Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium



1
Institute of Anatomy and Cell Biology, Friedrich-Alexander University Erlangen-Nürnberg, Krankenhausstraße 9, D-91054 Erlangen, Germany
2
Department of Neurology, Klinikum St. Marien Amberg, Mariahilfbergweg 7, D-92224 Amberg, Germany
3
Institute of Experimental and Clinical Pharmacology and Toxicology, Friedrich-Alexander University Erlangen-Nürnberg, Fahrstraße 17, D-91054 Erlangen, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(5), 1663; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051663
Submission received: 6 February 2020
/
Revised: 16 February 2020
/
Accepted: 26 February 2020
/
Published: 28 February 2020
(This article belongs to the Special Issue CNS Drug Action in Neurodegenerative Diseases)
Abstract
:Calcium ions are vital for maintaining the physiological and biochemical processes inside cells. The central nervous system (CNS) is particularly dependent on calcium homeostasis and its dysregulation has been associated with several neurodegenerative disorders including Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD), as well as with multiple sclerosis (MS). Hence, the modulation of calcium influx into the cells and the targeting of calcium-mediated signaling pathways may present a promising therapeutic approach for these diseases. This review provides an overview on calcium channels in neurons and glial cells. Special emphasis is put on MS, a chronic autoimmune disease of the CNS. While the initial relapsing-remitting stage of MS can be treated effectively with immune modulatory and immunosuppressive drugs, the subsequent progressive stage has remained largely untreatable. Here we summarize several approaches that have been and are currently being tested for their neuroprotective capacities in MS and we discuss which role calcium could play in this regard.
1. Role of Calcium in the Central Nervous System (CNS)
1.1. Role of Calcium under Physiological Conditions
Calcium is one of the most important second messengers regulating numerous cellular processes such as signal transduction, proliferation, muscle contraction and neurotransmitter release. The calcium concentration differs greatly between the milieu outside (2 mM) and inside (0.00001 mM) the cell [1,2].
Perhaps the most prominent example of a calcium-mediated process in the brain is neurotransmitter release. Yet, not only neurons, but also glia cells, which are estimated to be 10 times more frequent than neurons in the CNS, depend on calcium [3].
Oligodendrocytes are essential for the function and survival of neurons. Their main purpose is the formation of the myelin sheath [4,5], which is complex and requires different stimuli. The influx of calcium is important for the maturation of oligodendrocyte precursor cells (OPC) and the induction of myelination [6,7,8]. Additionally, the local calcium concentration seems to be essential for myelin elongation and sheath development [9,10,11].
Astrocytes are star-shaped glia cells in the brain, which are characterized by their expression of glial fibrillary acidic protein (GFAP). Astrocytes were first thought to be mainly important for providing passive structural support to neurons, but are now well acknowledged to actively participate in the regulation of brain functions [12,13]. They are part of the blood–brain barrier (BBB) [14] and are able to take up sodium and glutamate from the synaptic cleft following the initiation of an action potential [15]. It has been described that an increase in astrocytic intracellular calcium triggers the release of gliotransmitters (neurotransmitters secreted by glia cells) like adenosine triphosphate (ATP), glutamate, D-serine and gamma-aminobutyric acid (GABA) [16]. However, calcium signaling in astrocytes has remained a subject to debate and there are some controversies which have not yet been clarified (reviewed in [16]).
Microglia are the immune cells of the CNS and play a crucial role after tissue damage by mediating inflammatory processes. An increased extracellular calcium concentration can be recognized by microglia via specific channels, which triggers their migration to the site of the lesion [17].
Apart from neurons and glia, many other cells in the CNS depend on calcium. It is clearly beyond the scope of this review to further elaborate on all calcium-mediated cellular processes, but it should be noted that, e.g., the permeability of the BBB, which consists not only of astrocytes but also of endothelial cells and tight junctions, is regulated by intracellular calcium levels [18]. In addition, other immune cells such as T cells, which physiologically populate the human brain, rely on calcium as a second messenger for intracellular processes [19].
1.2. Role of Calcium under Pathological Conditions
Dysregulation of calcium homeostasis has been proposed in the pathogenesis of several CNS diseases (reviewed in [20]). One of the general concepts of calcium-induced cell death after damage is excitotoxicity. The term describes induced cell death following an overload of stimulating agents such as glutamate, and the subsequent increase in the intracellular calcium concentration. Excitotoxicity is known to occur in several neurodegenerative diseases including Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) [20,21,22]. An increased intracellular calcium concentration has been shown to induce superoxide formation and to trigger the release of proapoptotic proteins. It has also been associated with decreased production of ATP and activation of reactive oxygen species (ROS), which can trigger neuronal cell death [23,24]. Additionally, an increased calcium concentration activates calmodulin-dependent kinases and the production of nitric oxide (NO) [23,24,25,26].
2. Calcium Channels Expressed by the Different Cell Types of the CNS
2.1. Intracellular Calcium Channels
Intracellularly, there are three major groups of channels which are responsible for the release of calcium from the endoplasmic reticulum (ER): ryanodine receptors (RyR) [2], inositol-3-phosphate receptors (IP3R) [2,27] and two-pore channels (TPC) [28].
2.1.1. RyR
RyR are named after their most prominent binding partner, ryanodine. RyR activity is mainly triggered by an increased intracellular calcium concentration originating from the extracellular side [1]. The three different isotypes of RyR (RyR1-3) are all expressed in the brain. While RyR3 is widely distributed inside the brain [29], RyR2 seems to be the most abundant receptor type [30,31]. All RyR are present in neurons, but their distribution is different regarding cellular localization, brain area and stage of development [32]. For example, in granule cells of the cerebellum only RyR2 has been found [33], whereas Purkinje cells express all RyR subtypes [34]. In OPC, only RyR3 is present, which is then downregulated during development [35]. In mature oligodendrocytes, all RyR have been reported to be present [36]. The pattern of RyR expression in astrocytes is unclear. One study found that only RyR3 is expressed in astrocytes [37]. In contrast, another study has observed all RyR subtypes in rat brain astrocytes [35]. Microglia are known to express functional RyR1 and RyR2 channels, while RyR3 is only detectable in fetal stages of microglia during brain development [38].
2.1.2. IP3R
IP3R are named after their agonist inositol trisphosphate (IP3). IP3R activation is mainly mediated by G-protein coupled receptors (GPCR) [1]. Three different isotypes of IP3R exist: IP3R1, IP3R2 and IP3R3. Every brain cell type contains at least one type of IP3R [2]. Most of the neurons in the CNS contain IP3R1 [39], which is expressed throughout the cell, and IP3R3, which is expressed only in the soma [40,41]. IP3R2 is exclusively located in glia cells [41], especially in astrocytes [42].
2.1.3. TPC
TPC are nonselective cation channels conducting sodium and calcium [43] They are mainly located in the membrane of acidic vesicles [44], where they mediate calcium homeostasis after activation by nicotinic acid adenine dinucleotide phosphate (NAADP) [28]. In neuronal cells, the channel has been described to mediate differentiation [45] and it also seems to be important for autophagic processes in astrocytes [46,47].
2.2. Plasma Membrane Calcium Channels
There are several different calcium channels within the plasma membrane which are important for the function of CNS cells, including voltage-gated calcium channels (VGCC) [48], ionotropic glutamate receptors [49,50], transient receptor potential (TRP) channels [51], calcium release-activated calcium (CRAC) channels [52] and purinergic P2X receptors [53].
2.2.1. VGCC
VGCC comprise a range of different channels: L-type (CaV1.1–CaV1.4), P/Q-type (CaV2.1), N-type (CaV2.2), R-type (CaV2.3) and T-type (CaV3.1–CaV3.3) channels. The biophysical properties of each class are determined by the pore-forming α-subunit and associated auxiliary subunits (reviewed e.g., in [48,54]). All VGCC are activated by depolarization of the membrane potential. VGCC are widely distributed in neurons and glia cells [48].
CaV1.2 and CaV1.3 are the most abundant L-type channels in the brain. They are located at postsynaptic dendrites and the soma [55] and control neuronal excitability and gene transcription [56]. CaV2.1 and CaV2.2 are located at the presynapse, where they mediate transmitter release [57], whereas CaV2.3 is mainly located on neuronal spines [58]. T-type channels are expressed on the cell body and control the rhythmic burst firing of thalamic neurons, which is important for the generation of neuronal oscillations under physiological (sleep states) and pathophysiological (absence epilepsy) conditions [48,54].
The expression profile of VGCC changes during development from OPC to mature oligodendrocytes [59,60]. VGCC are expressed on OPC and mature oligodendrocytes, while being absent on immature oligodendrocytes [61]. Whereas low VGCC (probably T-type) are present at OPC extensions, high VGCC (probably L-type) are mostly located at the soma of OPC [60]. In mice, it has not only been shown that after knockout of CaV1.2 less myelination of axons occurred and the maturation from OPC to oligodendrocytes was disturbed [7], but also remyelination was impaired [62]. Remarkably, the expression of myelin basic protein (MBP), an essential myelin protein, has been demonstrated to inhibit the influx of calcium via VGCC, whereas the so-called golli forms (precursors of MBP, which are present in OPC) enhanced the influx of calcium via VGCC [63,64]. Additionally, the N-type CaV2.2 was detected on mature oligodendrocytes [65].
Furthermore, astrocytes express VGCC [66]. One of their functions in astrocytes is the release of gliotransmitters like glutamate [67]. However, there are several studies which provide contradictory results regarding the expression profile of VGCC on astrocytes. All studies have shown the expression of CaV1.2. In addition, Cheli et al. have demonstrated the expression of CaV2.1 and CaV1.3 on mouse primary astrocytes [68]. Another study was unable to detect CaV2.1 on rat primary astrocytes but observed the expression of CaV1.3, and of CaV2.2, CaV2.3 and CaV3.1 [69]. However, D’Ascenzo et al. did not detect CaV1.3, but CaV2.1, CaV2.2 and CaV2.3 on primary astrocytes from rats [70]. Taken together, it seems reasonable that the expression of VGCC on astrocytes is species-specific and dynamic, brain region- and/or development-dependent.
Microglia are predicted to express at least L-type VGCC, but the exact isoform has not been identified so far [71].
2.2.2. Ionotropic Glutamate Receptors
The three ionotropic glutamate receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), N-methyl-d-aspartate (NMDA) and kainate (KA) receptors [72], are essential for synaptic transmission and plasticity [73,74,75]. For a more detailed overview on their molecular structure, their subcellular compartment and their physiological properties, please refer to comprehensive reviews on AMPA [76], NMDA [77] and KA [78] receptors. All three receptors have been detected on oligodendrocytes [79,80], and excitotoxicity in these cells seems to be mediated by AMPA and KA receptors [81,82]. Astrocytes have AMPA and KA, but no NMDA receptors [83,84]. Microglia express AMPA and KA receptors [85] and at least a subunit of NMDA receptors with unknown function [86].
