Comparative Analysis of Complete Mitochondrial Genomes of Three Gerres Fishes (Perciformes: Gerreidae) and Primary Exploration of Their Evolution History

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
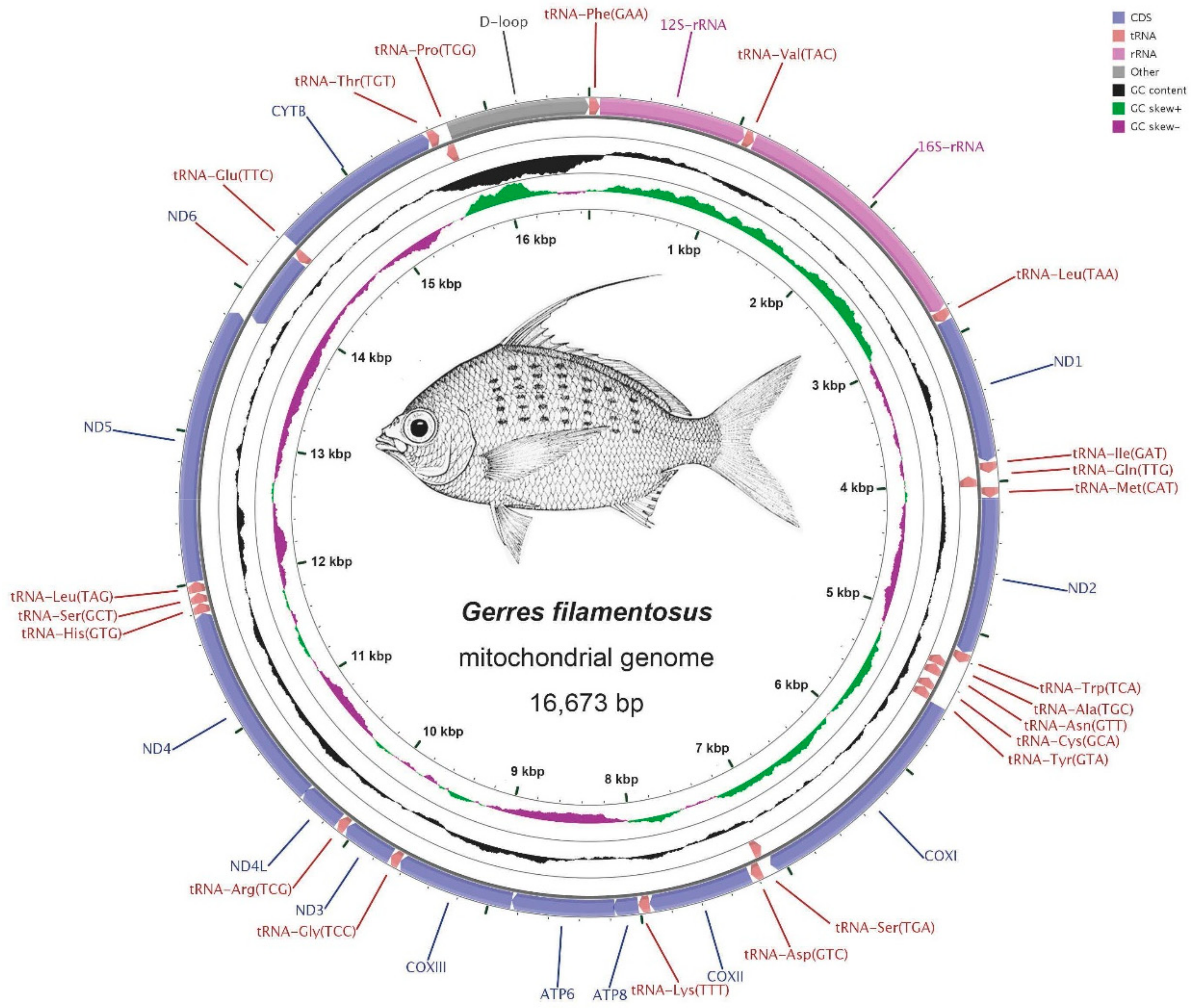
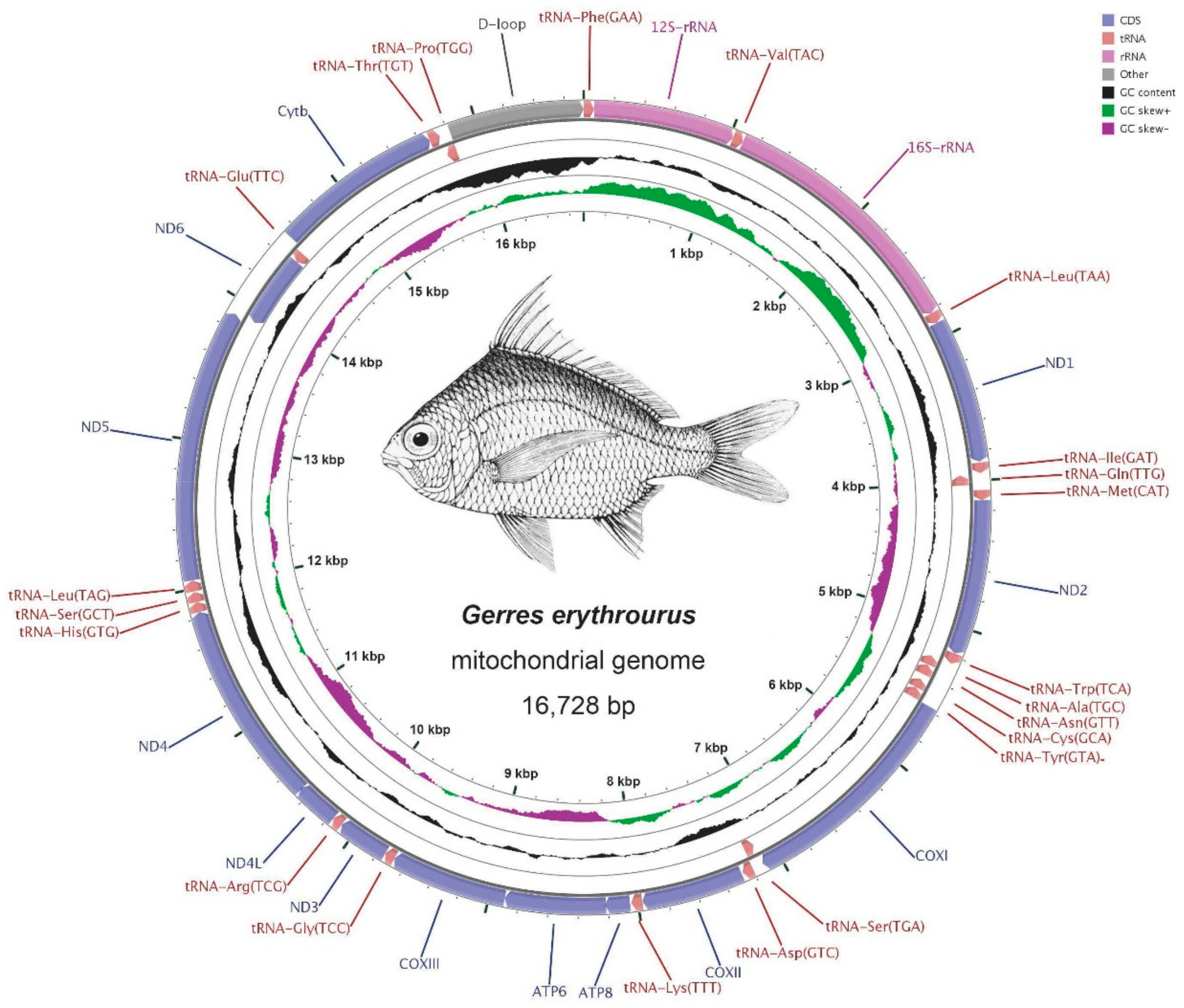
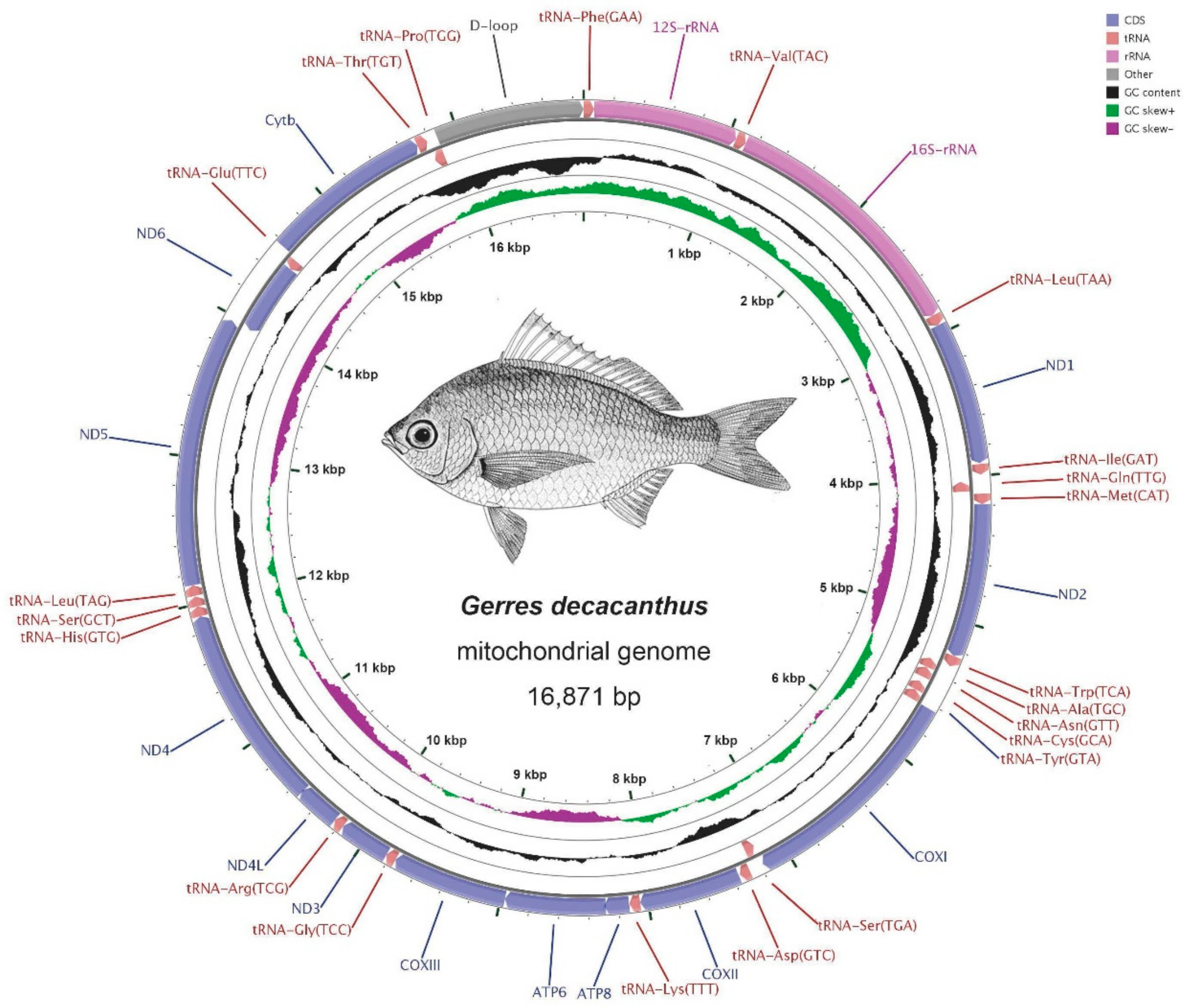
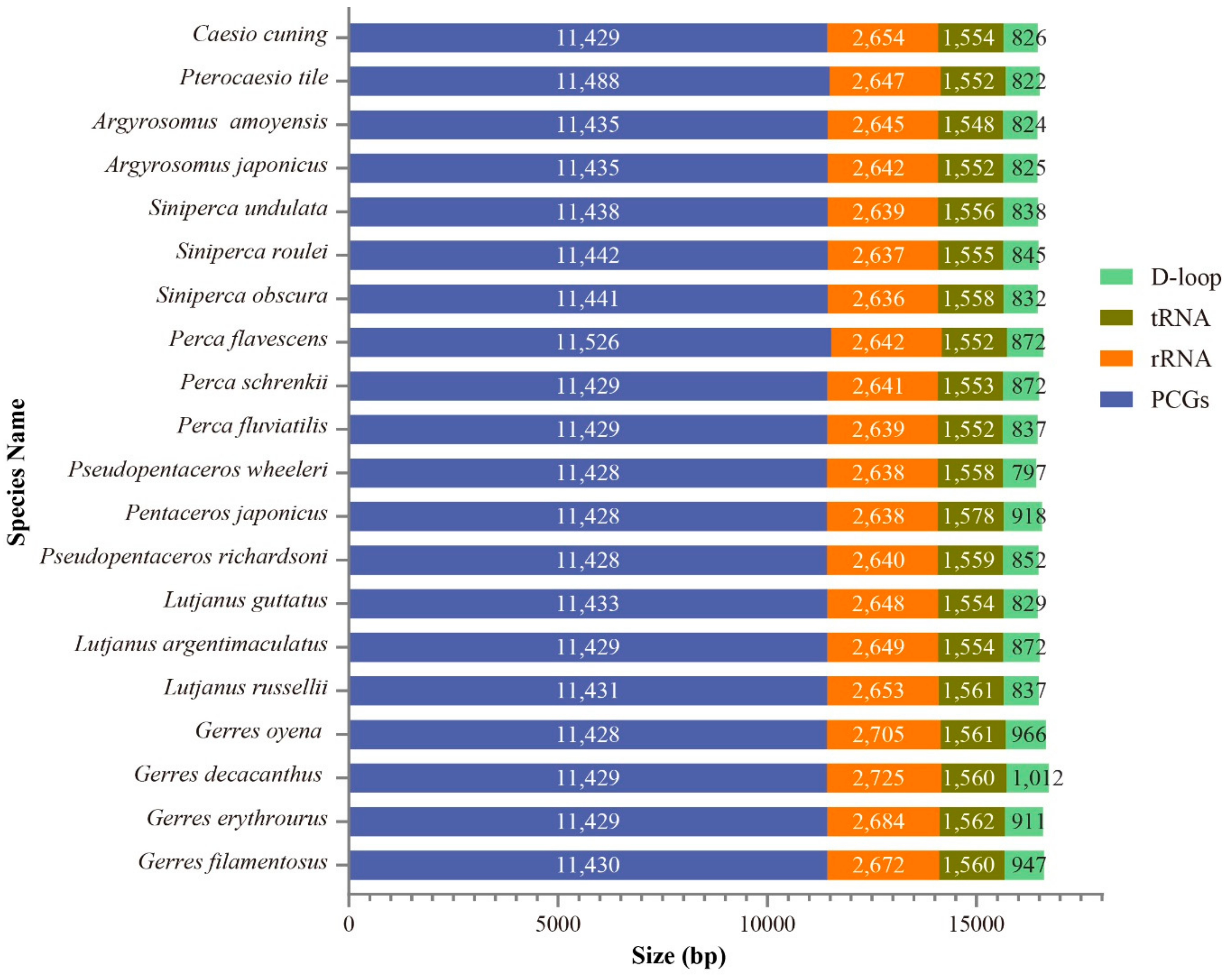
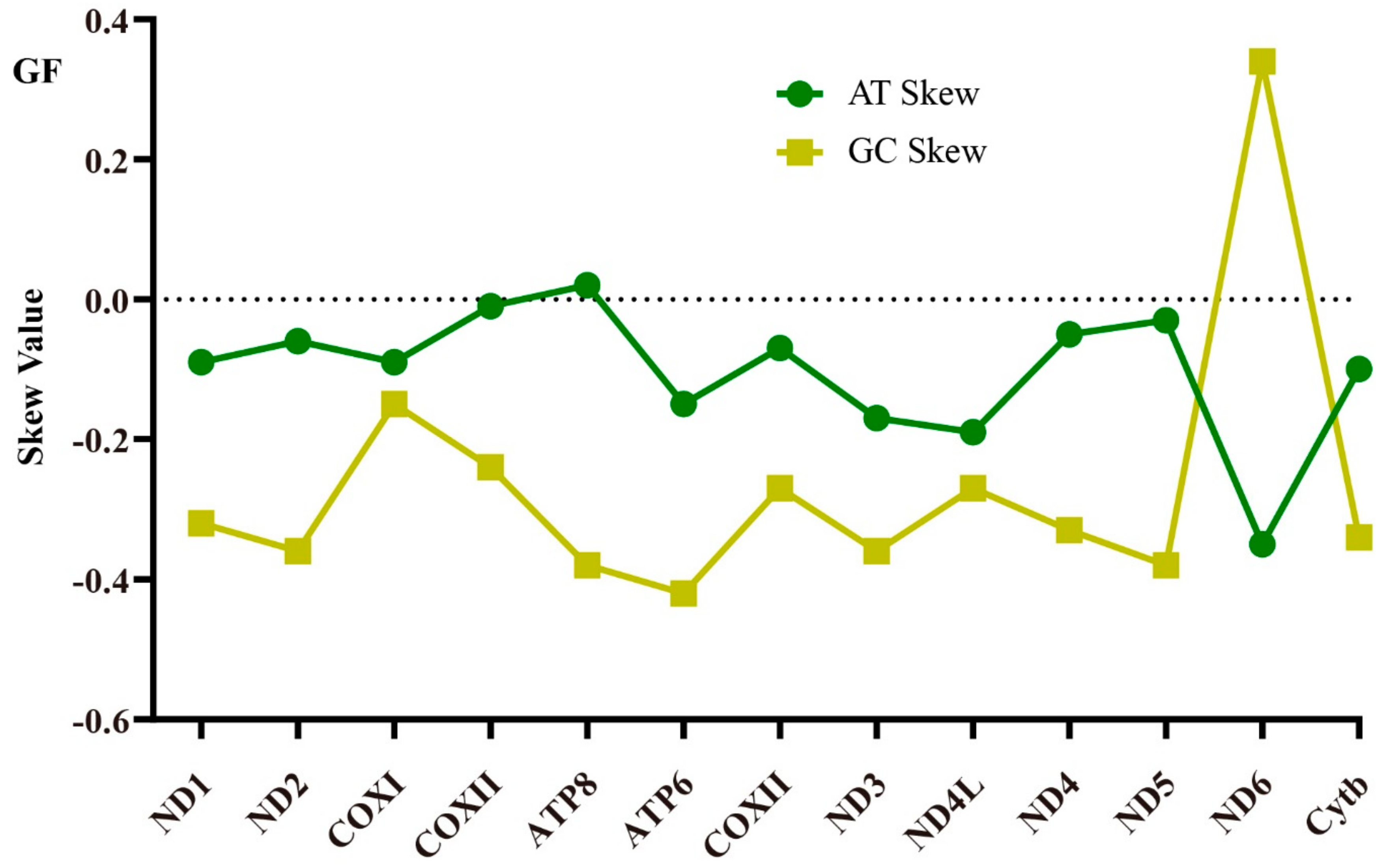
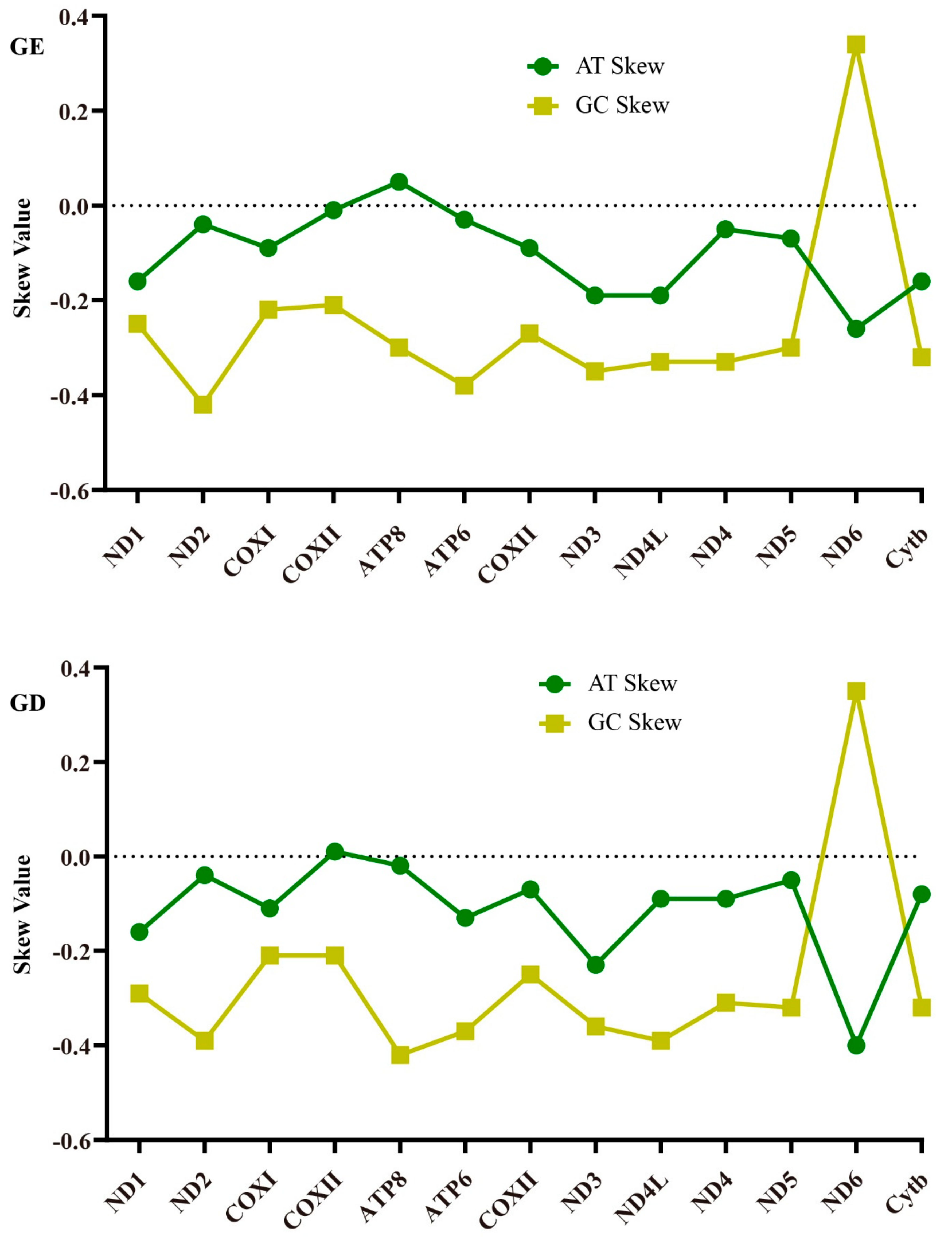
2.1. Genome Structure and Nucleotide Composition
2.2. RNA Genes
2.3. Intergenic Spacers and Overlapping
2.4. Protein Coding Genes (PCGs)
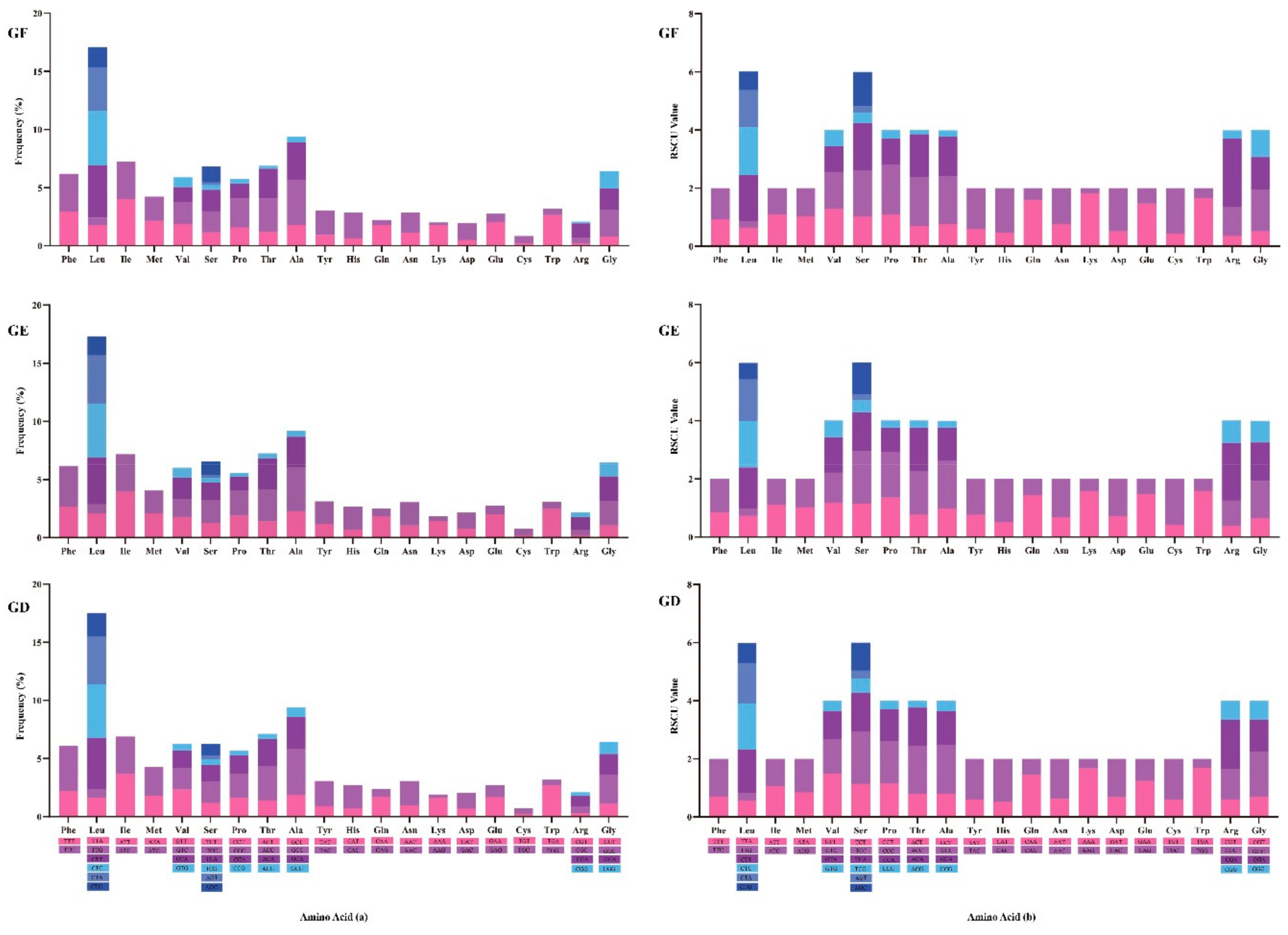
2.5. Usage of Mitogenome Codon
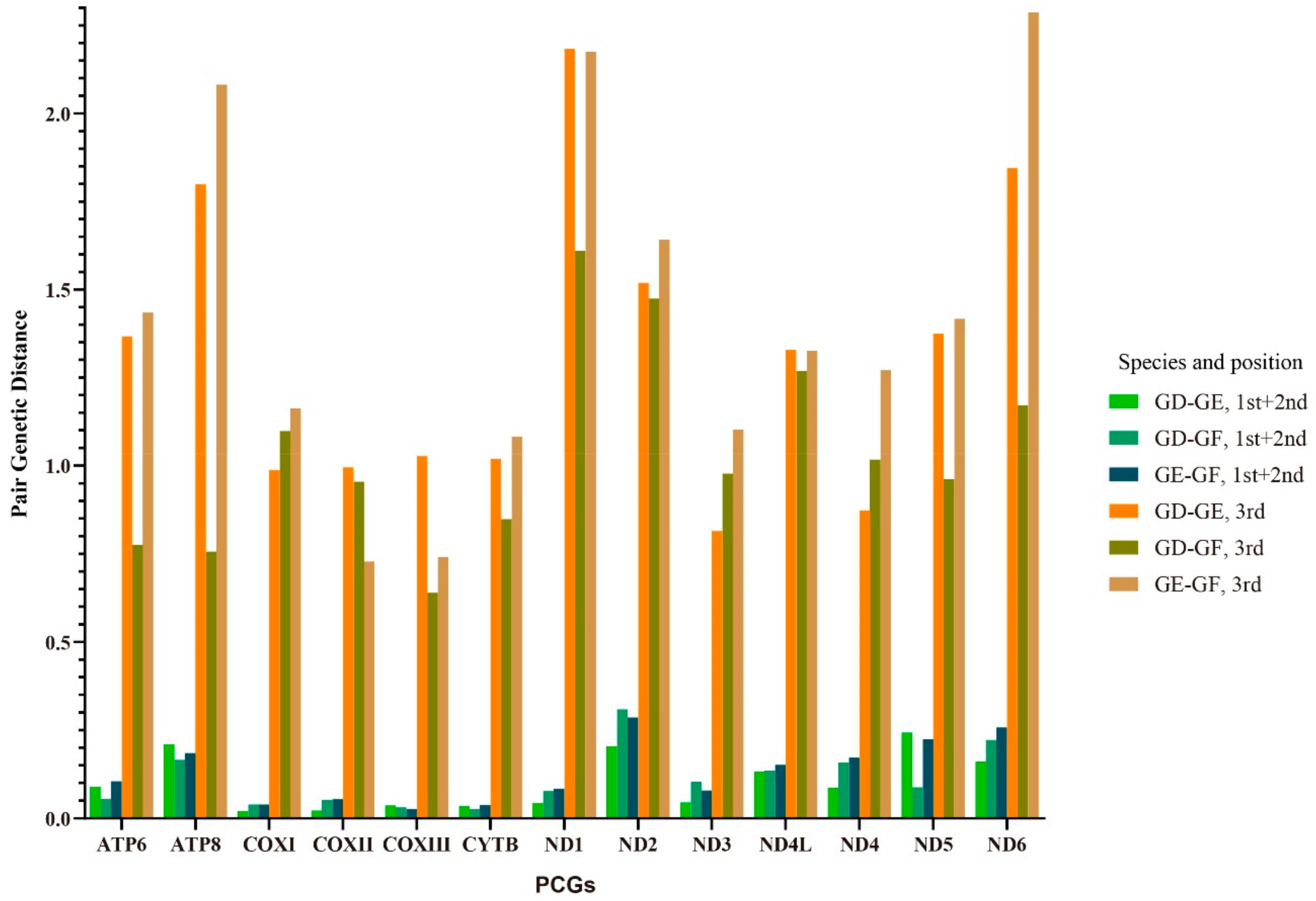
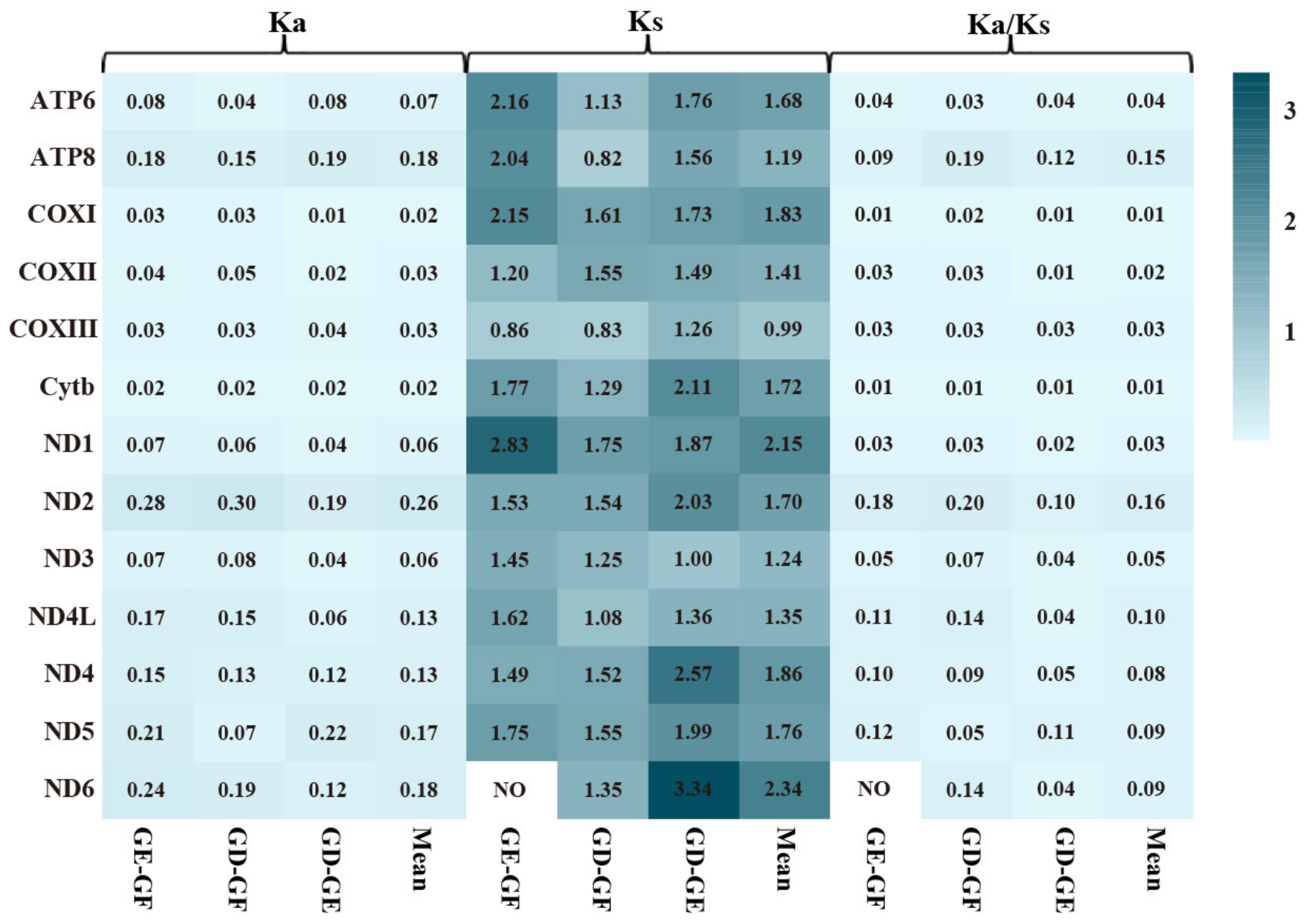
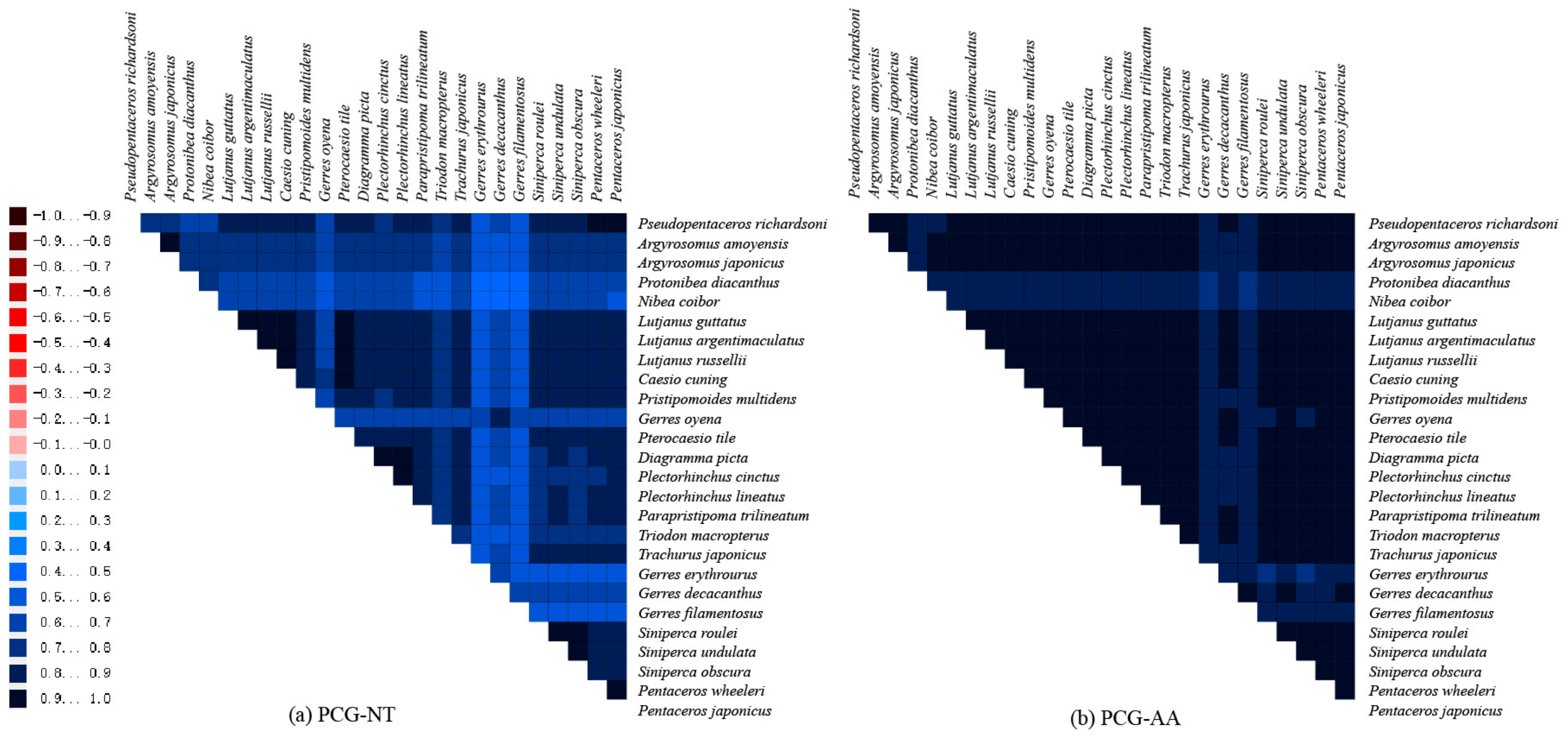
2.6. Variations, Genetic Distance, and Evolution Rates of PCGs
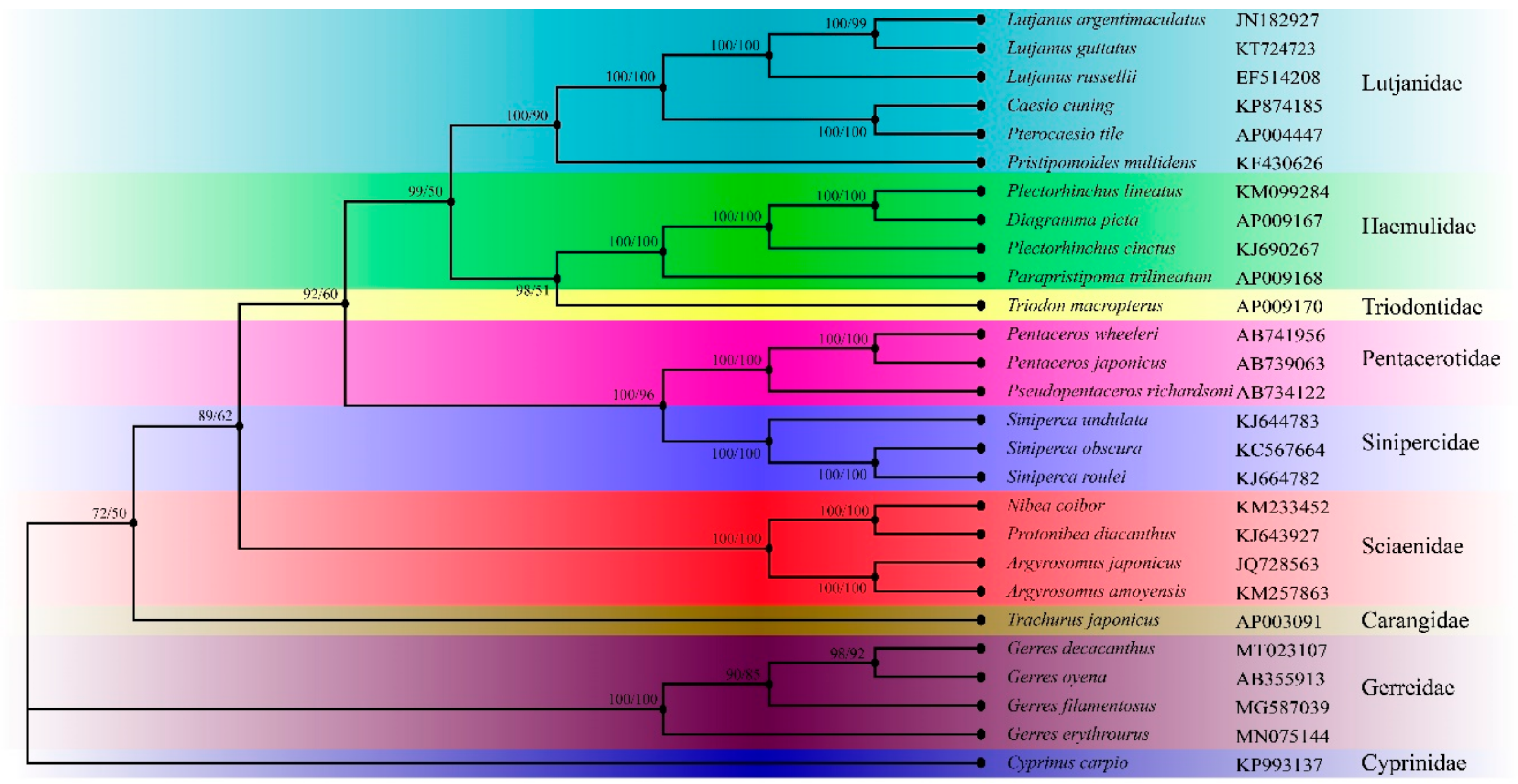
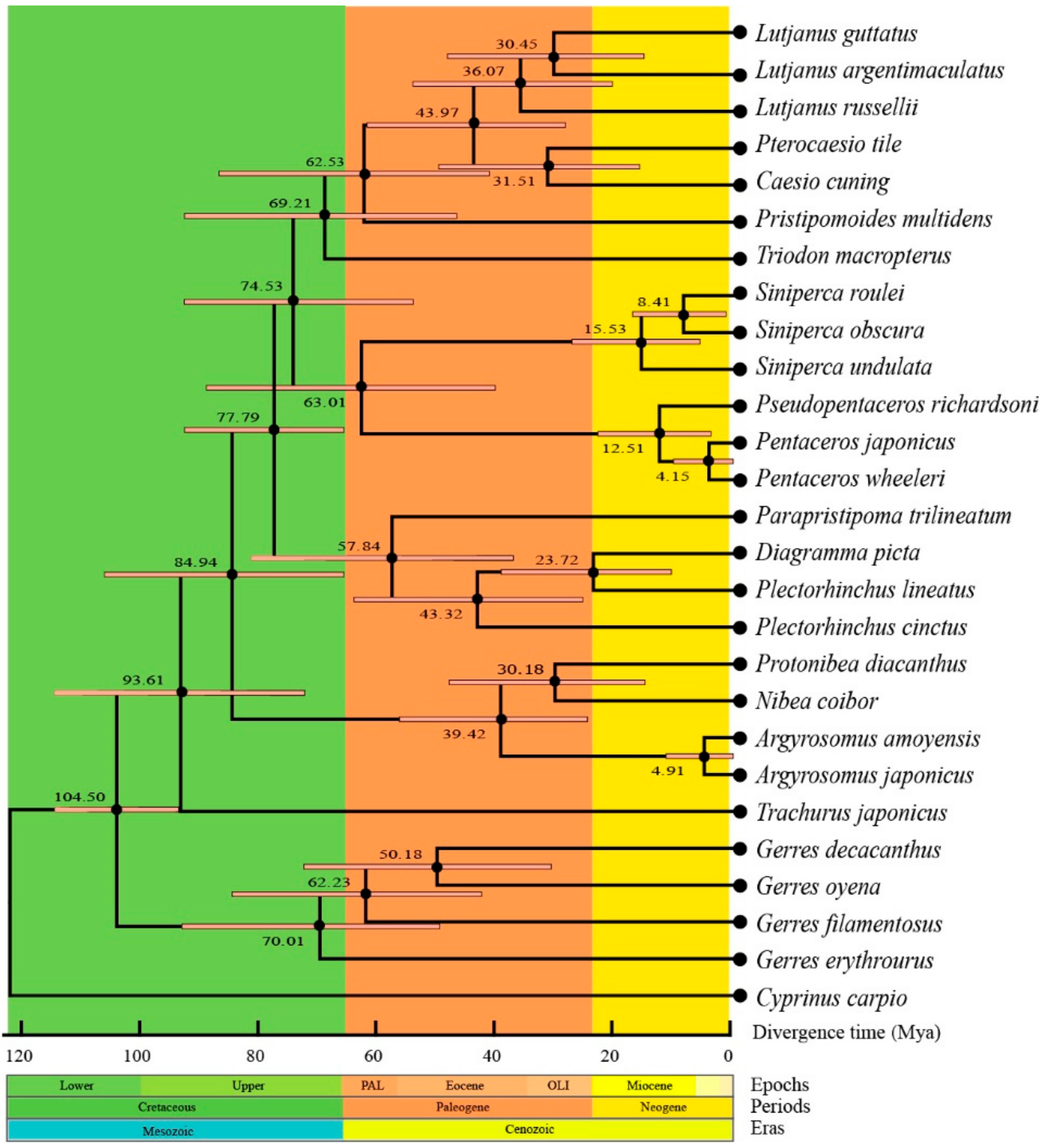
2.7. Phylogenetic and Divergence Times
3. Materials and Methods
3.1. Samples and DNA Extraction
3.2. Library Construction and High-Throughput Sequencing
3.3. Sequence Assembly, Annotation, and Analysis
3.4. Phylogenetic Analyses
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Zeng, L.; Wen, J.; Fan, S.; Chen, Z.; Xu, Y.; Sun, Y.; Chen, D.; Zhao, J. Species identification of fish maw (Porcupinefish) products sold on the market using DNA sequencing of 16S rRNA and COI genes. Food Control 2018, 86, 159–162. [Google Scholar] [CrossRef]
- Cutarelli, A.; Galiero, G.; Capuano, F.; Corrado, F. Species identification by means of mitochondrial cytochrome b DNA sequencing in processed anchovy, sardine and tuna products. Food Nutr. Sci. 2018, 9, 369–375. [Google Scholar]
- Zardoya, R.; Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 1996, 13, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Giusti, A.; Tinacci, L.; Sotelo, C.G.; Acutis, P.L.; Ielasi, N.; Armani, A. Authentication of ready-to-eat anchovy products sold on the Italian market by BLAST analysis of a highly informative cytochrome b gene fragment. Food Control 2019, 97, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.D.; Hanner, R.; Hebert, P.D. The campaign to DNA barcode all fishes, fish-biol. J. Fish Biol. 2009, 74, 329–356. [Google Scholar] [CrossRef]
- Cawthorn, D.M.; Duncan, J.; Kastern, C.; Francis, J.; Hoffman, L.C. Fish species substitution and misnaming in South Africa: An economic, safety and sustainability conundrum revisited. Food Chem. 2015, 185, 165–181. [Google Scholar] [CrossRef]
