagonists | Not tested | [228] | |
| KML29 [1,1,1,3,3,3-hexafluoropropan-2-yl 4-(bis(benzo[d][1,3]dioxol-5-yl)(hydroxy)methyl)piperidine-1-carboxylate] | Sciatic nerve CCI in mice | Systemic | Attenuation of mechanical and cold allodynia | CB1 receptor antagonist | Did not elicit catalepsy, hypothermia and hypo-motility | [224] |
| Attenuation of mechanical and cold allodynia | Not tested | Not tested | [225] | |||
| MJN110 [2,5-dioxopyrrolidin-1-yl 4-(bis(4-chlorophenyl) methyl)piperazine-1- carboxylate] | Sciatic nerve CCI injury in mice | Systemic | Attenuation of mechanical allodynia and thermal hyperalgesia | Mechanical allodynia antagonized by both CB1 and CB2 receptor antagonists and thermal hyperalgesia antagonized by CB1 receptor antagonist | Did not elicit hypo-motility, catalepsy and hypothermia | [226] |
| Streptozotocin-induced diabetic neuropathy in rats | Systemic | Attenuation of mechanical allodynia | Not tested | Not tested | [211] | |
| Paclitaxel- induced neuropathy in mice | Systemic | Attenuation of mechanical allodynia | CB1 and CB2 receptor antagonists, ineffective in both CB1 and CB2 knockout mice | Not tested | [227] |
| Compounds | Neuropathic Pain Models | Route of Administration | Anti-Nociceptive Effects | Antagonized by | Cannabimimetic Side Effects | Ref. |
|---|---|---|---|---|---|---|
| WIN 55,212-2 (synthetic cannabinoid) | Infra-orbital nerve CCI in rats | Systemic | Attenuation of mechanical allodynia and thermal hyperalgesia | CB1 but not CB2 antagonist | Not tested | [84] |
| HU210 (synthetic cannabinoid) | Infra-orbital nerve CCI in rats | Systemic | Attenuation of mechanical allodynia and thermal hyperalgesia | Not tested | Not tested | [84] |
| AM1241 (selective CB2 agonist) | Nitroglycerin- induced migraine model in rats | Systemic | Attenuation of nocifensive behavior induced by formalin injection into paw | Not tested | Not tested | [256] |
| URB597 (FAAH inhibitor) | Nitroglycerin- induced migraine model in mice | Systemic | Attenuation of mechanical hyperalgesia | CB1 receptor antagonist | Not tested | [248] |
| PF3845 (FAAH inhibitor) | Nitroglycerin- induced migraine model in mice | Systemic | Attenuation of mechanical hyperalgesia | CB1 receptor antagonist | Not tested | [248] |
| URB937 (FAAH inhibitor) | Nitroglycerin- induced migraine model in rats | Systemic | Attenuation of nocifensive behavior induced by formalin injection into lip and paw | Not tested | Not tested | [257] |
| JZL184 (MAGL inhibitor) | Partial transection of infra-orbital nerve in mice | Systemic | Attenuation of mechanical allodynia | Not tested | Not tested | [258] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.Z.; Ando, H.; Unno, S.; Kitagawa, J. Targeting Peripherally Restricted Cannabinoid Receptor 1, Cannabinoid Receptor 2, and Endocannabinoid-Degrading Enzymes for the Treatment of Neuropathic Pain Including Neuropathic Orofacial Pain. Int. J. Mol. Sci. 2020, 21, 1423. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041423
Hossain MZ, Ando H, Unno S, Kitagawa J. Targeting Peripherally Restricted Cannabinoid Receptor 1, Cannabinoid Receptor 2, and Endocannabinoid-Degrading Enzymes for the Treatment of Neuropathic Pain Including Neuropathic Orofacial Pain. International Journal of Molecular Sciences. 2020; 21(4):1423. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041423
Chicago/Turabian StyleHossain, Mohammad Zakir, Hiroshi Ando, Shumpei Unno, and Junichi Kitagawa. 2020. "Targeting Peripherally Restricted Cannabinoid Receptor 1, Cannabinoid Receptor 2, and Endocannabinoid-Degrading Enzymes for the Treatment of Neuropathic Pain Including Neuropathic Orofacial Pain" International Journal of Molecular Sciences 21, no. 4: 1423. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041423