2.2.3. TRP Channels
TRP channels have a broad distribution profile and are non-selective cation channels, with some of them being permeable for calcium. The superfamily of TRP channels consists of 28 members in humans, which are organized in six subgroups: TRPA, with A for “ankyrin”; TRPC, with C for “canonical”, TRPM, with M for “melastatin”, TRPML, with ML for “mucolipidin”, TRPP, with P for “polycystin” and TRPV, with V for “vanilloid” [87].
The TRPA class consists of only one receptor type: TRPA1. TRPA1 is expressed in all cortical layers as well as in the hippocampus of rodents, and it has been shown that TRPA1 can affect neuronal circuits [88]. Additionally, there are reports on TRPA1 expression on oligodendrocytes [89] and astrocytes [90].
Channels of the TRPC family which are known to be expressed in the brain are TRPC1, TRPC4, TRPC5 [91], TRPC3 and TRPC6 [92]. While TRPC1, TRPC4 and TRPC5 are activated via IP3, TRPC2, TRPC3, TRPC6 and TRPC7 are activated via diacylglycerol (DAG) [93]. TRPC1 expression has been described on OPC, where it mediates OPC differentiation [94]. TRPC3 is located on oligodendrocytes [95]. TRPC1 [96], TRPC3 [97] and TRPC6 [98] are present on astrocytes, while TRPC3, especially, seems to be involved in gliosis after neuronal damage [97]. Microglia are known to increase their expression of TRPC3 after brain-derived neurotrophic factor (BDNF) stimulation [99] and TRPC6 activation by amyloid β (Aβ) has been reported [100].
TRPM1 is found in very small quantities in the CNS and its function has remained unknown [92]. TRPM2 channels are expressed on neurons. Here, they mediate synaptic plasticity as well as oxidative stress and their expression is increased after interleukin stimulation [101]. TRPM3 and TRPM5 are expressed on Purkinje cells, whereby the former is predicted to play a role in early brain development [102,103]. TRPM7 is also highly expressed in the brain [92], where it plays an important role in learning and memory processes in hippocampal neurons [104]. Oligodendrocytes or at least OPC are known to express TRPM3, which can be actively involved in myelination [105]. TRPM2, TRPM4 and TRPM6 are present on astrocytes [106]. TRPM2 expression is increased after interleukin stimulation [101]. Reactive astrocytes express high levels of TRPM7, which seems to be important for gliotic scar formation [107]. Like TRPC3, TRPM1 is upregulated after stimulation of microglia with Aβ [108], whereas TRPM2 is involved in microglia activation [109] after interleukin stimulation [101]. TRPM7 is important for migration properties [110]. Additionally, the TRPM4 channel was found on microglia, but its function remains unclear [111].
TRPV channels mediate pain transduction in nociceptors. All TRPV channels (TRPV1 to TRPV6) are expressed in the mouse brain [112]. While TRPV1 to TRPV4 are expressed on neurons, TRPV5 and TRPV6 are mainly found in epithelial tissue [113]. For OPC, only the expression of TRPV4 has been reported, and channel activity was shown to increase OPC proliferation [114]. Mature oligodendrocytes also express TRPV1 [115]. Astrocytes express four types of TRPV channels: TRPV1, which is involved in astrogliosis [116], TRPV2, which is important for the regulation of the lipid metabolism [117], TRPV4, which is osmosensitive and regulates brain homoeostasis [118] and TRPV6 with unknown function [106]. Microglia or at least the BV2 microglia cell line are known to express TRPV1 and TRPV2, which mediate phagocytosis [119]. TRPV4 is additionally involved in microglia activation [120].
2.2.4. Calcium Release-Activated Calcium Channels
The family of CRAC channels consists of three different members: Orai1, Orai2 and Orai3 [52]. The influx of extracellular calcium into the cytosol as well as the release of internal calcium from the ER mediated by Orai channels drives exocytosis, stimulates mitochondrial metabolism, activates gene expression, and promotes proliferation [121]. The stromal interaction molecule 1 (STIM1) is important for the function of Orai1. It is located in the membrane of the ER and acts as a calcium sensor for increased calcium concentration in the cytoplasm. STIM1 interacts with Orai1, which is indispensable for the pore opening of the Orai1 channel [122]. This process is termed store-operated calcium entrance (SOCE). SOCE occurs in neurons upon Orai1 and Orai2 interaction with STIM1 and/or STIM2 [123], and this process appears to be essential for neuronal growth [124]. Oligodendrocytes and astrocytes (at least in the optic nerve of mice) also express Orai1 as well as STIM1 and STIM2 [125]. Microglia express Orai1 and STIM1 [126]. For a detailed review on Orai and STIM function see [127].
2.2.5. Purinergic P2X Channels
P2X receptors belong to the family of purinergic receptors. They are ionotropic and activated by ATP [128,129]. P2X channels are located at presynaptic and postsynaptic nerve terminals [129]. Regarding the different subtypes of P2X channels in neuronal cells see [53,128]. A detailed summary of P2X receptors is provided in [130]. A functional analysis of purinergic receptors in oligodendrocytes and microglia is provided in [131] and [132], respectively.
Figure 1 summarizes the expression profile of calcium channels by the different cell types of the CNS.
3. Pharmacological Blockers of Calcium Channels in the CNS
3.1. Dihydropyridines (DHP)
In 1969, the German physiologist Albrecht Fleckenstein described the effect of Bay a 1040 on calcium influx in the heart as “Ca++-antagonism”, later known to be due to a specific block of L-type VGCC [133]. This compound was named nifedipine and is the first in the drug class of DHP. Further drugs in this series included amlodipine, nicardipine, nimodipine and lercanidipine [134]. DHP block all L-type calcium channels (CaV 1.1–CaV 1.4), whereas P-, N-, and R-type (CaV 2.1–CaV 2.3) channels are relatively unaffected [48]. Some DHP, including nifedipine, amlodipine and lercanidipine, are important anti-hypertensive drugs [135], while other DHP like isradipine or nimodipine may have a beneficial effect in CNS-related diseases [48]. A proposed general effect of DHP action in CNS disorders could be the stimulation of autophagic processes and the clearance of toxic components like mutated huntingtin or tau protein [136]. A recent review summarizes the potential effects for each DHP and for each channel in detail in the context of CNS diseases including AD, HD, PD and psychotic disorders [48].
3.2. Other Calcium Channel Blockers Modulators Affecting the CNS
Besides DHP, other drugs act on L-type VGCC. Phenylalkylamines like verapamil, which have a very limited BBB permeability, and benzothiazepines like diltiazem, block L-type calcium channels [137]. In addition, several antiepileptic drugs including ethosuximide, valproic acid and zonisamide, and even some antipsychotic drugs like pimozide and penfluridol block T-type calcium channels, which may explain at least parts of their clinical effects [138]. Gabapentine and pregabalin, two anticonvulsant drugs which are also used for neuropathic pain treatment, block VGCC via binding to the α2δ subunit [139]. Not only chemically synthesized small molecules interact with VGCC. A highly specific blocking action has been found for various peptide toxins including ω-conotoxin and ω-agatoxins. A list with some toxins which bind to VGCC channels can be found in [140]. For other pharmacologically active substances that modulate calcium homeostasis, please refer to reviews on TRP channels [141,142], purinergic receptors [143], IP3R and RyR [144].
4. General Overview of Multiple Sclerosis (MS)
4.1. Clinical Course of MS
MS is the most common neurological CNS disease in young adults [145]. Due to irreversible deficits, premature retirement, long-term medication and nursing, MS causes a high socioeconomic burden [146]. The disease is characterized by an inflammatory response against oligodendrocytes, which eventually leads to demyelination and axonal damage and loss [147,148]. Around 85% of all patients initially show a relapsing-remitting course of MS (RRMS). Furthermore, 15% of MS patients suffer from the primary progressive disease subtype (PPMS), which is characterized by constant worsening of clinical symptoms [147]. Most patients with RRMS transit into a secondary progressive form of MS (SPMS) after 8 to 20 years [147]. While RRMS is treatable using anti-inflammatory and immune modulatory drugs, treatment options for SPMS are currently limited to the drug siponimod, which has recently been shown to have neuroprotective capacity in a phase 3 clinical trial [149].
4.2. Etiology of MS
The cause of MS is unknown. Yet, there are several theories regarding the factors that could trigger MS. These include (1) genetic predisposition and single nucleotide polymorphisms in genes which are involved in immune function [150], (2) viral infections, in particular with Epstein-Barr virus or bacteria including Helicobacter pylori [151], (3) vitamin D deficiency [152] as well as (4) a dysbalanced microbiome and pathology of the enteric nervous system [153].
4.3. Pathophysiology of MS
The pathophysiology of MS differs in patients with early stage RRMS compared to SPMS [154]. In RRMS, T cells, in particular of the TH1 and TH17 type, are assumed to be involved in early disease development [154]. CD4+ cells are presumably activated in the periphery before they cross the BBB to initiate the immune response in the CNS. After breakdown of the BBB, other immune cells like CD8+ T cells, B cells and macrophages are attracted, causing edema and a diversification of the immune response [155]. In later stages of the disease, meningeal B cell aggregates may locally contribute to the immunopathology [156]. In SPMS, infiltration from the periphery gradually wanes and neurodegenerative processes inside the brain prevail. The absence of clinical, imaging, immunological and clear-cut pathological criteria that define the transition from relapsing-remitting to progressive disease explains why SPMS can only be diagnosed retrospectively in most of the cases [157] and why there is still a lack of treatment strategies for late-stage MS [158].
4.4. Treatment of MS
While MS has remained incurable, RRMS has become treatable [158]. The main goal in the treatment of RRMS is anti-inflammation, immune modulation and the inhibition of immune cell infiltration into the CNS. This is achieved by disease-modifying therapies (DMT). The use of DMT is not uniformly regulated and guidelines for MS treatment can differ between countries. In the following, the drugs which are most commonly used in Europe are listed in alphabetical order: alemtuzumab, a monoclonal anti-CD52 antibody [159]; cladribine, which depletes both B and T cells; dimethyl fumarate, which is proposed to inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling and to alter immune cell activation; fingolimod, a sphingosin-1-phosphate receptor (S1PR) agonist which inhibits immune cell emigration from secondary lymphoid organs [160]; glatiramer acetate [161] and interferon-β [162], two immune modulatory drugs; natalizumab, a potent α4-integrin inhibitor which prevents lymphocyte migration over the BBB [160]; mitoxantrone, a cytostatic agent [163]; ocrelizumab, a monoclonal anti-CD20 antibody which depletes B cells; and teriflunomide, which has anti-proliferative action on immune cells [160].
5. The Role of Calcium in MS
The focus of research and treatment in MS has been on the reduction of immune cell infiltration into the CNS for a long time, leading to the discovery and development of several DMT. Yet, there is urgent need for the development of neuroprotective and neuroreparative strategies to prevent long-term disease progression and disability, and with that the socioeconomic burden.
Several treatment strategies along these lines have already been and are currently being tested in MS and other neurodegenerative diseases. While in many instances promising results were obtained in preclinical studies using cell culture and animal models, so far only the drug siponimod has made its way into clinical application for treatment of SPMS patients with its recent approval in the US and in Europe [147].
In the following we would like to review and discuss why calcium may be a reasonable therapeutic target when it comes to neuroprotection in MS.
