Antisense Oligonucleotide in LNA-Gapmer Design Targeting TGFBR2—A Key Single Gene Target for Safe and Effective Inhibition of TGFβ Signaling


Abstract
:1. Introduction
2. Results
2.1. Discovery Process
2.2. In Preparation to the First Experimental Round of Screening a Total of 118 Specific and Cross-Species Reactive Antisense Oligonucleotides Against TGFBR2 Were Designed In-Silico
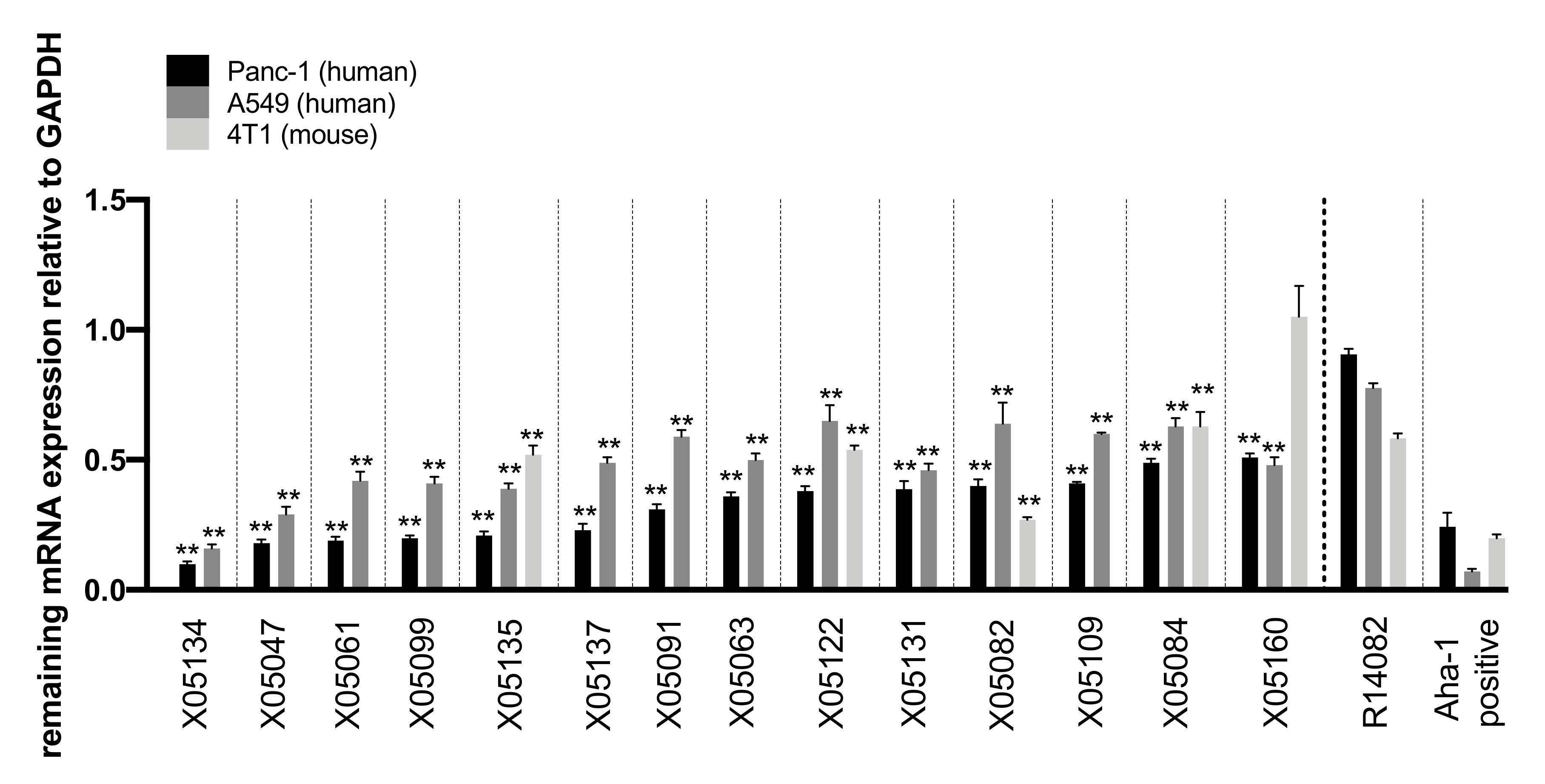
2.3. In-Vitro Screening Identified 14 Highly Active Antisense Oligonucleotides Against TGFBR2
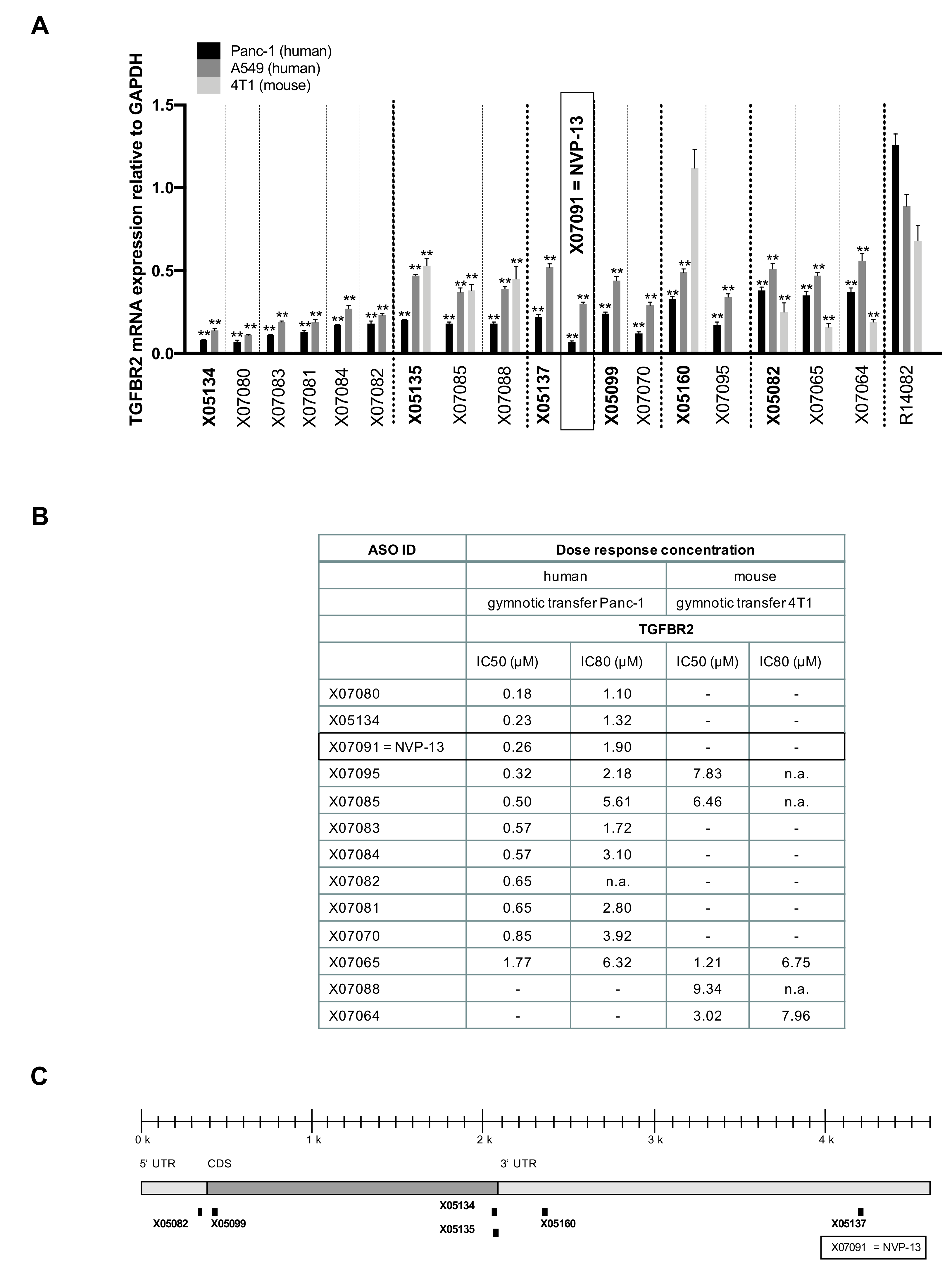
2.4. LNA Pattern Modifications Changed Activity of Antisense Oligonucleotides against TGFBR2
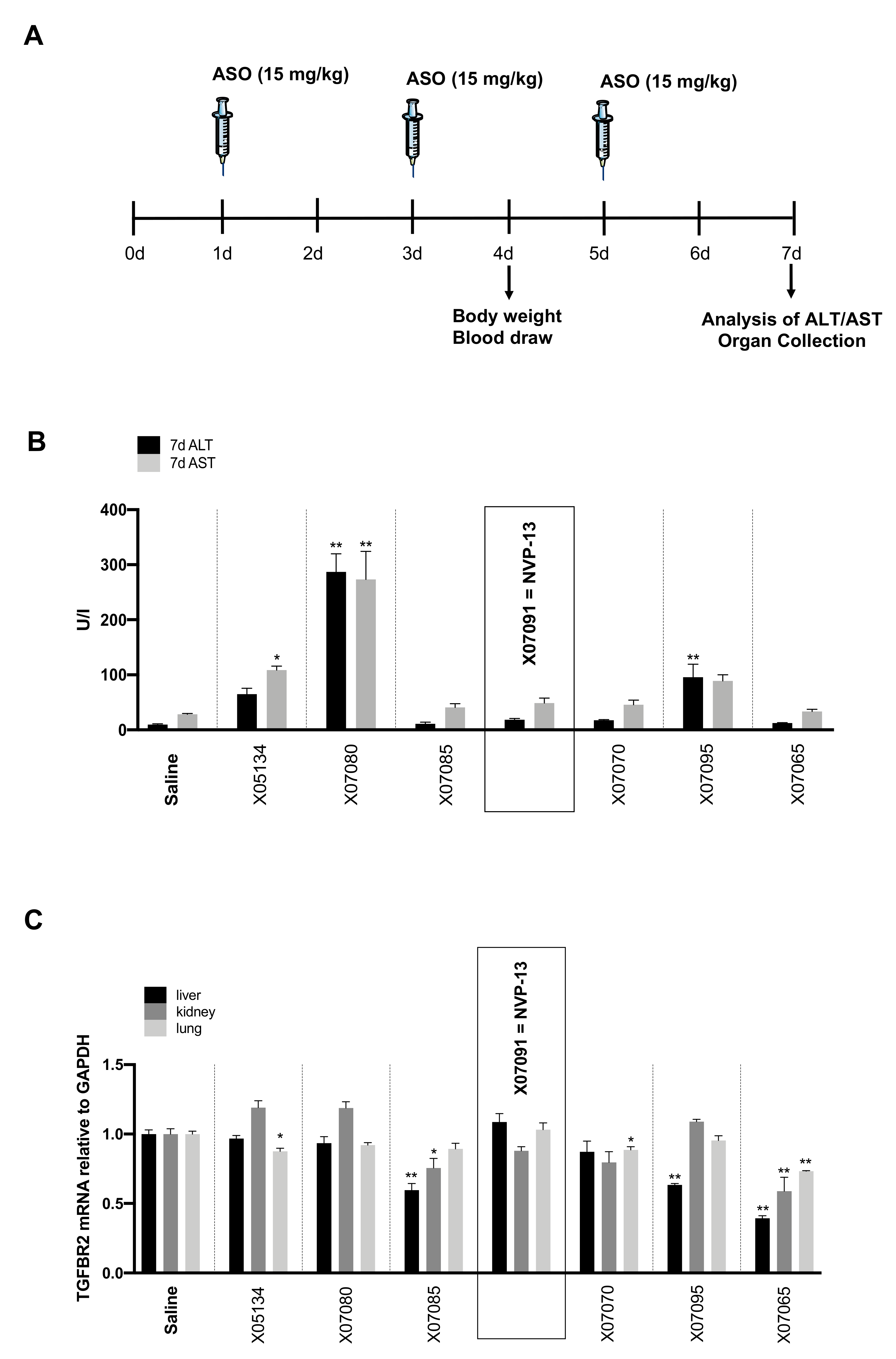
2.5. Early In-Vivo / In-Vitro Toxicity Assessment Indicates Probable Non-Toxic Candidates
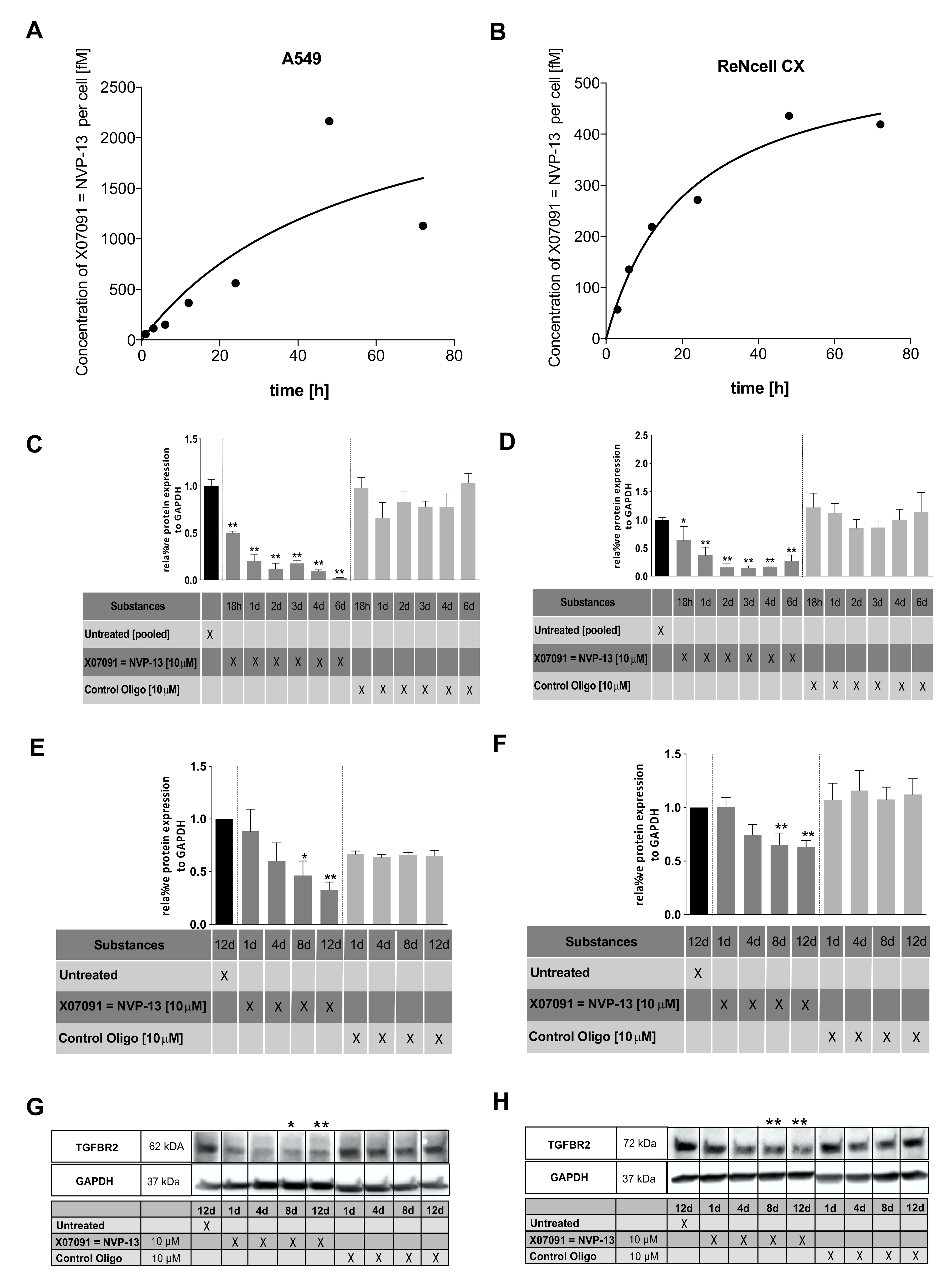