- Morin, P.A.; Archer, F.I.; Foote, A.D.; Vilstrup, J.; Allen, E.E.; Wade, P.; Durban, J.; Parsons, K.; Pitman, R.; Li, L. Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Res. 2010, 20, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, C.S.; Hui, C.F.; Shen, S.C.; Huang, P.C. The complete nucleotide sequence of the Crossostoma lacustre mitochondrial genome: Conservation and variations among vertebrates. Nucleic Acids Res. 1992, 20, 4853–4858. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhou, L.; Zhou, X.Y.; Yang, W.T.; Zhang, J.; Zhang, X.J.; Wang, Y.; Gui, J.F. Unusual AT-skew of Sinorhodeus microlepis mitogenome provides new insights into mitogenome features and phylogenetic implications of bitterling fishes. Int. J. Biol. Macromol. 2019, 129, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Nevatte, R.J.; Clark, J.A.; Williamson, J.E.; Gillings, M.R. The complete mitochondrial genome of the Epaulette Shark, Hemiscyllium ocellatum (Bonnaterre, 1788). Mitochondrial DNA Part B 2019, 4, 534–535. [Google Scholar] [CrossRef]
- FishBase. Available online: http://www.fishbase.org (accessed on 25 August 2019).
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2016; p. 433. [Google Scholar]
- Teixeira, R.L.; Helmer, J.L. Ecology of young mojarras (Pisces: Gerreidae) occupying the shallow waters of a tropical estuary. Revista Brasileira De Biol. 1997, 57, 637–646. [Google Scholar]
- Chollet-Villalpando, J.; De La Cruz-Agüero, J.; Garcia-Rodriguez, F.J. Comparison of urohyal bone morphology among gerreid fish (Perciformes: Gerreidae). Italian J. Zool. 2014, 81, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Fisheries and Aquaculture Department. Available online: http://www.fao.org/fishery/ (accessed on 26 August 2019).
- Aziz, M.; Ambily, V. Age and growth of Gerres filamentosus (Cuvier, 1829) from Kodungallur, Azhikode Estuary, Kerala. Afric. J. Agric. Res. 2013, 8, 4007–4014. [Google Scholar]
- Kanak, M.K.; Tachihara, K. Age and growth of Gerres sp (Perciformes: Gerreidae) in Okinawa Island of Southern Japan. Fisheries Sci. 2006, 72, 932–938. [Google Scholar] [CrossRef]
- Liu, J.; Yan, Y.R. A new species, Gerres septemfasciatus (Perciformes: Gerreidae) from the Chinese coastal waters of the South China Sea. Chin. J. Oceanol. Limn. 2009, 27, 555–557. [Google Scholar] [CrossRef]
- Iwatsuki, Y.; Heemstra, P.C. A new Gerreid fish species and redescription of Gerres maldivensis Regan, 1902 from the Indian Ocean (Perciformes: Gerreidae). Copeia 2007, 85–92. [Google Scholar] [CrossRef]