5.1. Calcium and Excitotoxicity
As described above, excitotoxicity is known to occur under pathological conditions and has been associated with both experimental autoimmune encephalomyelitis (EAE) and MS [164]. AMPA and KA receptors are involved in the pathological pathway of excitotoxicity, and an inhibition of these receptors can decrease EAE severity [24]. One reason for increased glutamate receptor activity in EAE could be that T cells either interfere directly with the receptors or enhance glutamate transmission through the release of tumor necrosis factor α (TNF-α) [165]. TNF-α also activates glutamate release from microglia which can further trigger excitotoxicity [166]. Another concept of excitotoxicity suggests altered glutamate uptake by glia cells. Astrocytes have shown decreased glutamate uptake in the presence of interleukin 17, a signature cytokine in EAE and MS, and increased calcium-dependent release of glutamate [167]. In another study, disturbed glutamate uptake in oligodendrocytes, which can be induced by chemicals, could be prevented by AMPA antagonists [168]. In post-mortem brains of MS patients an accumulation of activated microglia was observed in areas in which glutamate uptake was disturbed [169]. Elevated levels of glutamate can, in turn, trigger an increase in the intracellular calcium concentration, which again causes excitotoxicity by ROS production and calcium overload in mitochondria [170]. Because the TRPM2 channel is very sensitive to oxidative stress and activation of microglia is mediated by this channel, targeting TRPM2 might reduce excitotoxic processes [109].
5.2. Calcium and BBB Integrity
The BBB consists anatomically of smooth muscle cells, endothelial cells and astrocytes, forming a natural barrier that protects the brain. It also plays a role in regulating calcium homeostasis. The tight and adherent junctions, which are present between the endothelial cells of the BBB as well as the actin cytoskeleton inside the cells, are dependent on the intracellular and extracellular calcium concentration [171,172]. After damage, the intracellular calcium concentration increases due to GPCR and IP3 signaling in endothelial cells, subsequent activation of SOCE and a direct influx of calcium via calcium channels. This leads to the reorganization and/or modification of the actin cytoskeleton and tight junctions with subsequent BBB leakage [18,173]. Interestingly, cannabinoid CB2 receptor agonists have been shown to enhance the formation of tight junctions and to reduce leakiness of the BBB [174]. In addition, activation of the calcium/calmodulin-dependent protein kinase (CaMK) has been reported to promote BBB repair after stroke in a mouse model, which might prevent immune cell infiltration [175].
5.3. Calcium and Immune Cell Activation
Calcium homeostasis in T cells is important for T cell action. The T cell receptor can trigger intracellular calcium release via SOCE. TRP channels act as regulators on T cells, VDCC channels modulate their activity and purinergic receptors amplify the T cell response [176]. The complex interplay of calcium channels on and in T cells is reviewed in [176]. Depletion of STIM1 and STIM2 has been shown to protect mice from EAE [177]. The same was observed after knockout of the CaV3.1 channel in T cells, which was accompanied by altered cytokine secretion and reduced gene transcription [178]. Furthermore, the effect of vitamin D is currently under investigation in MS patients who frequently display decreased vitamin D levels [179]. Vitamin D supplementation seems to reduce neuronal damage after oxidative stress in cell culture [180] and has also been investigated regarding its impact on immune cells [181]. While vitamin D supplementation and subsequent hypercalcemia was counterproductive in EAE mice, a lower concentration seemed to be slightly beneficial [182].
5.4. Drugs that Directly Interfere with Calcium Homeostasis and May Have Neuroprotective Properties in MS
5.4.1. Olesoxime
The drug olesoxime (TRO19622) has the potential to interfere with calcium homeostasis by binding to pore proteins on the outer mitochondrial membrane [183]. The drug has revealed a remyelinating effect in several demyelinating rodent models [184,185] and also in a mouse model of amyotrophic lateral sclerosis (ALS) [186]. However, a clinical trial in ALS patients failed [187]. In a phase 1 study for spinal muscle atrophy (SMA) the drug was well tolerated [188], and a long-term study was completed (NCT02628743), from which results are estimated soon. A MS study was additionally performed (NCT01808885), but so far results have not been published.
5.4.2. Quetiapine
Quetiapine is an antipsychotic agent, which is used for the treatment of schizophrenia and bipolar disorders. Quetiapine has been shown to attenuate EAE severity in mice due to a reduction in the number of T cells in peripheral lymphoid organs [189] and a positive effect on oligodendrocyte development and myelin regeneration [190,191,192]. Quetiapine is suggested to have an impact on calcium homeostasis by interacting with STIM1 [193] and to improve remyelination through this pathway [193,194]. Clinical phase 1 and 2 trials in MS patients are ongoing (NCT02087631).
5.4.3. DHP
The L-type VGCC antagonist nimodipine is an interesting candidate for neuroprotection in MS and effects on CNS and PNS have been reported early on [195]. As described above, calcium influx via VGCC is important for neuronal excitability, gene expression, regulation of transcription factors and post-transcriptional modifications. Some subunits of VGCC can even act as transcription factors themselves and several so-called “channelopathies“ have been connected to mutations in VGCC genes [196].
Nimodipine is approved for the prevention of vasospasms in patients with subarachnoid hemorrhage. It can cross the BBB and enter the CNS [197]. A recent study has demonstrated a direct correlation between the concentration of nimodipine in cerebrospinal fluid, but not in arterial blood, and the disease outcome after subarachnoid hemorrhage [198]. These data may indicate a protective effect of nimodipine.
Further studies on the effects of nimodipine in the CNS have been performed in animal models, demonstrating an improvement of working memory not only in old [199], but also in young rats [200]. In addition, cognitive dysfunction after surgery could be prevented by treating rats with nimodipine before surgery [201]. Nimodipine also increased cognitive function after cerebral ischemia in different animal models [202] and displayed a neuroprotective effect on dopaminergic neurons in mouse and rat cell cultures [197] indicating a potential usefulness for the treatment of PD [203]. Finally, nimodipine has been shown to induce remyelination in mouse models of MS [204,205]. This remyelinative effect was connected to microglia-specific apoptosis and diminished production of NO and ROS [204]. When nimodipine was administered preventively at the time point of disease induction, EAE severity was decreased and the extent of demyelination decreased [205]. In yet another study, a neuroprotective effect of nimodipine treatment was observed when the drug was prophylactically given before vestibular schwannoma surgery [206].
Recently, nimodipine has been reported to have positive effects on Schwann cells, astrocytes and neurons, which was associated with increased phosphorylation of proteinkinase B and the cyclic adenosine monophosphate response element-binding protein (CREB) [207,208,209]. Additionally, the pro-apoptotic protein caspase 3 and the calcium-dependent protein calpain were downregulated upon nimodipine treatment, whereas calbindin expression was upregulated, indicating that modulation of calcium homeostasis and prevention of calcium overload might be responsible for the neuroprotective properties of nimodipine [207,210].
5.5. Potential Neuroprotective Drugs that Indirectly Interfere with Calcium Homeostasis via GPCR
GPCR are the most important family of receptors that activate calcium signaling upon external stimuli, making them ideal drug targets. In the following, several GPCR that could be modulated towards neuroprotection in MS are discussed along with potential drug candidates.
5.5.1. Interference with Cannabinoid CB1 and CB2 Receptors
Cannabidiol, which is an agonist at cannabinoid CB1 and CB2 receptors, has been shown to protect proteins from oxidative stress and to increase mitochondrial activity (reviewed in [212]). Moreover, the synthetic cannabinoids WIN 55,212-2, arachidonyl-2-chloroethylamide (ACEA), and JWH-015 have been reported to decrease inflammatory responses and to increase remyelination in the Theiler’s murine encephalomyelitis virus model [213]. WIN 55,212-2, a cannabinoid CB1 receptor agonist, has additionally been demonstrated to have a protective effect on granule cells of the dentate gyrus and it also interferes directly with CaV2.2 and TRPA1 at high concentrations [214]. The effect of ACEA is potentially mediated by inhibition of the mitochondrial permeability transition pore via cannabinoid CB1 receptor signaling [215]. For JWH-015, a cannabinoid CB2 receptor agonist, a similar mode of action is assumed [216], and an effect on potentially neuroprotective genes has been described [217]. The synthetic compound Yhhu4952 has been demonstrated to interact with OPC possibly via the cannabinoid CB2 receptor, enhancing their maturation [218].
5.5.2. Antagonism of Histamine Receptors
Clemastine, a histamine H1 receptor antagonist, has been investigated for remyelination in mice [219]. Additionally, the ReBUILD study has investigated the drug in optic neuritis [220]. The ReCOVER study is currently ongoing (NCT02521311). In addition, the two histamine H3 antagonists GSK239512 [221] and GSK247246 [222] were investigated for their remyelinating properties in mice.
5.5.3. Stimulation of the κ Opioid Receptor (KOR)
Activation of KOR has been connected to an inhibition of N-type calcium channels [223]. Moreover, the KOR agonist U-50488 directly blocks several low and high VGCC, however concentrations needed for this effect are much higher (100×) than for KOR activation [224].
Thus, KOR agonists are a potentially interesting target for controlling calcium homeostasis. Asimadoline, as well as U-50488, has been shown to induce remyelination in an EAE mouse model via KOR activation [225], however the suggested mechanism involved the MAP kinase pathway rather than a modulation of calcium channels [226].
5.5.4. Antagonism of Muscarinic Receptors
5.5.5. Vitamin D
Supplementation of vitamin D has been shown to have a beneficial impact on EAE outcome in mice [229]. This effect was attributed to an increase of anti-inflammatory and a decrease of pro-inflammatory cytokines [229]. In addition, vitamin D was reported to confer neuroprotection in parallel with downregulation of L-type VGCC in hippocampal neurons [230]. The potential benefits of vitamin D supplementation in MS patients are still under investigation [152].
6. Conclusions
In this review we aimed to illustrate the importance of calcium homeostasis in the CNS. This importance is not only reflected by numerous cellular processes which depend on calcium, but also by the sheer amount of calcium-associated channels and receptors, which are expressed by various CNS cell types. Dysregulation of calcium homeostasis is a pathogenic feature of several neurodegenerative diseases making it an interesting target for novel treatment approaches. MS is the most prevalent neurological disease of the CNS in young adults and there is hope that drugs interfering with calcium homeostasis may be a valuable option for the progressive disease, which is still untreatable, but causes high socioeconomic burden.
First data, in particular regarding desirable effects of DHP on brain pathology, exist. DHP encompass a well-characterized family of drugs, with several members showing BBB permeability. Future research will have to focus on the diseases which may benefit most from DHP treatment, going hand-in-hand with an in-depth characterization of drug-mediated effects and side effects. Nimodipine has shown neuroprotective effects in the EAE model [204] and in patients with vestibular schwannoma [206]. Since the drug has already been on the market for a long time and its safety profile is well established, a clinical trial in progressive MS seems feasible in the near future.
Beside DHP, the two drugs clemastine and quetiapine are currently being tested in SPMS patients. In addition, a trial on benzatropine could be worth considering since a remyelinating effect has also been observed in EAE.
Author Contributions
M.E. and S.K. wrote the manuscript with support from T.H. and A.L. All authors have read and agree to the published version of the manuscript.