2.6. X07091 = NVP-13 Shows Time-Dependent Cellular Uptake, as well as TGFBR2 mRNA and Protein Downregulation
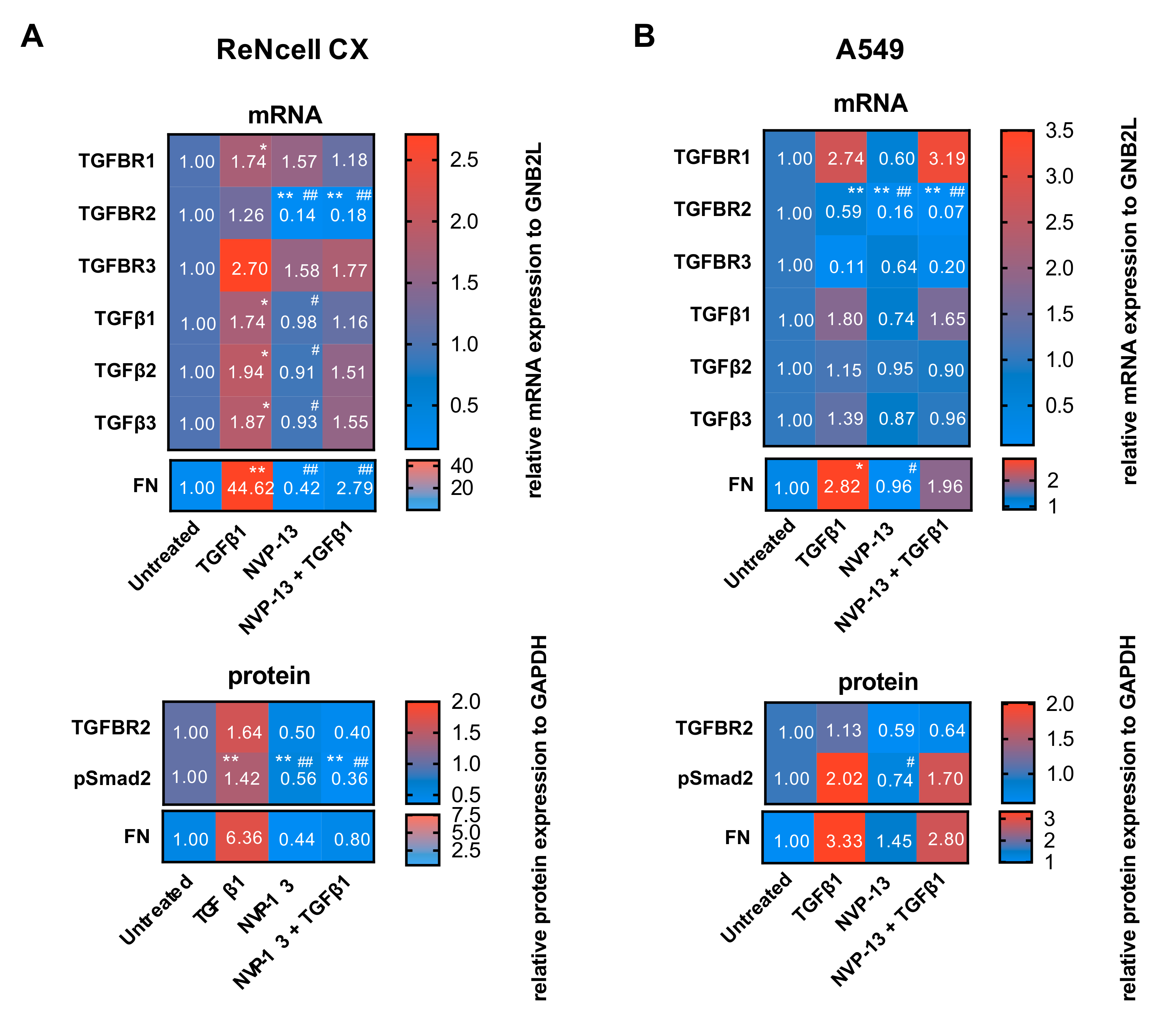
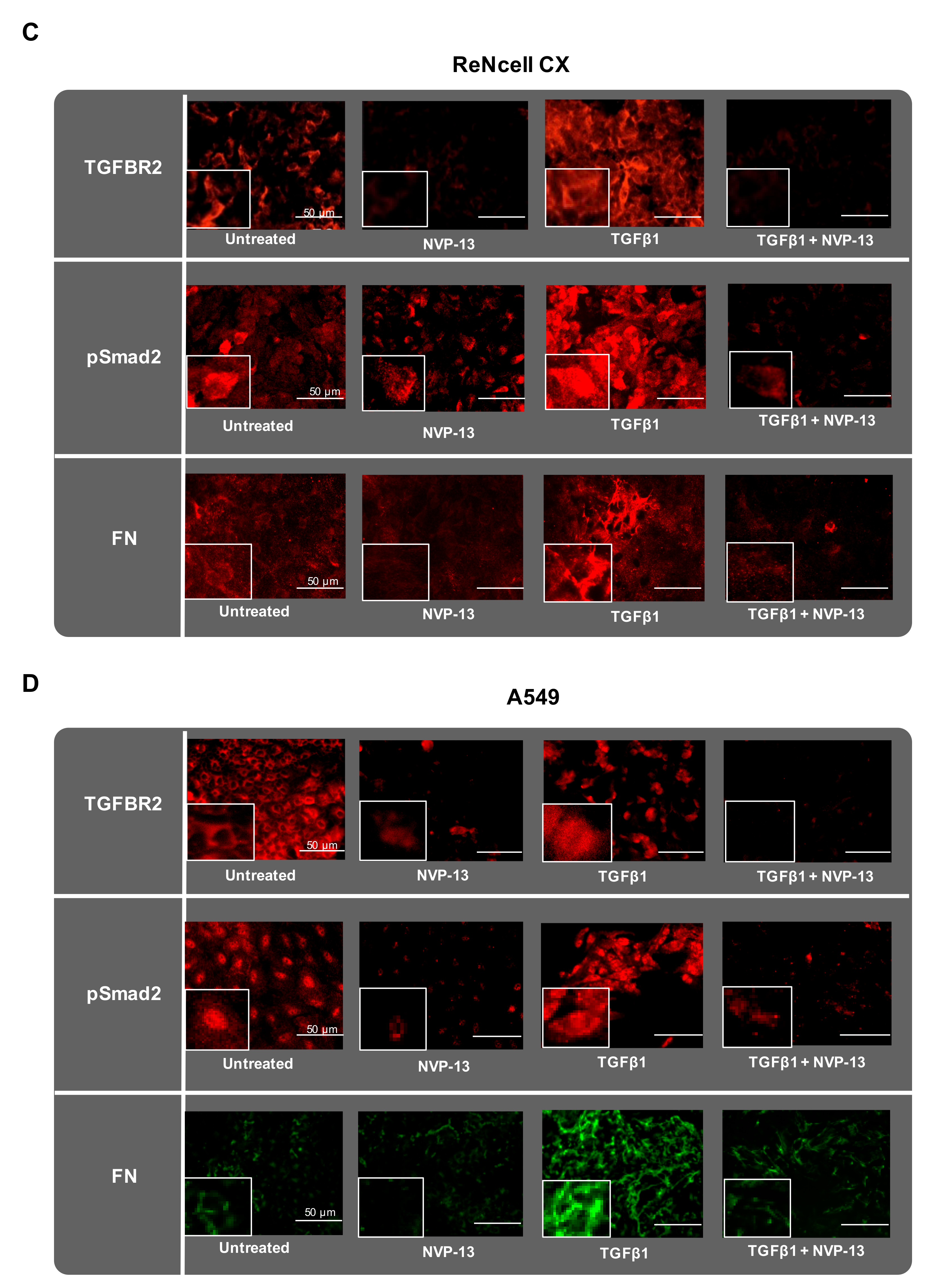
2.7. Gymnotic Transfer of NVP-13 in ReNcell CX® and A549 Cells is Efficacious even in Presence of TGFβ1
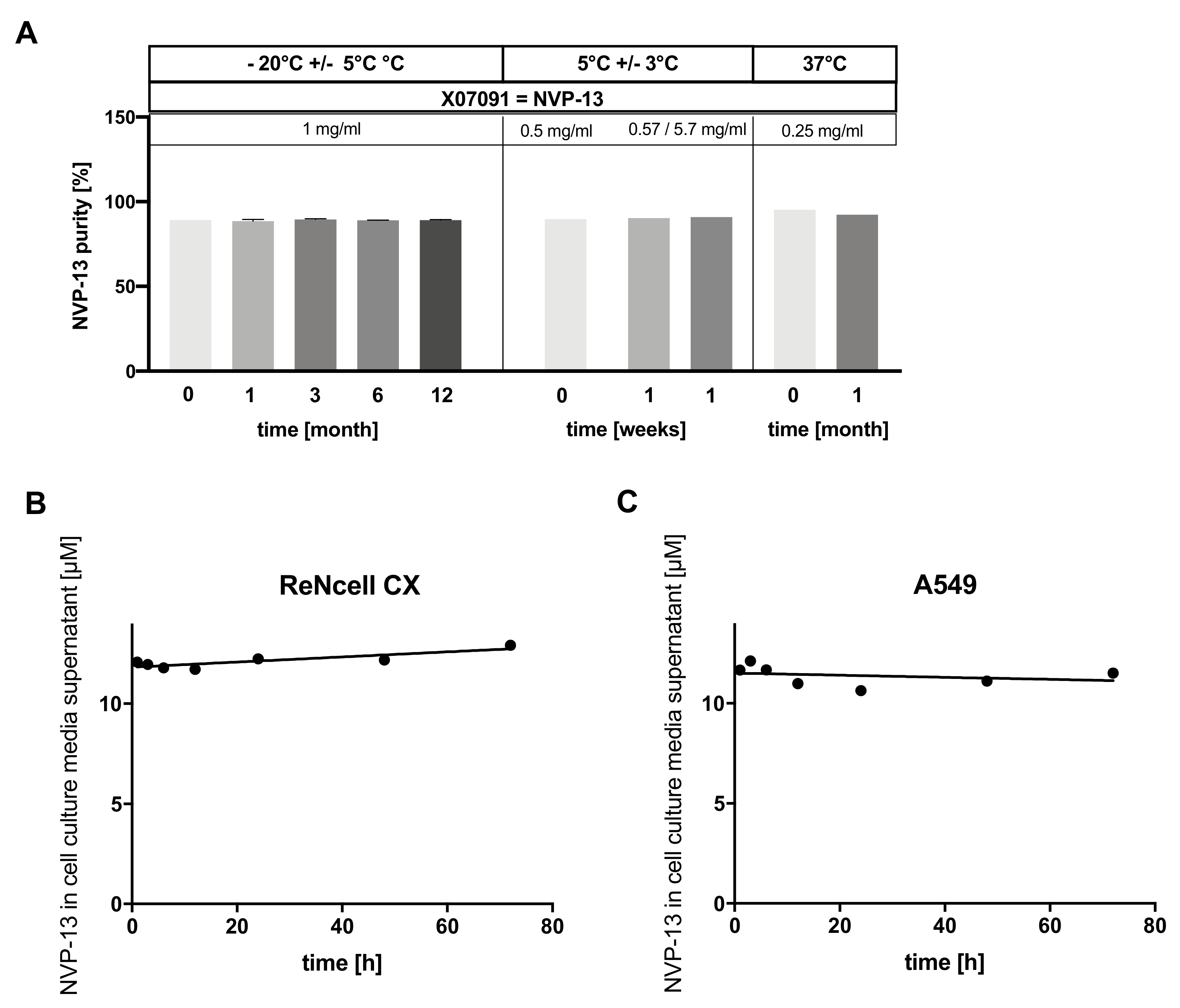
2.8. X07091 = NVP-13 Is Highly Stable in a Broad Range of Different Conditions
3. Discussion
4. Materials and Methods
4.1. TGFBR2 Antisense Oligonucleotide Design
4.2. Source of TGFBR2 Antisense Oligonucleotides
4.3. Cell Culture
4.4. Discovery Process
4.5. Dose-Response Analysis
4.6. Timeline Analysis
4.7. Gymnotic Transfer of ReNcell CX® and A549 Cells in Presence of TGFβ1
4.8. Determination of Cellular X07091 = NVP-13 Uptake
4.9. Branched DNA Assay (bDNA)
4.10. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.11. Western Blot
