Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana



Abstract
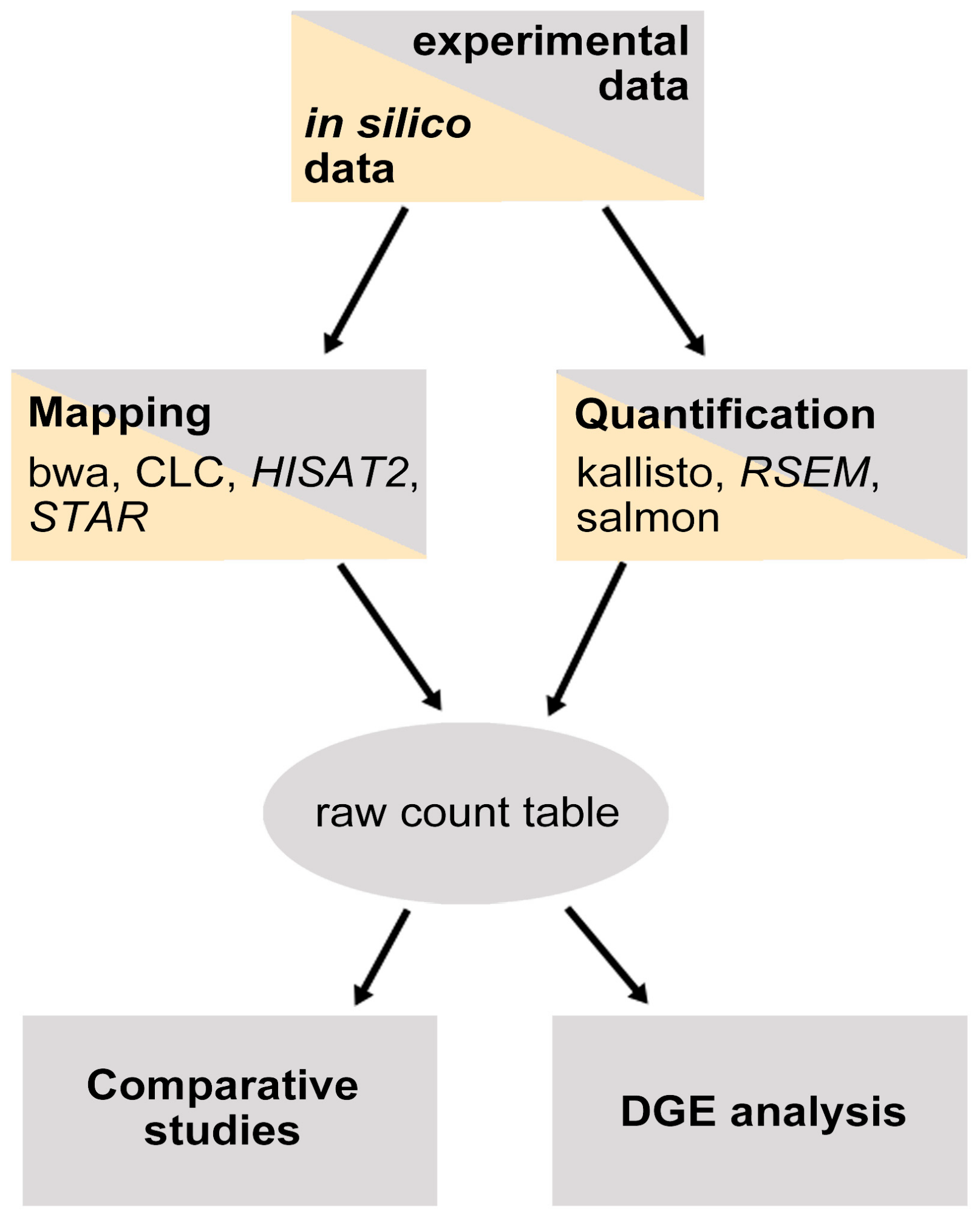
:1. Introduction
2. Results
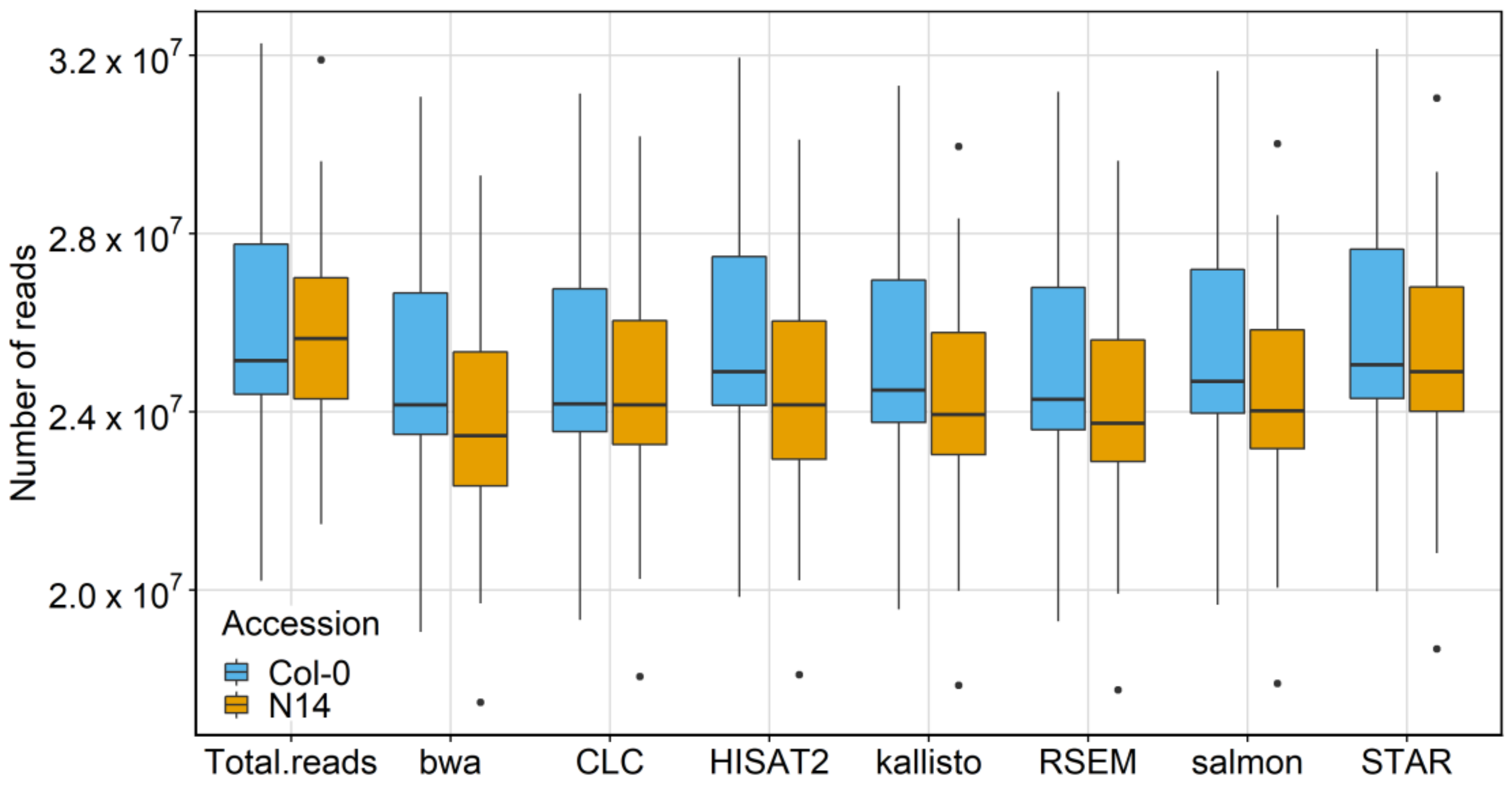
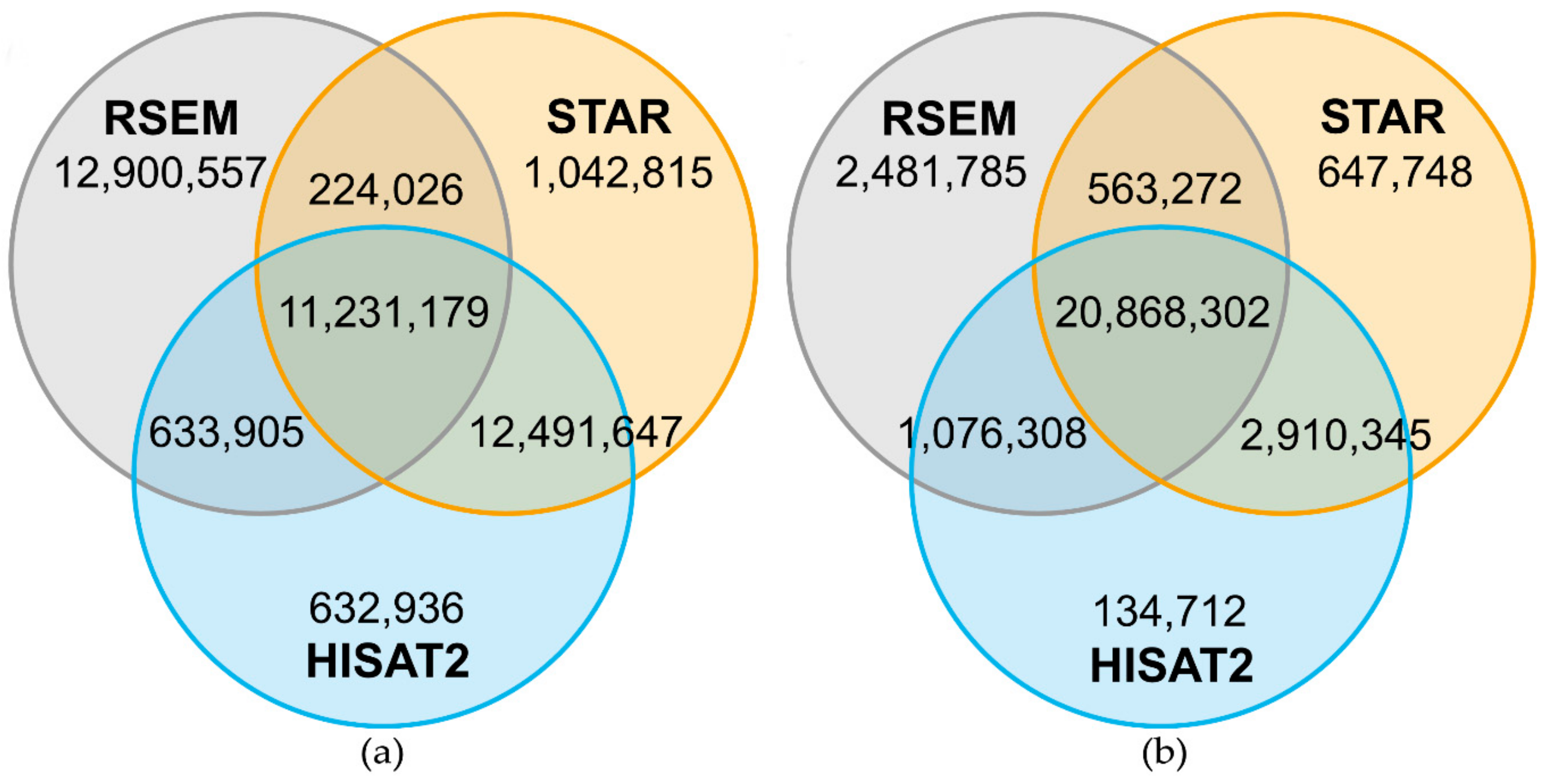
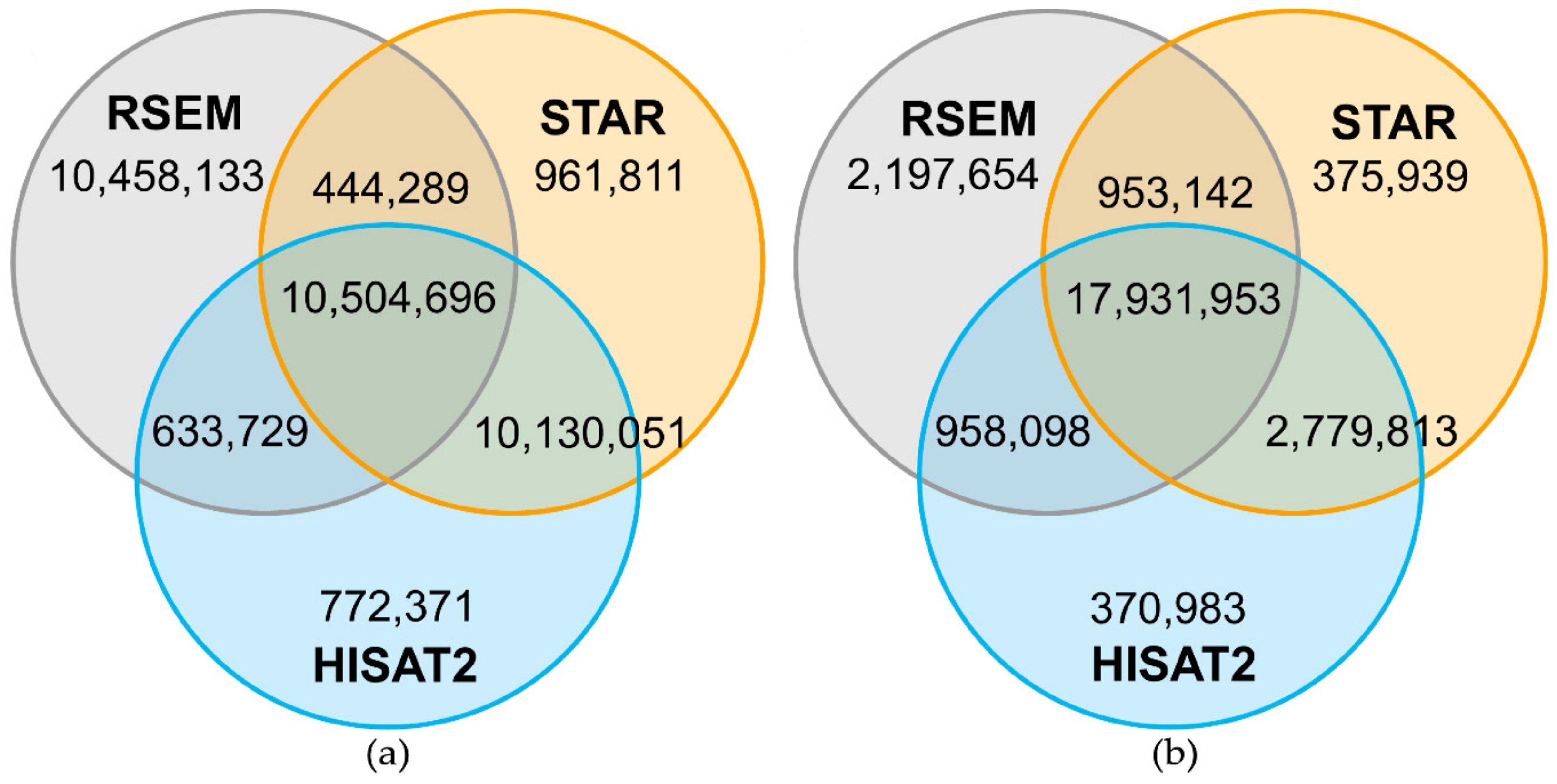
2.1. Mapping Statistics
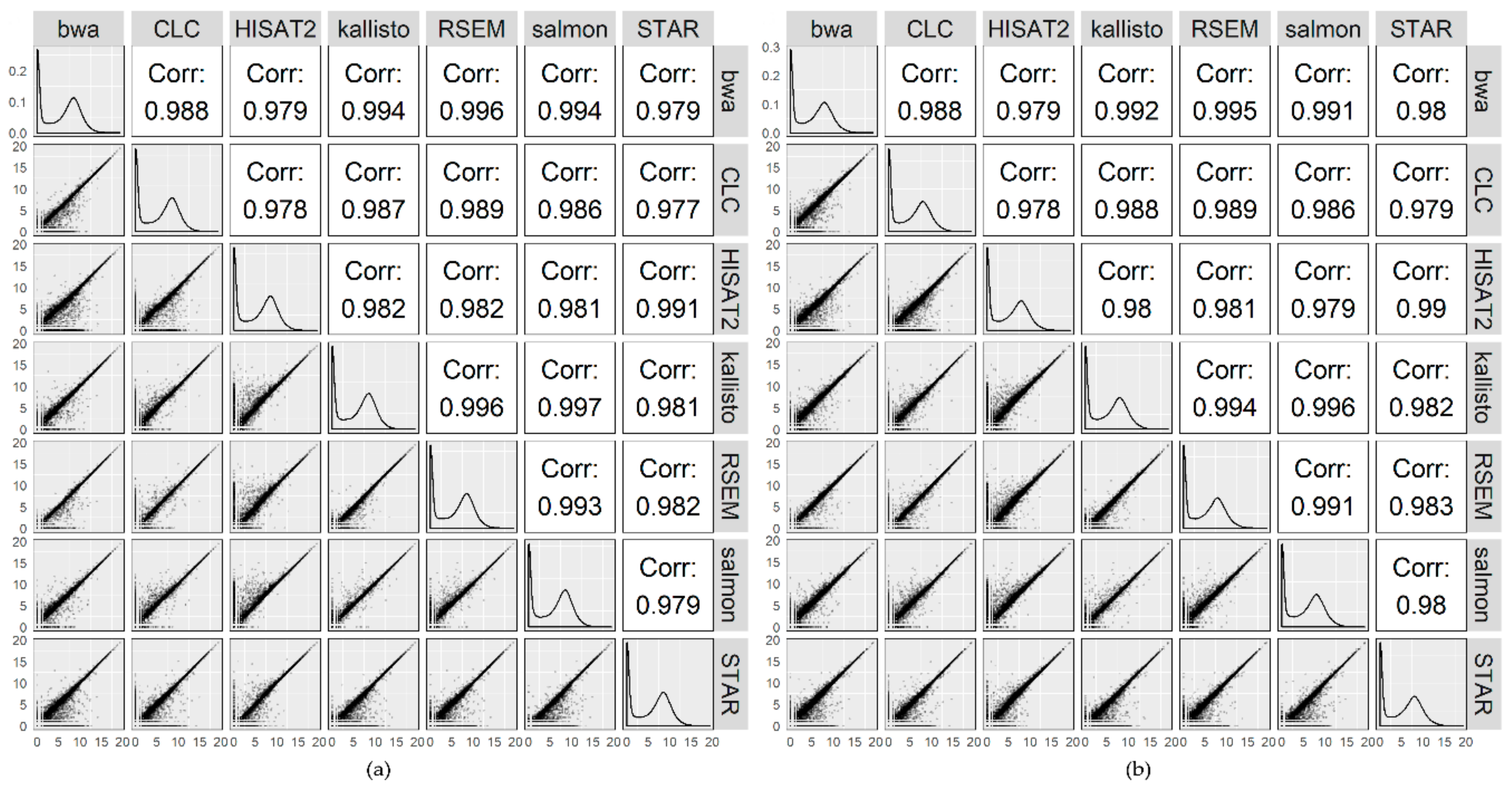
2.2. Raw Count Distribution for Individual Samples
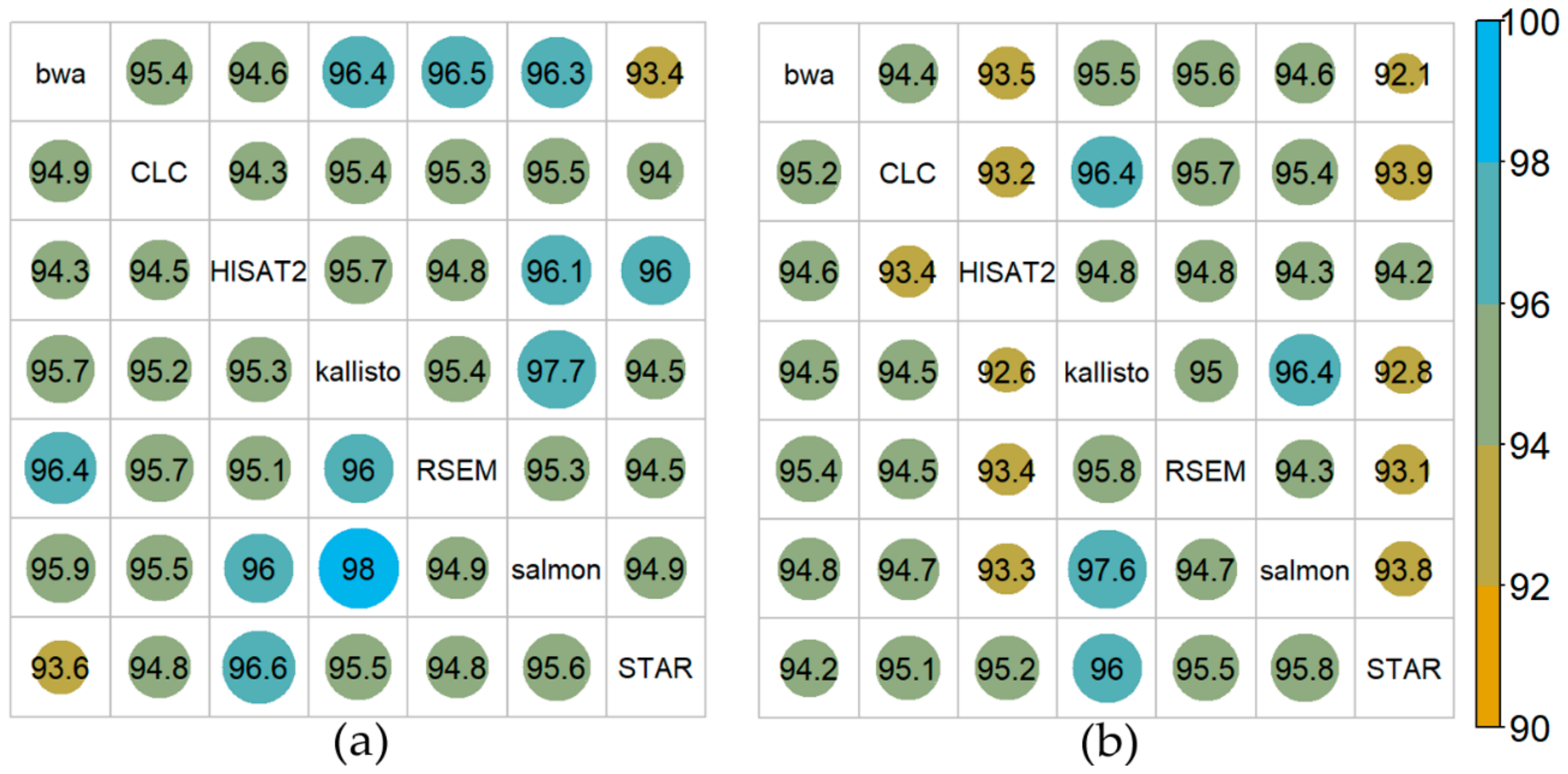
2.3. Overall Comparison of the Mappers
2.4. Mapping of in Silico Generated Reads
3. Discussion
4. Materials and Methods
4.1. Experimental Dataset
4.2. Mapping
4.3. Comparison Based on Expression Values
4.4. Differential Gene Expression
4.5. Mapping of in Silico Generated Reads
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA-Seq | RNA-sequencing |
| DGE | Differential Gene Expression |
| BWT | Burrows–Wheeler-Transformation |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Reads Raw Data | Number of Reads Pre-Processed Data |
|---|---|---|
| A | 26,551,078 | 25,965,205 |
| B | 24,160,253 | 23,723,408 |
| C | 24,987,211 | 24,631,398 |
| D | 24,679,891 | 24,314,564 |
| E | 32,902,966 | 32,265,838 |
| F | 25,343,870 | 24,962,434 |
| G | 25,633,391 | 25,255,295 |
| H | 24,767,056 | 24,276,316 |
| I | 22,434,138 | 22,074,152 |
| J | 27,102,013 | 26,738,311 |
| K | 29,909,220 | 29,473,355 |
| L | 30,039,895 | 29,625,213 |
| M | 25,373,173 | 25,045,811 |
| N | 27,401,911 | 27,059,316 |
| O | 32,172,339 | 31,758,225 |
| P | 26,713,325 | 26,326,809 |
| Q | 28,367,001 | 27,941,198 |
| R | 21,784,606 | 21,476,277 |
| S | 23,466,191 | 23,142,088 |
| T | 25,002,989 | 24,642,826 |