- Chen, W.J.; Ruiz-Carus, R.; Ortí, G. Relationships among four genera of mojarras (Teleostei: Perciformes: Gerreidae) from the western Atlantic and their tentative placement among percomorph fishes. J. Fish Biol. 2007, 70, 202–218. [Google Scholar] [CrossRef]
- De La Cruz-Agüero, J. Authorship, publication dates and prevailing usage of names for some species of the genus Eucinostomus Baird et Girard, 1855 (Perciformes: Gerreidae). J. Ichthyol. 2015, 55, 180–184. [Google Scholar] [CrossRef]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Randall, J.E. A checklist of the fishes of the South China Sea. Raffles Bull. Zool. Suppl. 2000, 8, 569–667. [Google Scholar]
- Li, Q.; Wang, Q.; Jin, X.; Chen, Z.; Xiong, C.; Li, P.; Zhao, J.; Huang, W. Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 2019, 121, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Naylor, G.J.; Collins, T.M.; Brown, W.M. Hydrophobicity and phylogeny. Nature 1995, 373, 565. [Google Scholar] [CrossRef]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef] [Green Version]
- Hardt, W.D.; Schlegl, J.; Erdmann, V.A.; Hartmann, R.K. Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes. Biochemistry 1993, 32, 13046–13053. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M. Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol. Biol. Evol. 1997, 14, 914–925. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Liu, Y.; Lai, M.; Song, H.; Wang, X.; Hu, Y.; Luo, J. Characterization of the Macropodus opercularis complete mitochondrial genome and family Channidae taxonomy using Illumina-based de novo transcriptome sequencing. Gene 2015, 559, 189–195. [Google Scholar] [CrossRef]
- Boore, J.L. Requirements and Standards for Organelle Genome Databases. OMICS: A J. Integrat. Biol. 2006, 10, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, B.E.; Warner, J.P.; Dunlop, N.; Fantes, J.; Intody, S.; Leek, J.; Markham, A.F.; Bonthron, D.T. Molecular genetics of the human glucokinase regulator-fructokinase (GCKR-KHK) region of chromosome 2p23. Biochem. Soc. T 1997, 25, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockman, S.A.; McFadden, C.S. The mitochondrial genome of Paraminabea aldersladei (Cnidaria: Anthozoa: Octocorallia) supports intramolecular recombination as the primary mechanism of gene rearrangement in octocoral mitochondrial genomes. Genome Biol. Evol. 2012, 4, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Song, N.; Yanagimoto, T.; Han, Z.; Shui, B.; Gao, T. Complete mitochondrial genome of three fish species (Perciformes: Amblyopinae): Genome description and phylogenetic relationships. Pakistan J. Zool. 2017, 49, 107–115. [Google Scholar] [CrossRef]