4.12. Immunocytochemistry
4.13. Peripheral Blood Mononuclear Cell Assay (PBMC Assay)
4.14. In-Vivo Toxicity Study (Mouse Model)
4.15. A bioprobe against X07091 = NVP-13
4.16. Stability
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeVos, S.L.; Miller, T.M. Antisense Oligonucleotides: Treating Neurodegeneration at the Level of RNA. Neurotherapeutics 2013, 10, 486–497. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Toonen, L.J.A.; van Roon-Mom, W.M.C. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoles, D.R.; Pulst, S.M. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 2018, 15, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 2018, 11, 175628641877693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 175628561875445. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.J.A.; Talbot, K.; Bowerman, M. Spinal muscular atrophy: Antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet. 2017, 26, R151–R159. [Google Scholar] [CrossRef]
- Darras, B.T.; Chiriboga, C.A.; Iannaccone, S.T.; Swoboda, K.J.; Montes, J.; Mignon, L.; Xia, S.; Bennett, C.F.; Bishop, K.M.; Shefner, J.M.; et al. Nusinersen in later-onset spinal muscular atrophy. Neurology 2019, 92, e2492–e2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigner, L.; Bogdahn, U. TGF-beta in neural stem cells and in tumors of the central nervous system. Cell Tissue Res. 2007, 331, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Kashima, R.; Hata, A. The role of TGF-β superfamily signaling in neurological disorders. Acta Biochim. Biophys. Sin. 2017, 50, 106–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckwalter, M.S.; Wyss-Coray, T. Modelling