| U | 25,470,737 | 25,137,081 |
| V | 32,322,582 | 31,890,842 |
| W | 31,880,034 | 31,451,153 |
| X | 28,614,863 | 28,223,380 |
| Y | 25,396,753 | 24,312,026 |
| Z | 25,402,962 | 24,351,761 |
| AA | 21,934,477 | 21,095,112 |
| AB | 29,068,271 | 27,924,700 |
| AC | 28,363,133 | 27,205,327 |
| AD | 27,538,807 | 26,446,048 |
| AE | 21,048,979 | 20,198,121 |
| AF | 22,915,893 | 21,786,356 |
| AG | 26,195,089 | 25,161,103 |
| AH | 23,710,160 | 22,348,705 |
| AI | 25,915,840 | 24,936,936 |
| AJ | 27,904,776 | 26,835,785 |
| Sample | bwa | CLC | HISAT2 | kallisto | RSEM | salmon | STAR |
|---|---|---|---|---|---|---|---|
| A | 24,990,288 | 25,070,332 | 25,727,064 | 25,202,788 | 25,068,400 | 25,488,500 | 25,877,150 |
| B | 22,235,860 | 22,831,185 | 22,831,427 | 22,489,984 | 22,450,834 | 22,625,100 | 23,535,895 |
| C | 23,568,631 | 23,650,969 | 24,398,527 | 23,911,331 | 23,729,822 | 24,096,400 | 24,545,823 |
| D | 22,665,145 | 23,292,374 | 23,392,011 | 23,182,011 | 23,001,552 | 23,294,400 | 24,114,936 |
| E | 31,067,889 | 31,136,183 | 31,948,360 | 31,315,586 | 31,186,635 | 31,651,600 | 32,144,692 |
| F | 22,079,186 | 23,828,249 | 22,529,274 | 23,226,055 | 22,975,469 | 23,289,400 | 24,362,368 |
| G | 24,360,053 | 24,368,639 | 25,003,451 | 24,630,743 | 24,435,931 | 24,818,000 | 25,152,392 |
| H | 22,607,768 | 23,256,060 | 23,230,510 | 22,983,234 | 22,847,497 | 23,135,800 | 23,972,434 |
| I | 20,887,128 | 20,897,575 | 21,647,744 | 21,094,905 | 21,052,741 | 21,301,500 | 21,759,724 |
| J | 25,002,889 | 25,748,821 | 25,729,980 | 25,530,361 | 25,258,258 | 25,626,800 | 26,525,228 |