- Aguilar, C.; Miller, M.J.; Loaiza, J.R.; Krahe, R.; De Leon, L.F. Mitochondrial genomes and phylogenetic analysis of Central American weakly-electric fishes: Apteronotus rostratus, Brachyhypopomus occidentalis and Sternopygus dariensis. bioRxiv 2018, 353565. [Google Scholar]
- Montoya, J.; Gaines, G.L.; Attardi, G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983, 34, 151–159. [Google Scholar] [CrossRef]
- Hogan, R.I.; Hopkins, K.; Wheeler, A.J.; Allcock, A.L.; Yesson, C. Novel diversity in mitochondrial genomes of deep-sea Pennatulacea (Cnidaria: Anthozoa: Octocorallia). Mitochondrial DNA Part A 2019, 30, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. Bmc Genomics 2016, 17, 719. [Google Scholar] [CrossRef] [Green Version]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470. [Google Scholar] [CrossRef]
- Lü, Z.; Zhu, K.; Jiang, H.; Lu, X.; Liu, B.; Ye, Y.; Jiang, L.; Liu, L.; Gong, L. Complete mitochondrial genome of Ophichthus brevicaudatus reveals novel gene order and phylogenetic relationships of Anguilliformes. Int. J. Biol. Macromol. 2019, 135, 609–618. [Google Scholar] [CrossRef]
- Yang, H.; Xia, J.; Zhang, J.-e.; Yang, J.; Zhao, H.; Wang, Q.; Sun, J.; Xue, H.; Wu, Y.; Chen, J. Characterization of the complete mitochondrial genome sequences of three croakers (perciformes, sciaenidae) and novel insights into the phylogenetics. Int. J. Mol. Sci. 2018, 19, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics 2010, 11, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Matassi, G. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 1994, 4, 851–860. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Tatarinova, T.V.; Alexandrov, N.N.; Bouck, J.B.; Feldmann, K.A. GC 3 biology in corn, rice, sorghum and other grasses. BMC Genomics 2010, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Zhu, K.C.; Liang, Y.Y.; Wu, N.; Guo, H.Y.; Zhang, N.; Jiang, S.G.; Zhang, D.C. Sequencing and characterization of the complete mitochondrial genome of Japanese Swellshark (Cephalloscyllium umbratile). Sci. Rep. 2017, 7, 15299. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Shen, X.; Pu, Z.; Chen, X.; Murphy, R.W.; Shen, Y. Convergent Evolution of Mitochondrial Genes in Deep-Sea Fishes. Front. Genet. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaack, S.; Ho, E.K.; Macrae, F. Disentangling the intertwined roles of mutation, selection and drift in the mitochondrial genome. Philos. Trans. R. Soc. B 2019, 375, 20190173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, D.; Shen, X.; Pu, Z.; Irwin, D.M.; Liao, M.; Shen, Y. Convergent evolution of human-isolated H7N9 avian influenza A viruses. J. Infect. Dis. 2018, 217, 1699–1707. [Google Scholar] [CrossRef]
- Barrientos, A.; Barros, M.H.; Valnot, I.; Rötig, A.; Rustin, P.; Tzagoloff, A. Cytochrome oxidase in health and disease. Gene 2002, 286, 53–63. [Google Scholar] [CrossRef]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, Q.; Li, M.; Wei, J.; Zhang, X.; Zhang, H. Comparative Mitogenomic Analysis of the Eurydema Genus in the Context of Representative Pentatomidae (Hemiptera: Heteroptera) Taxa. J. Insect Sci. 2019, 19, 20. [Google Scholar] [CrossRef]
- Wu, H.L.; Shao, G.Z.; Lai, C.F.; Zhuang, L.H.; Lin, P.L. Latin-Chinese Dictionary of Fish Names by Classification System; China Ocean University Press: Qingdao, China, 2017; p. 602. [Google Scholar]
- Puppo, P.; Curto, M.; Meimberg, H. Genetic structure of Micromeria (Lamiaceae) in Tenerife, the imprint of geological history and hybridization on within-island diversification. Ecol. Evol. 2016, 6, 3443–3460. [Google Scholar] [CrossRef] [Green Version]
- Catarino, D.; Knutsen, H.; Veríssimo, A.; Olsen, E.M.; Jorde, P.E.; Menezes, G.; Sannæs, H.; Stanković, D.; Company, J.B.; Neat, F. The Pillars of Hercules as a bathymetric barrier to gene flow promoting isolation in a global deep-sea shark (Centroscymnus coelolepis). Mol. Ecol. 2015, 24, 6061–6079. [Google Scholar] [CrossRef]
- Sweere, T.C.; Dickson, A.J.; Jenkyns, H.C.; Porcelli, D.; Elrick, M.; Boorn, S.H.; Henderson, G.M. Isotopic evidence for changes in the zinc cycle during Oceanic Anoxic Event 2 (Late Cretaceous). Geology 2018, 46, 463–466. [Google Scholar] [CrossRef]
- Yan, Y.; Yao, D.; Tian, Z.X.; Huang, C.Y.; Dilek, Y.; Clift, P.D.; Li, Z.A. Tectonic topography changes in Cenozoic East Asia: A landscape erosion-sediment archive in the South China Sea. Geochem. Geophys. Geosyst. 2018, 19, 1731–1750. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2007, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Kuck, P.; Meid, S.A.; Gross, C.; Wagele, J.W.; Misof, B. AliGROOVE-visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Element | Strand | Position Start/End | Size (bp) | GC Percentage | Codon Start/Stop | Intergenic Nucleotide* (bp) | Anti-Codon/One Letter Code | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GF | GE | GD | GF | GE | GD | GF | GE | GD | GF | GE | GD | GF | GE | GD | |||