Funding
This work funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 270949263/GRK2162.
Acknowledgments
We would like to thank Ursula Lebherz and Thomas Glaser for critical reading of the manuscript. Images in Figure 1 were obtained from https://smart.servier.com.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Aβ | Amyloid β |
| ACEA | Arachidonyl-2-chloroethylamide |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CRAC | Calcium release-activated calcium |
| CREB | Cyclic adenosine monophosphate response element-binding protein |
| DAG | Diacylglycerol |
| DHP | Dihydropyridines |
| DMT | Disease-modifying therapies |
| EAE | Experimental autoimmune encephalomyelitis |
| GABA | Gamma-aminobutyric acid |
| GFAP | Glial fibrillary acidic protein |
| GPCR | G-protein coupled receptors |
| DAG | Diacylglycerol |
| HD | Huntington’s disease |
| IP3 | Inositol-3-phosphate |
| IP3R | Inositol-3-phosphate receptor |
| KA | Kainate |
| KOR | κ Opioid receptor |
| MBP | Myelin basic protein |
| MS | Multiple sclerosis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NAADP | Nicotinic acid adenine dinucleotide phosphate |
| NMDA | N-methyl-d-aspartate |
| NO | Nitric oxide |
| OPC | Oligodendrocyte precursor cell |
| PD | Parkinson’s disease |
| PPMS | Primary progressive multiple sclerosis |
| ROS | Reactive oxygen species |
| RRMS | Relapsing-remitting multiple sclerosis |
| RyR | Ryanodine receptor |
| S1PR | Sphingosin-1-phosphate receptor |
| SMA | Spinal muscle atrophy |
| SOCE | Store-operated calcium entrance |
| STIM1/2 | Stromal interaction molecule 1/2 |
| SPMS | Secondary progressive multiple sclerosis |
| TPC | Two-pore channels |
| TRP | Transient receptor potential |
| TRPA | Transient receptor potential ankyrin channel |
| TRPC | Transient receptor potential canonical channel |
| TRPM | Transient receptor potential melastatin channel |
| TRPML | Transient receptor potential mucolipin channel |
| TRPP | Transient receptor potential polycystic channel |
| TRPV | Transient receptor potential vanilloid channel |
| VDAC | Voltage-dependent anion channels |
| VGCC | Voltage-gated calcium channels |
References
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Marks, A. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. CMP 2015, 8, 206–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartheld, C.S.v.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheli, V.T.; Santiago González, D.A.; Spreuer, V.; Paez, P.M. Voltage-gated Ca2+ entry promotes oligodendrocyte progenitor cell maturation and myelination in vitro. Exp. Neurol. 2015, 265, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Cheli, V.T.; Santiago González, D.A.; Namgyal Lama, T.; Spreuer, V.; Handley, V.; Murphy, G.G.; Paez, P.M. Conditional deletion of the L-Type calcium channel Cav1.2 in oligodendrocyte progenitor cells affects postnatal myelination in mice. J. Neurosci. 2016, 36, 10853–10869. [Google Scholar] [CrossRef] [Green Version]
- Pitman, K.A.; Ricci, R.; Gasperini, R.; Beasley, S.; Pavez, M.; Charlesworth, J.; Foa, L.; Young, K.M. The voltage-gated calcium channel Cav1.2 promotes adult oligodendrocyte progenitor cell survival in the mouse corpus callosum but not motor cortex. Glia 2020, 68, 376–392. [Google Scholar] [CrossRef]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 2018, 21, 19–23. [Google Scholar] [CrossRef]
- Krasnow, A.M.; Ford, M.C.; Valdivia, L.E.; Wilson, S.W.; Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 2018, 21, 24–28. [Google Scholar] [CrossRef]
- Itoh, T.; Beesley, J.; Itoh, A.; Cohen, A.S.; Kavanaugh, B.; Coulter, D.A.; Grinspan, J.B.; Pleasure, D. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J. Neurochem. 2002, 81, 390–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Tvrdik, P.; Kalani, M.Y.S. In vivo imaging of microglial calcium signaling in brain inflammation and injury. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Bock, M.d.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992, 6, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef]
- Berridge, M.J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Kilpatrick, B.S. Two-pore channels and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1678–1686. [Google Scholar] [CrossRef]
- Furuichi, T.; Furutama, D.; Hakamata, Y.; Nakai, J.; Takeshima, H.; Mikoshiba, K. Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. J. Neurosci. 1994, 14, 4794–4805. [Google Scholar] [CrossRef] [Green Version]
- McPherson, P.S.; Campbell, K.P. Characterization of the major brain form of the ryanodine receptor/Ca2+ release channel. J. Biol. Chem. 1993, 268, 19785–19790. [Google Scholar]
- Giannini, G.; Conti, A.; Mammarella, S.; Scrobogna, M.; Sorrentino, V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol. 1995, 128, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Omar, N.; Das, J.; Szeto, V.; Feng, Z.-P. Neuronal ryanodine receptors in development and aging. Mol. Neurobiol. 2018, 55, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Genazzani, A.A.; Carafoli, E.; Guerini, D. Calcineurin controls inositol 1,4,5-trisphosphate type 1 receptor expression in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 5797–5801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Wang, L.; Ma, T.; Wang, S.; Niu, J.; Li, H.; Xiao, L. Dynamic calcium release from endoplasmic reticulum mediated by ryanodine receptor 3 is crucial for oligodendroglial differentiation. Front. Mol. Neurosci. 2018, 11, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, A.; Matute, C.; Alberdi, E. Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010, 1, 54. [Google Scholar] [CrossRef] [Green Version]
- Matyash, M.; Matyash, V.; Nolte, C.; Sorrentino, V.; Kettenmann, H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 2002, 16, 84–86. [Google Scholar] [CrossRef]
- Klegeris, A.; Choi, H.B.; McLarnon, J.G.; McGeer, P.L. Functional ryanodine receptors are expressed by human microglia and THP-1 cells: Their possible involvement in modulation of neurotoxicity. J. Neurosci. Res. 2007, 85, 2207–2215. [Google Scholar] [CrossRef]
- Furuichi, T.; Simon-Chazottes, D.; Fujino, I.; Yamada, N.; Hasegawa, M.; Miyawaki, A.; Yoshikawa, S.; Guénet, J.L.; Mikoshiba, K. Widespread expression of inositol 1,4,5-trisphosphate receptor type 1 gene (Insp3r1) in the mouse central nervous system. Recept. Channels 1993, 1, 11–24. [Google Scholar]
- Johenning, F.W.; Zochowski, M.; Conway, S.J.; Holmes, A.B.; Koulen, P.; Ehrlich, B.E. Distinct intracellular calcium transients in neurites and somata integrate neuronal signals. J. Neurosci. 2002, 22, 5344–5353. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.H.; Nucifora, F.C.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugo, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Hertle, D.N.; Yeckel, M.F. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 2007, 150, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, T.; Cai, X.; Brailoiu, G.C.; Abood, M.E.; Brailoiu, E.; Patel, S. Two-pore channels provide insight into the evolution of voltage-gated Ca2+ and Na+ channels. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Hooper, R.; Cai, X.; Brailoiu, G.C.; Keebler, M.V.; Dun, N.J.; Marchant, J.S.; Patel, S. An ancestral deuterostome family of two-pore channels mediates nicotinic acid adenine dinucleotide phosphate-dependent calcium release from acidic organelles. J. Biol. Chem. 2010, 285, 2897–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Churamani, D.; Pandey, V.; Brailoiu, G.C.; Tuluc, F.; Patel, S.; Dun, N.J. Messenger-specific role for nicotinic acid adenine dinucleotide phosphate in neuronal differentiation. J. Biol. Chem. 2006, 281, 15923–15928. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef] [Green Version]
- Schampel, A.; Kuerten, S. Danger: High voltage – The role of voltage-gated calcium channels in central nervous system pathology. Cells 2017, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wong, A.H.C.; Liu, F. Ligand-gated ion channel interacting proteins and their role in neuroprotection. Front. Cell. Neurosci. 2014, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Connolly, C.N.; Wafford, K.A. The Cys-loop superfamily of ligand-gated ion channels: The impact of receptor structure on function. Biochem. Soc. Trans. 2004, 32, 529–534. [Google Scholar] [CrossRef]
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Puerto, A.; Wandosell, F.; Garrido, J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Front. Cell. Neurosci. 2013, 7, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Ortner, N.; Pinggera, A. Pharmacology of L-type calcium channels: Novel drugs for old targets? CMP 2015, 8, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallin, W.J.; Greenberg, M.E. Calcium regulation of gene expression in neurons: The mode of entry matters. Curr. Opin. Neurobiol. 1995, 5, 367–374. [Google Scholar] [CrossRef]
- Dolphin, A.C. Calcium channel auxiliary α2δ and β subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef]
- Bloodgood, B.L.; Sabatini, B.L. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 2007, 53, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.N.; Trotter, J.; Kettenmann, H. Cultured glial precursor cells from mouse cortex express two types of calcium currents. Neurosci. Lett. 1990, 112, 194–198. [Google Scholar] [CrossRef]
- Kirischuk, S.; Scherer, J.; Möller, T.; Verkhratsky, A.; Kettenmann, H. Subcellular heterogeneity of voltage-gated Ca2+ channels in cells of the oligodendrocyte lineage. Glia 1995, 13, 1–12. [Google Scholar] [CrossRef]
- Blankenfeld, G.v.; Verkhratsky, A.N.; Kettenmann, H. Ca 2+ Channel Expression in the Oligodendrocyte Lineage. Eur. J. Neurosci. 1992, 4, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Santiago González, D.A.; Cheli, V.T.; Zamora, N.N.; Lama, T.N.; Spreuer, V.; Murphy, G.G.; Paez, P.M. Conditional deletion of the L-type calcium channel Cav1.2 in NG2-positive cells impairs remyelination in mice. J. Neurosci. 2017, 37, 10038–10051. [Google Scholar] [CrossRef] [PubMed]
- Paez, P.M.; Spreuer, V.; Handley, V.; Feng, J.-M.; Campagnoni, C.; Campagnoni, A.T. Increased expression of golli myelin basic proteins enhances calcium influx into oligodendroglial cells. J. Neurosci. 2007, 27, 12690–12699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.S.T.; Paez, P.M.; Spreuer, V.; Campagnoni, C.W.; Boggs, J.M.; Campagnoni, A.T.; Harauz, G. Classical 18.5-and 21.5-kDa isoforms of myelin basic protein inhibit calcium influx into oligodendroglial cells, in contrast to golli isoforms. J. Neurosci. Res. 2011, 89, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, S.K.; Nashmi, R.; Fehlings, M.G. Role of L- and N-type calcium channels in the pathophysiology of traumatic spinal cord white matter injury. Neuroscience 2000, 99, 179–188. [Google Scholar] [CrossRef]