| K | 28,251,892 | 28,394,902 | 29,083,561 | 28,728,018 | 28,398,031 | 28,924,400 | 29,340,134 |
| L | 27,691,133 | 28,565,611 | 28,456,640 | 28,333,833 | 27,965,330 | 28,411,700 | 29,380,081 |
| M | 24,027,754 | 24,158,404 | 24,771,967 | 24,370,150 | 24,159,388 | 24,539,200 | 24,947,419 |
| N | 25,448,518 | 26,128,347 | 26,116,046 | 25,859,912 | 25,708,968 | 25,908,500 | 26,872,750 |
| O | 30,483,322 | 30,538,741 | 31,426,082 | 30,970,549 | 30,650,488 | 31,145,900 | 31,631,436 |
| P | 23,748,275 | 24,471,940 | 24,562,932 | 24,318,422 | 24,070,551 | 24,406,800 | 25,332,412 |
| Q | 26,863,089 | 26,968,681 | 27,679,891 | 27,157,401 | 26,977,076 | 27,405,000 | 27,843,106 |
| R | 19,700,000 | 20,245,101 | 20,218,359 | 19,970,836 | 19,918,383 | 20,052,800 | 20,826,196 |
| S | 22,171,458 | 22,274,280 | 22,902,423 | 22,444,868 | 22,280,936 | 22,624,300 | 23,035,647 |
| T | 23,165,182 | 23,815,751 | 23,748,284 | 23,543,789 | 23,398,523 | 23,638,100 | 24,456,937 |
| U | 24,145,575 | 24,192,319 | 24,905,595 | 24,499,411 | 24,279,099 | 24,667,800 | 25,057,610 |
| V | 29,305,198 | 30,181,899 | 30,105,423 | 29,951,050 | 29,635,355 | 30,012,700 | 31,037,834 |
| W | 30,171,991 | 30,240,229 | 31,135,272 | 30,619,320 | 30,314,724 | 30,820,900 | 31,321,391 |
| X | 26,417,781 | 27,215,579 | 27,146,639 | 26,999,068 | 26,701,971 | 27,089,600 | 28,004,846 |
| Y | 23,467,493 | 23,523,457 | 24,062,637 | 23,713,957 | 23,548,791 | 23,915,800 | 24,211,437 |
| Z | 22,939,890 | 23,531,637 | 23,488,307 | 23,241,258 | 23,186,262 | 23,333,700 | 24,169,828 |
| AA | 20,347,062 | 20,333,841 | 20,891,798 | 20,594,460 | 20,425,031 | 20,742,500 | 21,011,777 |