| tRNA-Phe | H | 1/71 | 1/70 | 1/71 | 71 | 70 | 71 | 42.25% | 48.57% | 42.25% | 0 | 0 | 0 | GAA/F | |||
| 12S-rRNA | H | 72/1053 | 71/1022 | 72/1053 | 982 | 952 | 982 | 48.98% | 48.63% | 49.90% | 0 | 0 | 0 | ||||
| tRNA-Val | H | 1054/1122 | 1023/1093 | 1054/1124 | 69 | 71 | 71 | 43.48% | 43.66% | 40.85% | 1 | 0 | 0 | TAC/V | |||
| 16S-rRNA | H | 1124/2813 | 1094/2825 | 1125/2867 | 1690 | 1732 | 1743 | 45.50% | 46.42% | 45.44% | 0 | 0 | 0 | ||||
| tRNA-Leu | H | 2814/2887 | 2826/2899 | 2868/2941 | 74 | 74 | 74 | 52.70% | 45.95% | 51.35% | 0 | 0 | 0 | TAA/L | |||
| ND1 | H | 2888/3862 | 2900/3874 | 2942/3916 | 975 | 975 | 975 | 47.59% | 49.44% | 50.67% | ATG/TAA | ATG/TAA | ATG/TAG | 7 | 5 | 6 | |
| tRNA-Ile | H | 3870/3940 | 3880/3951 | 3923/3993 | 71 | 72 | 71 | 53.52% | 54.17% | 60.56% | 11 | 5 | 40 | GAT/I | |||
| tRNA-Gln | L | 3952/4026 | 3957/4027 | 4034/4104 | 75 | 71 | 71 | 44.00% | 46.48% | 46.48% | 9 | 37 | 18 | TTG/Q | |||
| tRNA-Met | H | 4036/4104 | 4065/4133 | 4123/4193 | 69 | 69 | 71 | 40.58% | 40.58% | 43.66% | 0 | 0 | 0 | CAT/M | |||
| ND2 | H | 4105/5151 | 4134/5179 | 4194/5239 | 1047 | 1046 | 1047 | 48.42% | 48.09% | 52.58% | ATG/TAG | ATG/TA | ATG/TAA | 0 | 0 | 0 | |
| tRNA-Trp | H | 5152/5222 | 5180/5251 | 5241/5312 | 71 | 72 | 72 | 49.30% | 50.00% | 54.17% | 1 | 1 | 1 | TCA/W | |||
| tRNA-Ala | L | 5224/5292 | 5253/5321 | 5314/5382 | 69 | 69 | 69 | 40.58% | 37.68% | 40.58% | 1 | 2 | 2 | TGC/A | |||
| tRNA-Asn | L | 5294/5366 | 5324/5396 | 5385/5457 | 73 | 73 | 73 | 49.32% | 47.95% | 49.32% | 35 | 37 | 36 | GTT/N | |||
| tRNA-Cys | L | 5402/5468 | 5434/5501 | 5494/5560 | 67 | 68 | 67 | 49.25% | 45.59% | 44.78% | 0 | 0 | 0 | GCA/C | |||
| tRNA-Tyr | L | 5469/5539 | 5502/5572 | 5561/5631 | 71 | 71 | 71 | 47.89% | 46.48% | 46.48% | 1 | 1 | 1 | GTA/Y | |||
| COXI | H | 5541/7091 | 5574/7124 | 5633/7183 | 1551 | 1551 | 1551 | 46.74% | 46.16% | 47.32% | GTG/TAA | GTG/TAA | GTG/TAA | 0 | 0 | 0 | |
| tRNA-Ser | L | 7092/7162 | 7125/7195 | 7184/7254 | 71 | 71 | 71 | 53.52% | 52.11% | 50.70% | 3 | 3 | 29 | TGA/S | |||
| tRNA-Asp | H | 7166/7237 | 7199/7270 | 7284/7354 | 72 | 72 | 71 | 40.28% | 45.83% | 50.70% | 9 | 10 | 9 | GTC/D | |||
| COXII | H | 7247/7937 | 7281/7971 | 7364/8054 | 691 | 691 | 691 | 46.45% | 43.99% | 45.30% | ATG/T | ATG/T | ATG/T | 0 | 0 | 0 | |
| tRNA-Lys | H | 7938/8011 | 7972/8046 | 8055/8128 | 74 | 75 | 74 | 52.70% | 50.67% | 55.41% | 1 | 8 | 1 | TTT/K | |||