neuroinflammatory phenotypes in vivo. J. Neuroinflamm 2004, 1, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Endo, F.; Yamanaka, K. Astrocytic TGF-β1: Detrimental factor in ALS. Oncotarget 2015, 6, 15728–15729. [Google Scholar] [CrossRef]
- Peters, S.; Zitzelsperger, E.; Kuespert, S.; Iberl, S.; Heydn, R.; Johannesen, S.; Petri, S.; Aigner, L.; Thal, D.R.; Hermann, A.; et al. The TGF-β System As a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 669. [Google Scholar] [CrossRef] [Green Version]
- Wachs, F.-P.; Winner, B.; Couillard-Despres, S.; Schiller, T.; Aigner, R.; Winkler, J.; Bogdahn, U.; Aigner, L. Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Khalil, N.; O’Connor, R.N.; Unruh, H.W.; Warren, P.W.; Flanders, K.C.; Kemp, A.; Bereznay, O.H.; Greenberg, A.H. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1991, 5, 155–162. [Google Scholar] [CrossRef]
- Alvarez, M.A.; Freitas, J.P.; Mazher Hussain, S.; Glazer, E.S. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J. Gastrointest Cancer 2019, 50, 207–213. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiong, W.; Xiaofeng, G.; Junfang, W. Transforming growth factor-β1 (TGF-β1) induces mouse precartilaginous stem cell differentiation through TGFRII-CK1ε-β-catenin signalling. Int J. Exp. Path. 2018, 99, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Xi, Q. TGF-β control of stem cell differentiation genes. FEBS Lett. 2012, 586, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-β/Smad signaling. FEBS Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef] [Green Version]