| AB | 26,183,324 | 26,842,810 | 26,903,033 | 26,663,997 | 26,539,902 | 26,769,800 | 27,709,817 |
| AC | 26,065,885 | 26,102,795 | 26,890,847 | 26,358,644 | 26,235,209 | 26,562,400 | 27,054,414 |
| AD | 24,904,560 | 25,532,006 | 25,483,657 | 25,267,366 | 25,201,022 | 25,348,100 | 26,234,545 |
| AE | 19,055,414 | 19,320,692 | 19,842,597 | 19,566,392 | 19,295,597 | 19,670,300 | 19,967,391 |
| AF | 17,469,949 | 18,053,590 | 18,090,815 | 17,854,059 | 17,755,997 | 17,898,700 | 18,672,118 |
| AG | 24,163,365 | 24,161,812 | 24,876,952 | 24,468,971 | 24,283,108 | 24,682,500 | 25,047,179 |
| AH | 20,953,174 | 21,498,746 | 21,436,520 | 21,310,760 | 21,215,866 | 21,379,400 | 22,109,670 |
| AI | 23,823,058 | 23,916,429 | 24,617,944 | 24,223,766 | 23,973,245 | 24,383,700 | 24,792,766 |
| AJ | 25,023,005 | 25,804,958 | 25,767,495 | 25,481,098 | 25,325,866 | 25,563,800 | 26,594,060 |
| Col-0 % | 95.9 | 96.2 | 98.9 | 97.2 | 96.4 | 97.9 | 99.5 |
| N14 % | 92.4 | 95.2 | 94.9 | 94.2 | 93.6 | 94.6 | 98.1 |
| Total % | 94.1 | 95.7 | 96.9 | 95.7 | 95.0 | 96.3 | 98.8 |
| Experiment 1 | Experiment 2 | Experiment 3 | |||||
|---|---|---|---|---|---|---|---|
| Cond. | Acc. | Sample | Cond. | Acc. | Sample | Cond. | Acc. |
| C28 | Col-0 | M | C28 | Col-0 | Y | C28 | Col-0 |
| C28 | N14 | N | C28 | N14 | Z | C28 | N14 |
| C28P3 | Col-0 | O | C28P3 | Col-0 | AA | C28P3 | Col-0 |
| C28P3 | N14 | P | C28P3 | N14 | AB | C28P3 | N14 |
| C35 | Col-0 | Q | C35 | Col-0 | AC | C35 | Col-0 |
| C35 | N14 | R | C35 | N14 | AD | C35 | N14 |
| C28P3L7 | Col-0 | S | C28P3L7 | Col-0 | AE | C28P3L7 | Col-0 |