| ATP8 | H | 8013/8180 | 8055/8222 | 8130/8297 | 168 | 168 | 168 | 47.62% | 42.26% | 45.24% | ATG/TAA | ATG/TAA | ATG/TAA | −10 | −7 | −10 | |
| ATP6 | H | 8171/8854 | 8216/8895 | 8288/8970 | 684 | 680 | 683 | 46.20% | 45.29% | 47.29% | ATG/TAA | ATA/TA | ATG/TA | −1 | 0 | 0 | |
| COXIII | H | 8854/9638 | 8896/9680 | 8971/9755 | 785 | 785 | 785 | 49.17% | 46.50% | 48.41% | ATG/TA | ATG/TA | ATG/TA | 0 | 1 | 0 | |
| tRNA-Gly | H | 9639/9708 | 9682/9751 | 9756/9825 | 70 | 70 | 70 | 37.14% | 42.86% | 38.57% | 0 | 0 | 0 | TCC/G | |||
| ND3 | H | 9709/10057 | 9752/10100 | 9826/10174 | 349 | 349 | 349 | 50.72% | 45.85% | 49.00% | ATG/T | ATG/T | ATG/T | 0 | 0 | 0 | |
| tRNA-Arg | H | 10058/10126 | 10101/10169 | 10175/10243 | 69 | 69 | 69 | 36.23% | 30.43% | 34.78% | 0 | 0 | 0 | TCG/R | |||
| ND4L | H | 10127/10423 | 10170/10466 | 10244/10540 | 297 | 297 | 297 | 49.49% | 51.52% | 49.49% | ATG/TAA | ATG/TAA | ATG/TAA | −7 | −7 | −7 | |
| ND4 | H | 10417/11797 | 10460/11840 | 10534/11914 | 1381 | 1381 | 1381 | 49.67% | 49.31% | 48.95% | ATG/T | GTG/T | ATG/T | 0 | 0 | 0 | |
| tRNA-His | H | 11798/11866 | 11841/11909 | 11915/11983 | 69 | 69 | 69 | 36.23% | 34.78% | 30.43% | 0 | 0 | 0 | GTG/H | |||
| tRNA-Ser | H | 1186711934 | 11910/11977 | 11984/12051 | 68 | 68 | 68 | 45.59% | 51.47% | 52.94% | 4 | 5 | 5 | GCT/S | |||
| tRNA-Leu | H | 11939/12011 | 11983/12056 | 12057/12129 | 73 | 74 | 73 | 42.47% | 45.95% | 42.47% | 0 | 1 | 0 | TAG/L | |||
| ND5 | H | 12012/13850 | 12058/13898 | 12130/13968 | 1839 | 1841 | 1839 | 46.28% | 46.50% | 45.89% | ATG/TAA | ATG/TA | ATG/TAA | −4 | 10 | −4 | |
| ND6 | L | 13847/14368 | 13909/14430 | 13965/14486 | 522 | 522 | 522 | 46.55% | 50.00% | 49.23% | ATG/TAG | ATG/TAA | ATG/TAG | 0 | 0 | 0 | |
| tRNA-Glu | L | 14369/14437 | 14431/14500 | 14487/14555 | 69 | 70 | 69 | 46.38% | 48.57% | 42.03% | 4 | 30 | 19 | TTC/E | |||
| CYTB | H | 14442/15582 | 14531/15671 | 14575/15715 | 1141 | 1141 | 1141 | 46.45% | 45.05% | 46.45% | ATG/T | ATG/T | ATG/T | 0 | 0 | 0 | |
| tRNA-Thr | H | 15583/15655 | 15672/15744 | 15716/15787 | 73 | 73 | 72 | 54.79% | 54.79% | 56.94% | −1 | 1 | −1 | TGT/T | |||
| tRNA-Pro | L | 15655/15726 | 15746/15817 | 15787/15859 | 72 | 72 | 73 | 34.72% | 38.89% | 38.36% | 0 | 0 | 0 | TGG/P | |||
| D-loop | H | 15727/16673 | 15818/16728 | 15860/16871 | 947 | 911 | 1012 | 34.21% | 35.35% | 39.03% | 0 | 0 | 0 | ||||