- Shigetomi, E.; Patel, S.; Khakh, B.S. Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 2016, 26, 300–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaguchi, T.; Nishizaki, T. Extracellular high K+ stimulates vesicular glutamate release from astrocytes by activating voltage-dependent calcium channels. J. Cell. Physiol. 2010, 225, 512–518. [Google Scholar] [CrossRef]
- Cheli, V.T.; Santiago González, D.A.; Smith, J.; Spreuer, V.; Murphy, G.G.; Paez, P.M. L-type voltage-operated calcium channels contribute to astrocyte activation In vitro. Glia 2016, 64, 1396–1415. [Google Scholar] [CrossRef] [Green Version]
- Latour, I.; Hamid, J.; Beedle, A.M.; Zamponi, G.W.; Macvicar, B.A. Expression of voltage-gated Ca2+ channel subtypes in cultured astrocytes. Glia 2003, 41, 347–353. [Google Scholar] [CrossRef]
- D’Ascenzo, M.; Vairano, M.; Andreassi, C.; Navarra, P.; Azzena, G.B.; Grassi, C. Electrophysiological and molecular evidence of L-(Cav1), N- (Cav2.2), and R- (Cav2.3) type Ca2+ channels in rat cortical astrocytes. Glia 2004, 45, 354–363. [Google Scholar] [CrossRef]
- Colton, C.A.; Jia, M.; Li, M.X.; Gilbert, D.L. K+ modulation of microglial superoxide production: Involvement of voltage-gated Ca2+ channels. Am. J. Physiol. 1994, 266, C1650–C1655. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Finkbeiner, S. NMDA and AMPA receptors: Old channels, new tricks. Trends Neurosci. 2007, 30, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, Z.A.; Clarke, V.R.; Delany, C.M.; Parry, M.C.; Smolders, I.; Vignes, M.; Ho, K.H.; Miu, P.; Brinton, B.T.; Fantaske, R.; et al. Kainate receptors are involved in synaptic plasticity. Nature 1999, 402, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Diering, G.H.; Huganir, R.L. The AMPA receptor code of synaptic plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, G.J.; Popescu, G.K. NMDA receptors: Linking physiological output to biophysical operation. Nat. Rev. Neurosci. 2017, 18, 236–249. [Google Scholar] [CrossRef]
- Evans, A.J.; Gurung, S.; Henley, J.M.; Nakamura, Y.; Wilkinson, K.A. Exciting times: New advances towards understanding the regulation and roles of kainate receptors. Neurochem. Res. 2019, 44, 572–584. [Google Scholar] [CrossRef] [Green Version]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- Patneau, D.K.; Wright, P.W.; Winters, C.; Mayer, M.L.; Gallo, V. Glial cells of the oligodendrocyte lineage express both kainate- and AMPA-preferring subtypes of glutamate receptor. Neuron 1994, 12, 357–371. [Google Scholar] [CrossRef]
- Mcdonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat. Med. 1998, 4, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Gómez, M.V.; Matute, C. AMPA and kainate receptors each mediate excitotoxicity in oligodendroglial cultures. Neurobiol. Dis. 1999, 6, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, D.; Grooms, S.Y.; Araneda, R.C.; Johnson, A.B.; Dobrenis, K.; Kessler, J.A.; Zukin, R.S. AMPA receptor protein expression and function in astrocytes cultured from hippocampus. J. Neurosci. Res. 1999, 57, 557–571. [Google Scholar] [CrossRef]
- Matthias, K.; Kirchhoff, F.; Seifert, G.; Hüttmann, K.; Matyash, M.; Kettenmann, H.; Steinhäuser, C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J. Neurosci. 2003, 23, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA–kainate subtypes of glutamate receptor in rat cerebral microglia. J. Neurosci. 2000, 20, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef]
- Dietrich, A. Transient receptor potential (TRP) channels in health and disease. Cells 2019, 8, 413. [Google Scholar] [CrossRef] [Green Version]
- Kheradpezhouh, E.; Choy, J.M.C.; Daria, V.R.; Arabzadeh, E. TRPA1 expression and its functional activation in rodent cortex. Open Biol. 2017, 7, 160314. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, N.B.; Kolodziejczyk, K.; Kougioumtzidou, E.; Attwell, D. Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 2016, 529, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Shigetomi, E.; Tong, X.; Kwan, K.Y.; Corey, D.P.; Khakh, B.S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat. Neurosci. 2011, 15, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Minard, A.; Bauer, C.C.; Wright, D.J.; Rubaiy, H.N.; Muraki, K.; Beech, D.J.; Bon, R.S. Remarkable progress with small-molecule modulation of TRPC1/4/5 channels: Implications for understanding the channels in health and disease. Cells 2018, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 2006, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harteneck, C.; Leuner, K. TRP channels in neuronal and glial signal transduction. In Neurochemistry; Heinbockel, T., Ed.; InTech: Rijeka, Croatia, 2014. [Google Scholar]
- Paez, P.M.; Fulton, D.; Spreuer, V.; Handley, V.; Campagnoni, A.T. Modulation of canonical transient receptor potential channel 1 in the proliferation of oligodendrocyte precursor cells by the golli products of the myelin basic protein gene. J. Neurosci. 2011, 31, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, F.R.; Martorana, A.; Giampà, C.; March, Z.d.; Vacca, F.; Tozzi, A.; Longone, P.; Piccirilli, S.; Paolucci, S.; Sancesario, G.; et al. Cellular localization of TRPC3 channel in rat brain: Preferential distribution to oligodendrocytes. Neurosci. Lett. 2004, 365, 137–142. [Google Scholar] [CrossRef]
- Li, B.; Dong, L.; Fu, H.; Wang, B.; Hertz, L.; Peng, L. Effects of chronic treatment with fluoxetine on receptor-stimulated increase of Ca2+i in astrocytes mimic those of acute inhibition of TRPC1 channel activity. Cell Calcium 2011, 50, 42–53. [Google Scholar] [CrossRef]
- Shirakawa, H.; Sakimoto, S.; Nakao, K.; Sugishita, A.; Konno, M.; Iida, S.; Kusano, A.; Hashimoto, E.; Nakagawa, T.; Kaneko, S. Transient receptor potential canonical 3 (TRPC3) mediates thrombin-induced astrocyte activation and upregulates its own expression in cortical astrocytes. J. Neurosci. 2010, 30, 13116–13129. [Google Scholar] [CrossRef]
- Shirakawa, H.; Katsumoto, R.; Iida, S.; Miyake, T.; Higuchi, T.; Nagashima, T.; Nagayasu, K.; Nakagawa, T.; Kaneko, S. Sphingosine-1-phosphate induces Ca2+ signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 2017, 65, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, Y.; Kato, T.A.; Seki, Y.; Ohgidani, M.; Sagata, N.; Horikawa, H.; Yamauchi, Y.; Sato-Kasai, M.; Hayakawa, K.; Inoue, R.; et al. Brain-derived neurotrophic factor (BDNF) induces sustained intracellular Ca2+ elevation through the up-regulation of surface transient receptor potential 3 (TRPC3) channels in rodent microglia. J. Biol. Chem. 2014, 289, 18549–18555. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhuang, Y.; Zhou, Z.; Zhao, J.; Chen, Q.; Zheng, J. NF-κB dependent up-regulation of TRPC6 by Aβ in BV-2 microglia cells increases COX-2 expression and contributes to hippocampus neuron damage. Neurosci. Lett. 2017, 651, 1–8. [Google Scholar] [CrossRef]
- Wang, J.; Jackson, M.F.; Xie, Y.F. Glia and TRPM2 channels in plasticity of central nervous system and Alzheimer’s diseases. Neural Plast. 2016, 2016, 1680905. [Google Scholar] [CrossRef] [Green Version]
- Zamudio-Bulcock, P.A.; Everett, J.; Harteneck, C.; Valenzuela, C.F. Activation of steroid-sensitive TRPM3 channels potentiates glutamatergic transmission at cerebellar Purkinje neurons from developing rats. J. Neurochem. 2011, 119, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kang, E.; Makino, Y.; Park, S.; Shin, J.H.; Song, H.; Launay, P.; Linden, D.J. Characterizing the conductance underlying depolarization-induced slow current in cerebellar Purkinje cells. J. Neurophysiol. 2013, 109, 1174–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, C.; Liu, Y.; Li, W.; Wang, Z.; Sun, Q.; Zhou, H.; Chen, X.; Yu, Y.; Wang, Y.; et al. TRPM7 is required for normal synapse density, learning, and memory at different developmental stages. Cell Rep. 2018, 23, 3480–3491. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Grimm, C.; Kraft, R.; Goldbaum, O.; Wrede, A.; Nolte, C.; Hanisch, U.K.; Richter-Landsberg, C.; Brück, W.; Kettenmann, H.; et al. TRPM3 is expressed in sphingosine-responsive myelinating oligodendrocytes. J. Neurochem. 2010, 114, 654–665. [Google Scholar] [CrossRef]
- Choi, H.J.; Sun, D.; Jakobs, T.C. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol. Vis. 2015, 21, 749–766. [Google Scholar]
- Kamermans, A.; Planting, K.E.; Jalink, K.; van Horssen, J.; Vries, H.E.d. Reactive astrocytes in multiple sclerosis impair neuronal outgrowth through TRPM7-mediated chondroitin sulfate proteoglycan production. Glia 2019, 67, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Raj, D.; Saiepour, N.; van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef]
- Malko, P.; Syed Mortadza, S.A.; McWilliam, J.; Jiang, L.H. TRPM2 channel in microglia as a new player in neuroinflammation associated with a spectrum of central nervous system pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, T.; Lively, S.; Ferreira, R.; Wong, R.; Schlichter, L.C. Expression and contributions of TRPM7 and KCa2.3/SK3 channels to the increased migration and invasion of microglia in anti-inflammatory activation states. PLoS ONE 2014, 9, e106087. [Google Scholar] [CrossRef]
- Beck, A.; Penner, R.; Fleig, A. Lipopolysaccharide-induced down-regulation of Ca2+ release-activated Ca2+ currents (I CRAC) but not Ca2+-activated TRPM4-like currents (I CAN) in cultured mouse microglial cells. J. Physiol. 2008, 586, 427–439. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, O.; Singh, U.; Goswami, C.; Singru, P.S. Transient receptor potential vanilloid 1-6 (Trpv1-6) gene expression in the mouse brain during estrous cycle. Brain Res. 2018, 1701, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A.; Gibson, H.E. Hot flash: TRPV channels in the brain. Trends Neurosci. 2009, 32, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Deyashiki, A.; Miyake, T.; Nagayasu, K.; Shibasaki, K.; Shirakawa, H.; Kaneko, S. TRPV4 is functionally expressed in oligodendrocyte precursor cells and increases their proliferation. Pflugers Arch. 2018, 470, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Reyes, L.E.; Ladas, T.P.; Chiang, C.C.; Durand, D.M. TRPV1 antagonist capsazepine suppresses 4-AP-induced epileptiform activity in vitro and electrographic seizures in vivo. Exp. Neurol. 2013, 250, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.W.; Lambert, W.S.; Calkins, D.J. Activation of the TRPV1 cation channel contributes to stress-induced astrocyte migration. Glia 2014, 62, 1435–1451. [Google Scholar] [CrossRef] [Green Version]
- Shibasaki, K.; Ishizaki, Y.; Mandadi, S. Astrocytes express functional TRPV2 ion channels. Biochem. Biophys. Res. Commun. 2013, 441, 327–332. [Google Scholar] [CrossRef]