| C28P3L7 | N14 | T | C28P3L7 | N14 | AF | C28P3L7 | N14 |
| C35P3 | Col-0 | U | C35P3 | Col-0 | AG | C35P3 | Col-0 |
| C35P3 | N14 | V | C35P3 | N14 | AH | C35P3 | N14 |
| C28P3L7T3 | Col-0 | W | C28P3L7T3 | Col-0 | AI | C28P3L7T3 | Col-0 |
| C28P3L7T3 | N14 | X | C28P3L7T3 | N14 | AJ | C28P3L7T3 | N14 |
References
- Collins, F.S.; Morgan, M.; Patrinos, A. The Human Genome Project: Lessons from large-scale biology. Science 2003, 300, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Dillies, M.-A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2012, 14, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, R95. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, A.M.; Nichols, M.; Burke, T.W.; Ginsburg, G.S.; Lucas, J.E. Comparing reference-based RNA-Seq mapping methods for non-human primate data. BMC Genom. 2014, 15, 570. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Golovnina, K.; Chen, Z.X.; Lee, H.N.; Negron, Y.L.; Sultana, H.; Oliver, B.; Harbison, S.T. Comparison of normalization and differential expression analyses using RNA-Seq data from 726 individual Drosophila melanogaster. BMC Genom. 2016, 17, 28. [Google Scholar] [CrossRef] [Green Version]
- Amin, S.; Prentis, P.J.; Gilding, E.K.; Pavasovic, A. Assembly and annotation of a non-model gastropod (Nerita melanotragus) transcriptome: A comparison of De novo assemblers. BMC Res. Notes 2014, 7, 488. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.B.; Zadlock, F.J.I.V.; Zhang, Z.; Murphy, W.R.; Bentivegna, C.S. Comparison of de novo transcriptome assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS ONE 2016, 11, e0153104. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Meth. 2017, 14, 417. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Zuther, E.; Schaarschmidt, S.; Fischer, A.; Erban, A.; Pagter, M.; Mubeen, U.; Giavalisco, P.; Kopka, J.; Sprenger, H.; Hincha, D.K. Molecular signatures associated with increased freezing tolerance due to low temperature memory in Arabidopsis. Plant Cell Environ. 2019, 42, 854–873. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Baggerly, K.A.; Deng, L.; Morris, J.S.; Aldaz, C.M. Differential expression in SAGE: Accounting for normal between-library variation. Bioinformatics 2003, 19, 1477–1483. [Google Scholar] [CrossRef]
- Baruzzo, G.; Hayer, K.E.; Kim, E.J.; Di Camillo, B.; FitzGerald, G.A.; Grant, G.R. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Meth. 2016, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Luypaert, M.; Maag, J.L.V.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Wan, Y.-W.; Liu, Z. Comprehensive evaluation of RNA-seq quantification methods for linearity. BMC Bioinform. 2017, 18 (Suppl. 4), 117. [Google Scholar] [CrossRef] [Green Version]
- Sahraeian, S.M.E.; Mohiyuddin, M.; Sebra, R.; Tilgner, H.; Afshar, P.T.; Au, K.F.; Bani Asadi, N.; Gerstein, M.B.; Wong, W.H.; Snyder, M.P.; et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat. Commun. 2017, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Love, M.I.; Davis, C.A.; Djebali, S.; Dobin, A.; Graveley, B.R.; Li, S.; Mason, C.E.; Olson, S.; Pervouchine, D.; et al. Erratum to: A benchmark for RNA-seq quantification pipelines. Genome Biol. 2016, 17, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Meth. 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledo, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627. [Google Scholar] [CrossRef]