| G. filamentosus | Total Number | G. erythrourus | Total Number | G. decacanthus | Total Number | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | T | G | C | A + T | A | T | G | C | A + T | A | T | G | C | A + T | ||||
| Complete genome | 26.85 | 26.65 | 17.13 | 29.19 | 53.50 | 16,673 | 26.43 | 27.28 | 17.57 | 28.72 | 53.71 | 16,728 | 26.29 | 26.54 | 17.79 | 29.38 | 52.83 | 16,871 |
| Protein-coding genes | ||||||||||||||||||
| first | 24.76 | 21.24 | 26.41 | 27.59 | 46.00 | 3813 | 24.73 | 21.45 | 26.51 | 27.30 | 46.81 | 3813 | 24.52 | 20.72 | 26.78 | 27.98 | 45.24 | 3813 |
| second | 17.96 | 40.64 | 14.12 | 27.28 | 58.60 | 3809 | 18.27 | 13.89 | 40.69 | 27.15 | 58.97 | 3809 | 18.14 | 41.03 | 13.70 | 27.12 | 59.18 | 3809 |
| third | 29.18 | 23.63 | 10.61 | 35.58 | 52.81 | 3808 | 29.12 | 24.65 | 11.04 | 35.19 | 53.77 | 3805 | 27.48 | 23.86 | 11.93 | 36.73 | 51.34 | 3806 |
| total | 23.96 | 28.50 | 17.05 | 30.48 | 52.47 | 11430 | 24.04 | 28.93 | 17.15 | 29.88 | 52.97 | 11427 | 23.38 | 28.54 | 17.47 | 30.61 | 51.92 | 11428 |
| tRNA | 27.37 | 27.44 | 24.04 | 21.15 | 54.81 | 1560 | 29.05 | 25.27 | 21.24 | 24.44 | 54.32 | 1563 | 26.97 | 27.05 | 24.17 | 21.99 | 53.85 | 1560 |
| rRNA | 30.88 | 22.34 | 20.66 | 26.12 | 53.22 | 2672 | 30.66 | 22.13 | 21.80 | 25.41 | 52.79 | 2684 | 29.98 | 22.97 | 21.14 | 25.91 | 52.95 | 2725 |
| D-loop | 34.74 | 31.05 | 14.68 | 19.54 | 65.79 | 947 | 31.72 | 32.93 | 14.05 | 21.30 | 64.65 | 911 | 33.99 | 26.98 | 16.80 | 22.23 | 60.97 | 1012 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, H.; Li, M.; Li, Z.; Huang, J.; Chen, W.; Sun, J.; Liu, L.; Zou, K. Comparative Analysis of Complete Mitochondrial Genomes of Three Gerres Fishes (Perciformes: Gerreidae) and Primary Exploration of Their Evolution History. Int. J. Mol. Sci. 2020, 21, 1874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051874
Ruan H, Li M, Li Z, Huang J, Chen W, Sun J, Liu L, Zou K. Comparative Analysis of Complete Mitochondrial Genomes of Three Gerres Fishes (Perciformes: Gerreidae) and Primary Exploration of Their Evolution History. International Journal of Molecular Sciences. 2020; 21(5):1874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051874
Chicago/Turabian StyleRuan, Huiting, Min Li, Zhenhai Li, Jiajie Huang, Weiyuan Chen, Jijia Sun, Li Liu, and Keshu Zou. 2020. "Comparative Analysis of Complete Mitochondrial Genomes of Three Gerres Fishes (Perciformes: Gerreidae) and Primary Exploration of Their Evolution History" International Journal of Molecular Sciences 21, no. 5: 1874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051874