- Benfenati, V.; Amiry-Moghaddam, M.; Caprini, M.; Mylonakou, M.N.; Rapisarda, C.; Ottersen, O.P.; Ferroni, S. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience 2007, 148, 876–892. [Google Scholar] [CrossRef]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, X.; Wang, L.; Wang, Y.; Dong, F.; Wu, J.; Qu, X.; Liu, Y.; Liu, Z.; Fan, H.; et al. TRPV4 inhibition improved myelination and reduced glia reactivity and inflammation in a cuprizone-induced mouse model of demyelination. Front. Cell. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papanikolaou, M.; Lewis, A.; Butt, A.M. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct. Funct. 2017, 222, 2993–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuma, A.; Kim, J.Y.; Kacimi, R.; Stauderman, K.; Dunn, M.; Hebbar, S.; Yenari, M.A. Microglial calcium release-activated calcium channel inhibition improves outcome from experimental traumatic brain injury and microglia-induced neuronal death. J. Neurotrauma 2019, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels (Austin) 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Pankratov, Y.; Lalo, U.; Nedergaard, M. P2X receptors in neuroglia. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1. [Google Scholar] [CrossRef] [Green Version]
- Welsh, T.G.; Kucenas, S. Purinergic signaling in oligodendrocyte development and function. J. Neurochem. 2018, 145, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Calovi, S.; Mut-Arbona, P.; Sperlágh, B. Microglia and the purinergic signaling system. Neuroscience 2019, 405, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.; Tritthart, H.; Döring, H.J.; Byon, K.Y. BAY a 1040 – ein hochaktiver Ca-antagonistischer Inhibitor der elektro-mechanischen Koppelungsprozesse im Warmblüter-Myokard. Arzneimittelforschung 1972, 22, 22–33. [Google Scholar] [PubMed]
- Wang, A.L.; Iadecola, C.; Wang, G. New generations of dihydropyridines for treatment of hypertension. J. Geriatr. Cardiol. 2017, 14, 67–72. [Google Scholar] [PubMed]
- Epstein, B.J.; Vogel, K.; Palmer, B.F. Dihydropyridine calcium channel antagonists in the management of hypertension. Drugs 2007, 67, 1309–1327. [Google Scholar] [CrossRef]
- Siddiqi, F.H.; Menzies, F.M.; Lopez, A.; Stamatakou, E.; Karabiyik, C.; Ureshino, R.; Ricketts, T.; Jimenez-Sanchez, M.; Esteban, M.A.; Lai, L.; et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 2019, 10, 1817. [Google Scholar] [CrossRef] [Green Version]
- Hockerman, G.H.; Peterson, B.Z.; Johnson, B.D.; Catterall, W.A. Molecular determinants of drug binding and action on L-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 361–396. [Google Scholar] [CrossRef] [Green Version]
- Kopecky, B.J.; Liang, R.; Bao, J. T-type calcium channel blockers as neuroprotective agents. Pflugers Arch. 2014, 466, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Gee, N.S.; Brown, J.P.; Dissanayake, V.U.; Offord, J.; Thurlow, R.; Woodruff, G.N. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 1996, 271, 5768–5776. [Google Scholar] [CrossRef] [Green Version]
- Nimmrich, V.; Gross, G. P/Q-type calcium channel modulators. Br. J. Pharmacol. 2012, 167, 741–759. [Google Scholar] [CrossRef] [Green Version]
- Harteneck, C.; Gollasch, M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr. Pharm. Biotechnol. 2011, 12, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.M. TRP channels as potential drug targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. The purinergic system as a pharmacological target for the treatment of immune-mediated inflammatory diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Koulen, P.; Thrower, E.C. Pharmacological Modulation of intracellular Ca2+ channels at the single-channel level. Mol. Neurobiol. 2001, 24, 65–86. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Hardy, T.A.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2017, 389, 1336–1346. [Google Scholar] [CrossRef]
- Kobelt, G.; Thompson, A.; Berg, J.; Gannedahl, M.; Eriksson, J. New insights into the burden and costs of multiple sclerosis in Europe. Mult. Scler. 2017, 23, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.-A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Multiple sclerosis review. P T 2012, 37, 175–184. [Google Scholar]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; Jager, P.L.d.; Bakker, P.I.W.d.; Gabriel, S.B.; Mirel, D.B.; Ivinson, A.J.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar]
- Marrodan, M.; Alessandro, L.; Farez, M.F.; Correale, J. The role of infections in multiple sclerosis. Mult. Scler. 2019, 25, 891–901. [Google Scholar] [CrossRef]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and multiple sclerosis: A comprehensive review. Neurol. Ther. 2018, 7, 59–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pröbstel, A.K.; Baranzini, S.E. The role of the gut microbiome in multiple sclerosis risk and progression: Towards characterization of the “MS microbiome”. Neurotherapeutics 2018, 15, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Rottlaender, A.; Kuerten, S. Stepchild or Prodigy? Neuroprotection in multiple sclerosis (MS) research. Int. J. Mol. Sci. 2015, 16, 14850–14865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [Green Version]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Angelis, F.d.; John, N.A.; Brownlee, W.J. Disease-modifying therapies for multiple sclerosis. BMJ 2018, 363, k4674. [Google Scholar] [CrossRef]
- Munari, L.; Lovati, R.; Boiko, A. Therapy with glatiramer acetate for multiple sclerosis. Cochrane Database Syst. Rev. 2004, CD004678. [Google Scholar]
- Filippini, G.; Munari, L.; Incorvaia, B.; Ebers, G.C.; Polman, C.; D’Amico, R.; Rice, G.P.A. Interferons in relapsing remitting multiple sclerosis: A systematic review. Lancet 2003, 361, 545–552. [Google Scholar] [CrossRef]
- Gajofatto, A.; Benedetti, M.D. Treatment strategies for multiple sclerosis: When to start, when to change, when to stop? WJCC 2015, 3, 545. [Google Scholar] [CrossRef] [PubMed]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majláth, Z.; Vécsei, L. Excitotoxins, mitochondrial and redox disturbances in multiple sclerosis. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, A.; Vito, F.d.; Fresegna, D.; Rizzo, F.R.; Bullitta, S.; Guadalupi, L.; Vanni, V.; Buttari, F.; Stampanoni Bassi, M.; Leuti, A.; et al. Peripheral T cells from multiple sclerosis patients trigger synaptotoxic alterations in central neurons. Neuropathol. Appl. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Thomas, A.G.; O’Driscoll, C.M.; Bressler, J.; Kaufmann, W.; Rojas, C.J.; Slusher, B.S. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Colic, M. IL-17 signalling in astrocytes promotes glutamate excitotoxicity: Indications for the link between inflammatory and neurodegenerative events in multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 11, 12–17. [Google Scholar] [CrossRef]
- Domercq, M.; Etxebarria, E.; Pérez-Samartín, A.; Matute, C. Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition. Glia 2005, 52, 36–46. [Google Scholar] [CrossRef]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: Correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.C.; Tewari, B.P.; Singh, M.; Joshi, P.G.; Joshi, N.B. AMPA receptor activation causes preferential mitochondrial Ca2+ load and oxidative stress in motor neurons. Brain Res. 2015, 1616, 1–9. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, 343–356. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Raya-Sandino, A.; González-González, L.; Hernández-Guzmán, C. Relationship between G proteins coupled receptors and tight junctions. Tissue Barriers 2018, 6, e1414015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, S.H.; Haskó, J.; Skuba, A.; Fan, S.; Dykstra, H.; McCormick, R.; Reichenbach, N.; Krizbai, I.; Mahadevan, A.; Zhang, M.; et al. Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood-brain barrier dysfunction under inflammatory conditions. J. Neurosci. 2012, 32, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Bu, F.; Min, J.W.; Munshi, Y.; Howe, M.D.; Liu, L.; Koellhoffer, E.C.; Qi, L.; McCullough, L.D.; Li, J. Inhibition of calcium/calmodulin-dependent protein kinase kinase (CaMKK) exacerbates impairment of endothelial cell and blood-brain barrier after stroke. Eur. J. Neurosci. 2019, 49, 27–39. [Google Scholar] [CrossRef]
- Trebak, M.; Kinet, J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Ma, J.; McCarl, C.A.; Khalil, S.; Lüthy, K.; Feske, S. T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur. J. Immunol. 2010, 40, 3028–3042. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Xue, L.; Xing, J.; Jouvin, M.H.; Putney, J.W.; Anderson, M.P.; Trebak, M.; Kinet, J.P. Low-voltage-activated Cav3.1 calcium channels shape T helper cell cytokine profiles. Immunity 2016, 44, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef] [Green Version]
- AlJohri, R.; AlOkail, M.; Haq, S.H. Neuroprotective role of vitamin D in primary neuronal cortical culture. eNeurologicalSci 2019, 14, 43–48. [Google Scholar] [CrossRef]
- Medrano, M.; Carrillo-Cruz, E.; Montero, I.; Perez-Simon, J.A. Vitamin D: Effect on haematopoiesis and immune system and clinical applications. Int. J. Mol. Sci. 2018, 19, 2663. [Google Scholar] [CrossRef] [Green Version]
- Häusler, D.; Torke, S.; Peelen, E.; Bertsch, T.; Djukic, M.; Nau, R.; Larochelle, C.; Zamvil, S.S.; Brück, W.; Weber, M.S. High dose vitamin D exacerbates central nervous system autoimmunity by raising T-cell excitatory calcium. Brain 2019, 142, 2737–2755. [Google Scholar] [CrossRef] [PubMed]
- Bordet, T.; Berna, P.; Abitbol, J.L.; Pruss, R.M. Olesoxime (TRO19622): A novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalon, K.; Zimmer, C.; Cayre, M.; Khaldi, J.; Bourbon, C.; Robles, I.; Tardif, G.; Viola, A.; Pruss, R.M.; Bordet, T.; et al. Olesoxime accelerates myelination and promotes repair in models of demyelination. Ann. Neurol. 2012, 71, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Han, W.; Hu, F.; Qian, Y.; Chen, Q. TRO19622 promotes myelin repair in a rat model of demyelination. Int. J. Neurosci. 2013, 123, 810–822. [Google Scholar] [CrossRef]
- Sunyach, C.; Michaud, M.; Arnoux, T.; Bernard-Marissal, N.; Aebischer, J.; Latyszenok, V.; Gouarné, C.; Raoul, C.; Pruss, R.M.; Bordet, T.; et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 2012, 62, 2346–2352. [Google Scholar] [CrossRef]
- Lenglet, T.; Lacomblez, L.; Abitbol, J.L.; Ludolph, A.; Mora, J.S.; Robberecht, W.; Shaw, P.J.; Pruss, R.M.; Cuvier, V.; Meininger, V. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 529–536. [Google Scholar] [CrossRef]
- Bertini, E.; Dessaud, E.; Mercuri, E.; Muntoni, F.; Kirschner, J.; Reid, C.; Lusakowska, A.; Comi, G.P.; Cuisset, J.M.; Abitbol, J.L.; et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 513–522. [Google Scholar] [CrossRef]
- Mei, F.; Guo, S.; He, Y.; Wang, L.; Wang, H.; Niu, J.; Kong, J.; Li, X.; Wu, Y.; Xiao, L. Quetiapine, an atypical antipsychotic, is protective against autoimmune-mediated demyelination by inhibiting effector T cell proliferation. PLoS ONE 2012, 7, e42746. [Google Scholar] [CrossRef] [Green Version]