- Hancock, A.M.; Brachi, B.; Faure, N.; Horton, M.W.; Jarymowycz, L.B.; Sperone, F.G.; Toomajian, C.; Roux, F.; Bergelson, J. Adaptation to climate across the Arabidopsis thaliana genome. Science 2011, 334, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Meinke, D.W.; Cherry, J.M.; Dean, C.; Rounsley, S.D.; Koornneef, M. Arabidopsis thaliana: A model plant for genome analysis. Science 1998, 282, 662–682. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.; Schüller, C.; Wambutt, R.; Murphy, G.; Volckaert, G.; Pohl, T.; Düsterhöft, A.; Stiekema, W.; Entian, K.D.; Terryn, N.; et al. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature 1999, 402, 769. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Meth. 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, N.A.; Marioni, J.; Brazma, A. RNA-Seq gene profiling—A systematic empirical comparison. PLoS ONE 2014, 9, e107026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.K.; Hoang, T.V.; Robinson, M.L.; Tsonis, P.A.; Liang, C. CADBURE: A generic tool to evaluate the performance of spliced aligners on RNA-Seq data. Sci. Rep. 2015, 5, 13443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; AE, L. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EnsemblPlants Arabidopsis Thaliana Assembly and Gene Annotation. Available online: http://plants.ensembl.org/info/website/ftp/index.html (accessed on 5 June 2016).