- Chandran, P.; Upadhyay, J.; Markosyan, S.; Lisowski, A.; Buck, W.; Chin, C.L.; Fox, G.; Luo, F.; Day, M. Magnetic resonance imaging and histological evidence for the blockade of cuprizone-induced demyelination in C57BL/6 mice. Neuroscience 2012, 202, 446–453. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H.; Jiang, W.; Xiao, L.; Yan, B.; He, J.; Wang, Y.; Bi, X.; Li, X.; Kong, J.; et al. Quetiapine alleviates the cuprizone-induced white matter pathology in the brain of C57BL/6 mouse. Schizophr. Res. 2008, 106, 182–191. [Google Scholar] [CrossRef]
- Zeng, Z.; Leng, T.; Feng, X.; Sun, H.; Inoue, K.; Zhu, L.; Xiong, Z.G. Silencing TRPM7 in mouse cortical astrocytes impairs cell proliferation and migration via ERK and JNK signaling pathways. PLoS ONE 2015, 10, e0119912. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Tian, Y.; Wu, X.; He, Y.; Li, C.; Namaka, M.; Kong, J.; Li, H.; Xiao, L. Quetiapine inhibits microglial activation by neutralizing abnormal STIM1-mediated intercellular calcium homeostasis and promotes myelin repair in a cuprizone-induced mouse model of demyelination. Front. Cell. Neurosci. 2015, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hu, Y.; Shazeeb, M.S.; Pedraza, C.E.; Pande, N.; Weinstock, D.; Polites, G.H.; Zhang, W.; Chandross, K.J.; Ying, X. In vivo optical imaging of myelination events in a myelin basic protein promoter-driven luciferase transgenic mouse model. ASN Neuro 2018, 10, 1759091418777329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bär, P.R.; Traber, J.; Schuurman, T.; Gispen, W.H. CNS and PNS effects of nimodipine. J. Neural Transm. Suppl. 1990, 31, 55–71. [Google Scholar] [PubMed]
- Barbado, M.; Fablet, K.; Ronjat, M.; Waard, M.d. Gene regulation by voltage-dependent calcium channels. Biochim. Biophys. Acta 2009, 1793, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
- Riva, R.; Pegoli, M.; Contin, M.; Perrone, A.; Mohamed, S.; Zanello, M. Cerebrospinal fluid concentrations of nimodipine correlate with long-term outcome in aneurysmal subarachnoid hemorrhage: Pilot study. Clin. Neuropharmacol. 2019, 42, 157–162. [Google Scholar] [CrossRef]
- Jong, G.I.d.; Weerd, H.d.; Schuurman, T.; Traber, J.; Luiten, P.G. Microvascular changes in aged rat forebrain. Effects of chronic nimodipine treatment. Neurobiol. Aging 1990, 11, 381–389. [Google Scholar]
- Levy, A.; Kong, R.M.; Stillman, M.J.; Shukitt-Hale, B.; Kadar, T.; Rauch, T.M.; Lieberman, H.R. Nimodipine improves spatial working memory and elevates hippocampal acetylcholine in young rats. Pharmacol. Biochem. Behav. 1991, 39, 781–786. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Bao, Y.; Yin, C.; Xin, X.; Guo, Y.; Gao, F.; Huo, S.; Wang, X.; Wang, Q. Pretreatment with nimodipine reduces incidence of POCD by decreasing calcineurin mediated hippocampal neuroapoptosis in aged rats. BMC Anesthesiol. 2018, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Taya, K.; Watanabe, Y.; Kobayashi, H.; Fujiwara, M. Nimodipine improves the disruption of spatial cognition induced by cerebral ischemia. Physiol. Behav. 2000, 70, 19–25. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Schampel, A.; Volovitch, O.; Koeniger, T.; Scholz, C.J.; Jörg, S.; Linker, R.A.; Wischmeyer, E.; Wunsch, M.; Hell, J.W.; Ergün, S.; et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 3295–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingwersen, J.; Santi, L.d.; Wingerath, B.; Graf, J.; Koop, B.; Schneider, R.; Hecker, C.; Schröter, F.; Bayer, M.; Engelke, A.D.; et al. Nimodipine confers clinical improvement in two models of experimental autoimmune encephalomyelitis. J. Neurochem. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, C.; Wienke, A.; Wurm, F.; Simmermacher, S.; Rampp, S.; Prell, J.; Rachinger, J.; Scheller, K.; Koman, G.; Strauss, C.; et al. Neuroprotective efficacy of prophylactic enteral and parenteral nimodipine treatment in vestibular schwannoma surgery: A comparative study. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2014, 75, 251–258. [Google Scholar] [PubMed]
- Leisz, S.; Simmermacher, S.; Prell, J.; Strauss, C.; Scheller, C. Nimodipine-dependent protection of Schwann cells, astrocytes and neuronal cells from osmotic, oxidative and heat stress is associated with the activation of AKT and CREB. Int. J. Mol. Sci. 2019, 20, 4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Liu, Z.; Lv, P.; Wang, H.; Zhu, Y.; Qi, Q.; Xu, J.; Gao, L. Nimodipine activates neuroprotective signaling events and inactivates autophages in the VCID rat hippocampus. Neurol. Res. 2017, 39, 904–909. [Google Scholar] [CrossRef]
- Herzfeld, E.; Strauss, C.; Simmermacher, S.; Bork, K.; Horstkorte, R.; Dehghani, F.; Scheller, C. Investigation of the neuroprotective impact of nimodipine on Neuro2a cells by means of a surgery-like stress model. Int. J. Mol. Sci. 2014, 15, 18453–18465. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Verma, P.; Raju, A.; Mohanakumar, K.P. Nimodipine attenuates the parkinsonian neurotoxin, MPTP-induced changes in the calcium binding proteins, calpain and calbindin. J. Chem. Neuroanat. 2019, 95, 89–94. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Ingram, G.; Pearson, O.R. Cannabis and multiple sclerosis. Pract. Neurol. 2019, 19, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Martín, Á.; Vela, J.M.; Molina-Holgado, E.; Borrell, J.; Guaza, C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci. 2003, 23, 2511–2516. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Kreutz, S.; Böttger, C.; Grabiec, U.; Ghadban, C.; Korf, H.W.; Dehghani, F. The cannabinoid WIN 55,212-2-mediated protection of dentate gyrus granule cells is driven by CB1 receptors and modulated by TRPA1 and Cav 2.2 channels. Hippocampus 2011, 21, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Niu, W.; Yang, S.; Tian, J.; Luan, H.; Cao, M.; Xi, W.; Tu, W.; Jia, J.; Lv, J. Inhibition of mitochondrial permeability transition pore opening contributes to cannabinoid type 1 receptor agonist ACEA-induced neuroprotection. Neuropharmacology 2018, 135, 211–222. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Jimenez-Del-Rio, M.; Lores-Arnaiz, S.; Bustamante, J. Protective effects of the synthetic cannabinoids CP55,940 and JWH-015 on rat brain mitochondria upon paraquat exposure. Neurochem. Res. 2010, 35, 1323–1332. [Google Scholar] [CrossRef]
- Yang, M.C.; Zhang, H.Z.; Wang, Z.; You, F.L.; Wang, Y.F. The molecular mechanism and effect of cannabinoid-2 receptor agonist on the blood-spinal cord barrier permeability induced by ischemia-reperfusion injury. Brain Res. 2016, 1636, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Cheng, G.; Zhang, L.; Zhang, Y.; Wang, Q.; Zhao, M.; Zeng, L.; Hu, Y.; Feng, L. N-Phenylquinazolin-2-amine Yhhu4952 as a novel promotor for oligodendrocyte differentiation and myelination. Sci. Rep. 2018, 8, 14040. [Google Scholar] [CrossRef]
- Li, Z.; He, Y.; Fan, S.; Sun, B. Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination. Neurosci. Bull. 2015, 31, 617–625. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbach, C.J.; Grove, R.A.; Brown, R.; Tompson, D.; Then Bergh, F.; Arnold, D.L. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: A randomised, single-blind, phase II study. J. Neurol. 2017, 264, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Rangon, C.M.; Schang, A.L.; van Steenwinckel, J.; Schwendimann, L.; Lebon, S.; Fu, T.; Chen, L.; Beneton, V.; Journiac, N.; Young-Ten, P.; et al. Myelination induction by a histamine H3 receptor antagonist in a mouse model of preterm white matter injury. Brain Behav. Immun. 2018, 74, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallent, M.; Dichter, M.A.; Bell, G.I.; Reisine, T. The cloned kappa opioid receptor couples to an N-type calcium current in undifferentiated PC-12 cells. Neuroscience 1994, 63, 1033–1040. [Google Scholar] [CrossRef]
- Berecki, G.; Motin, L.; Adams, D.J. Mechanism of direct Cav2.2 channel block by the κ-opioid receptor agonist U50488H. Neuropharmacology 2016, 109, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Duan, Y.; Wei, W.; Cai, Y.; Chai, H.; Lv, J.; Du, X.; Zhu, J.; Xie, X. Kappa opioid receptor activation alleviates experimental autoimmune encephalomyelitis and promotes oligodendrocyte-mediated remyelination. Nat. Commun. 2016, 7, 11120. [Google Scholar] [CrossRef] [Green Version]
- Borniger, J.C.; Hesp, Z.C. Enhancing Remyelination through a Novel Opioid-Receptor Pathway. J. Neurosci. 2016, 36, 11831–11833. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.K.; Nissen, J.C.; Pretory, A.; Tsirka, S.E. Tuftsin combines with remyelinating therapy and improves outcomes in models of CNS demyelinating disease. Front. Immunol. 2018, 9, 2784. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Haghmorad, D.; Yazdanpanah, E.; Jadid Tavaf, M.; Zargarani, S.; Soltanmohammadi, A.; Mahmoudi, M.B.; Mahmoudi, M. Prevention and treatment of experimental autoimmune encephalomyelitis induced mice with 1, 25-dihydroxyvitamin D3. Neurol. Res. 2019, 41, 943–957. [Google Scholar] [CrossRef]
- Brewer, L.D.; Thibault, V.; Chen, K.C.; Langub, M.C.; Landfield, P.W.; Porter, N.M. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J. Neurosci. 2001, 21, 98–108. [Google Scholar] [CrossRef]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Dupree, J.L.; Gacias, M.; Frawley, R.; Sikder, T.; Naik, P.; Casaccia, P. Clemastine enhances myelination in the prefrontal cortex and rescues behavioral changes in socially isolated mice. J. Neurosci. 2016, 36, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yang, Y.J.; Yang, N.; Chen, X.J.; Huang, N.X.; Zhang, J.; Wu, Y.; Liu, Z.; Gao, X.; Li, T.; et al. Enhancing oligodendrocyte myelination rescues synaptic loss and improves functional recovery after chronic hypoxia. Neuron 2018, 99, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhen, W.; Guo, T.; Zhao, Y.; Liu, A.; Rubio, J.P.; Krull, D.; Richardson, J.C.; Lu, H.; Wang, R. Histamine receptor 3 negatively regulates oligodendrocyte differentiation and remyelination. PLoS ONE 2017, 12, e0189380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, P.E.; Maderspach, K.; Hauser, K.F. Endogenous opioid system in developing normal and jimpy oligodendrocytes: Mu and kappa opioid receptors mediate differential mitogenic and growth responses. Glia 1998, 22, 189–201. [Google Scholar] [CrossRef]
- Mei, F.; Mayoral, S.R.; Nobuta, H.; Wang, F.; Desponts, C.; Lorrain, D.S.; Xiao, L.; Green, A.J.; Rowitch, D.; Whistler, J.; et al. Identification of the kappa-opioid receptor as a therapeutic target for oligodendrocyte remyelination. J. Neurosci. 2016, 36, 7925–7935. [Google Scholar] [CrossRef]
- Mahfoz, A.M.; Abdel-Wahab, A.F.; Afify, M.A.; Shahzad, N.; Ibrahim, I.A.A.; ElSawy, N.A.; Bamagous, G.A.; Al Ghamdi, S.S. Neuroprotective effects of vitamin D alone or in combination with lamotrigine against lithium-pilocarpine model of status epilepticus in rats. Naunyn Schmiedebergs. Arch. Pharmacol. 2017, 390, 977–985. [Google Scholar] [CrossRef]
- Saari, R.; Törmä, J.C.; Nevalainen, T. Microwave-assisted synthesis of quinoline, isoquinoline, quinoxaline and quinazoline derivatives as CB2 receptor agonists. Bioorg. Med. Chem. 2011, 19, 939–950. [Google Scholar] [CrossRef]
Figure 1.
Expression profile of calcium channels in mammalian central nervous system (CNS) cell types. The image shows neurons (yellow), oligodendrocytes (purple), astrocytes (green) and microglia (blue). For a detailed description please refer to the text.
Figure 1.
Expression profile of calcium channels in mammalian central nervous system (CNS) cell types. The image shows neurons (yellow), oligodendrocytes (purple), astrocytes (green) and microglia (blue). For a detailed description please refer to the text.


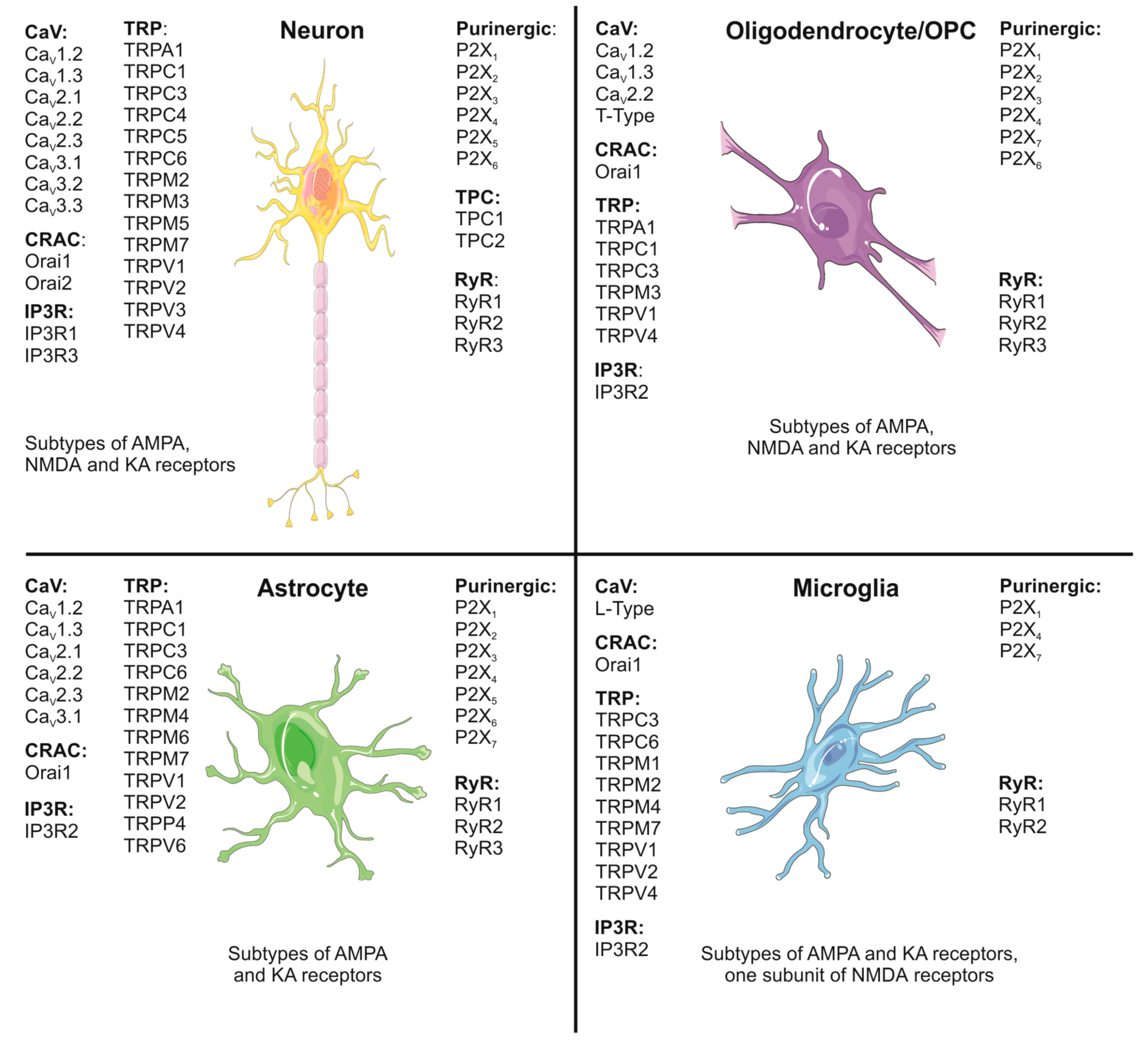{kind=link}
Table 1.
Drugs which directly interfere with calcium signaling and may potentially be neuroprotective in multiple sclerosis (MS).
Table 1.
Drugs which directly interfere with calcium signaling and may potentially be neuroprotective in multiple sclerosis (MS).
| Drug | Mode of Action | Proposed Effects | Disease (Model) | Species | NCT | Obvious Drawbacks | References |
|---|---|---|---|---|---|---|---|
| Nimodipine | Dihydropyridine blocking L-type voltage-gated calcium channels (VGCC) | Increased oligodendrocyte precursor cell (OPC) numbers, microglia-specific apoptosis, reduction of nitric oxide (NO) and reactive oxygen species (ROS) production, increased remyelination | Experimental autoimmune encephalomyelitis (EAE) | Mouse | - | - | [204,205] |
| Olesoxime | Cholesterol targeting voltage-dependent anion channels (VDAC) in the outer mitochondrial membrane | Neuroprotective agent affecting cytosolic calcium homoeostasis | EAE | Mouse, rat | NCT02628743 NCT01808885 | No long-term experience, not approved for any disease | [183,184,185,186,187,188] |
| Quetiapine | Antagonist at multiple G-protein coupled receptors (GPCR) (e.g., histamine H1, dopamine D2, 5-HT2A), interaction with mitochondrial calcium channel STIM1 | Increased maturation of oligodendrocytes | Cuprizone model, MS | Mouse, human | NCT02087631 | - | [190,191,192,193,194] |
Table 2.
Drugs which indirectly interfere with calcium signaling and may potentially be neuroprotective in MS.
Table 2.
Drugs which indirectly interfere with calcium signaling and may potentially be neuroprotective in MS.
| Drug | Mode of Action | Proposed Effects | Disease (Model) | Species | NCT | Drawback | References |
|---|---|---|---|---|---|---|---|
| Arachidonyl-2-chloroethylamide (ACEA) | Cannabinoid CB1 receptor agonist | Inhibition of the mitochondrial permeability transition pore leading to neuroprotection by decreased calcium influx into the cytosol | Theiler’s murine encephalomyelitis virus model | Mouse | - | No long-term experience | [213,215] |
| Asimadoline | κ Opioid Receptor (KOR) agonist | Induction of remyelination | EAE | Mouse | - | Low blood–brain barrier (BBB) permeability | [225] |
| Benzatropine | Muscarinic M1/M3 receptor antagonist | Enhanced remyelination via the induction of OPC differentiation | Cuprizone model, EAE | Mouse | - | - | [227,228,231] |
| Clemastine | Histamin H1 receptor antagonist | Induction of remyelination | Cuprizone model, patients with acute optic neuritis | Mouse, human | NCT02521311 | - | [219,220,232,233] |
| GSK239512 | Histamine H3 receptor antagonist | Small but positive effect on remyelination in a phase 2 study | Relapsing-remitting MS (RRMS) | Human | NCT01772199 | No long-term experience | [221] |
| GSK247246 | Histamine H3 receptor antagonist | Induction of remyelination | Cuprizone model | Mouse | - | No long-term experience | [222,234] |
| JWH-015 | Cannabinoid CB2 receptor agonist | Inhibition of the mitochondrial permeability transition pore leading to neuroprotection by decreased calcium influx into the cytosol | Theiler’s murine encephalomyelitis virus model | Mouse | - | Potentially psychoactive, illegal in some countries, no long-term experience | [213,216,217] |
| U-50488 | KOR agonist | Increased remyelination | EAE | Mouse | - | No long-term experience | [225,233,235,236] |
| Vitamin D | Vitamin D receptor stimulation | Unclear neuroprotective mechanism, evidence for the downregulation of L-type VGCC | EAE, hippocampal neurons, MS | Mouse, rat, human | - | Uncertain effects | [229,230,237] |
| WIN 55,212-2 | Cannabinoid CB1 receptor agonist | Potential neuroprotective effects by modulation of TRPA1 and CaV2.2 activity | Theiler’s murine encephalomyelitis virus model | Mouse | - | Potentially psychoactive, illegal in some countries, no long-term experience | [213,214] |
| Yhhu4952 | Cannabinoid CB2 receptor agonist | Improved remyelination, increased OPC maturation in culture by alteration of the Notch1 pathway, reduction of BBB leakiness | Cuprizone model | Neonatal rats, mouse | - | No long-term experience | [218,238] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Enders, M.; Heider, T.; Ludwig, A.; Kuerten, S. Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium. Int. J. Mol. Sci. 2020, 21, 1663. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051663
AMA Style
Enders M, Heider T, Ludwig A, Kuerten S. Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium. International Journal of Molecular Sciences. 2020; 21(5):1663. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051663
Chicago/Turabian StyleEnders, Michael, Thorsten Heider, Andreas Ludwig, and Stefanie Kuerten. 2020. "Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium" International Journal of Molecular Sciences 21, no. 5: 1663. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051663
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.