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Qiagen CLC Genomics Workbench. Available online: https://www.qiagenbioinformatics.com/ (accessed on 25 February 2019).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kazi-Aoual, F.; Hitier, S.; Sabatier, R.; Lebreton, J.-D. Refined approximations to permutations tests for multivariate inference. Comput. Stat. Data Anal. 1995, 20, 643–656. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Josse, J.; Husson, F.; Pagés, J. Testing the significance of the RV coefficient. Comput. Stat. Data Anal. 2007, 53, 82–91. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix. Available online: https://github.com/taiyun/corrplot (accessed on 3 July 2019).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Bwa | CLC | HISAT2 | Kallisto | RSEM | Salmon | STAR | |
|---|---|---|---|---|---|---|---|
| Before filtering | 32,243 | 33,602 | 33,602 | 32,243 | 33,602 | 32,243 | 33,602 |
| After filtering | 24,197 | 23,903 | 24,840 | 24,810 | 25,144 | 24,574 | 24,515 |
| DESeq2 | CLC | |||||
|---|---|---|---|---|---|---|
| Comparison | Accession | Baggerly | Overlap DESeq2 | EDGE | Overlap DESeq2 | |
| C28P3/C28 | Col-0 | 2014 | 3034 | 1013 | 2921 | 1006 |
| N14 | 2101 | 3414 | 1061 | 3311 | 1052 | |
| C28P3L7T3/C35P3 | Col-0 | 1 | 98 | 0 | 86 | 0 |
| N14 | 1 | 168 | 0 | 259 | 0 | |
| Bwa | CLC | HISAT2 | Kallisto | RSEM | Salmon | STAR | |
|---|---|---|---|---|---|---|---|
| Reference | |||||||
| Genome | X | X | X | ||||
| Transcriptome | X | X | X | X | X | X | X |
| Needs annotation | X | X | X | X | X | ||
| Specifications | |||||||
| Alignment-based | X | X | X | X | X | ||
| Pseudo-alignment | X | X | |||||
| Expression values | X | X | X | X | X | ||
| Splice aware | X | X | X | ||||
| Commercial software | X |
| HISAT2 | STAR | |||
|---|---|---|---|---|
| Col-0 | N14 | Col-0 | N14 | |
| Assigned to exon | 94.34 | 90.70 | 93.05 | 90.72 |
| Unmapped | 1.10 | 5.16 | 0.50 | 1.99 |
| Multimapped | 4.01 | 3.61 | 5.93 | 6.77 |
| No Feature (intergenic) | 0.55 | 0.53 | 0.51 | 0.53 |
| HISAT2 | in % | RSEM | in % | STAR | in % | |
|---|---|---|---|---|---|---|
| Primary | ||||||
| Mapped on right transcript | 57,981,570 | 98.7 | 58,072,536 | 98.9 | 58,000,379 | 98.8 |
| Mapped on wrong transcript | 689,541 | 1.2 | 658,699 | 1.1 | 668,909 | 1.1 |
| Unmapped | 18,022 | 0.031 | 773 | 0.001 | 19,526 | 0.033 |
| Mapped not on known exon | 42,875 | 0.073 | 0 | 0.0 | 43,194 | 0.1 |
| total reads | 58,732,008 | 100 | 58,732,008 | 100 | 58,732,008 | 100 |
| Secondary | ||||||
| Mapped on right transcript | 962,756 | 41.5 | 1,788,234 | 4.1 | 727,039 | 36.9 |
| Mapped on wrong transcript | 1,280,622 | 55.1 | 42,112,759 | 95.9 | 1,164,065 | 59.1 |
| mapped on different gene | 825,766 | 64.5 | 38,112,265 | 90.5 | 842,864 | 72.4 |
| mapped on paralog gene | 454,178 | 35.5 | 3,957,169 | 9.4 | 320,812 | 27.6 |
| mapped on different isoform | 678 | 0.1 | 43,325 | 0.1 | 389 | 0.0 |
| Mapped not on exon | 79,118 | 3.4 | 0 | 0.0 | 77,647 | 3.9 |
| total reads | 2,322,496 | 100 | 43,900,993 | 100 | 1,968,751 | 100 |
| Mapper | Version | Parameters | Reference |
|---|---|---|---|
| bwa aln | 0.7.13 | Default | Li and Durbin (2009) [11] |
| CLC | 9 | Default | Qiagen, Hilden, Germany [41] |
| kallisto quant | 0.42.5 | --single, -l 150 and -s 25 | Bray et al. (2016) [16] |
| HISAT2 | 2.1.0 | Default | Kim et al. (2019) [19] |
| RSEM | 1.2.30 | --bowtie2, --fragment-length-mean 150 & --fragment-length-sd 25 | Li and Dewey (2011) [14] |
| salmon quant | 0.6.0 | --type quasi, -k 31 --fldMean 150, --fldSD 25 and -l SF | Patro et al. (2017) [15] |
| STAR | 2.5.2a | --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 20 --alignSJDBoverhangMin 8 --outSAMunmapped Within --quantMode TranscriptomeSAM GeneCounts | Dobin et al. (2012) [12] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 1720. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051720
Schaarschmidt S, Fischer A, Zuther E, Hincha DK. Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana. International Journal of Molecular Sciences. 2020; 21(5):1720. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051720
Chicago/Turabian StyleSchaarschmidt, Stephanie, Axel Fischer, Ellen Zuther, and Dirk K. Hincha. 2020. "Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana" International Journal of Molecular Sciences 21, no. 5: 1720. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051720