- Orum, H.; Wengel, J. Locked nucleic acids: A promising molecular family for gene-function analysis and antisense drug development. Curr. Opin. Mol. Ther. 2001, 3, 239–243. [Google Scholar]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bondensgaard, K.; Petersen, M.; Singh, S.K.; Rajwanshi, V.K.; Kumar, R.; Wengel, J.; Jacobsen, J.P. Structural studies of LNA:RNA duplexes by NMR: Conformations and implications for RNase H activity. Chemistry 2000, 6, 2687–2695. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; De Hoyos, C.L.; Migawa, M.T.; Vickers, T.A.; Sun, H.; Low, A.; Bell, T.A.; Rahdar, M.; Mukhopadhyay, S.; Hart, C.E.; et al. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019, 37, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Chun, S.J.; Norris, D.A.; Hung, G.; Lee, S.; Matson, J.; Fey, R.A.; Gaus, H.; Hua, Y.; Grundy, J.S.; et al. Pharmacology of a Central Nervous System Delivered 2′- O-Methoxyethyl–Modified Survival of Motor Neuron Splicing Oligonucleotide in Mice and Nonhuman Primates. J. Pharmacol. Exp. Ther. 2014, 350, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense Masking of an hnRNP A1/A2 Intronic Splicing Silencer Corrects SMN2 Splicing in Transgenic Mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN Rx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; Benimetskaya, L.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2009, 38, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 23, 1–13. [Google Scholar] [CrossRef]
- Heindryckx, F.; Li, J.-P. Role of proteoglycans in neuro-inflammation and central nervous system fibrosis. Matrix Biol. 2018, 68, 589–601. [Google Scholar] [CrossRef]
- Tom, V.J.; Doller, C.M.; Malouf, A.T.; Silver, J. Astrocyte-associated fibronectin is critical for axonal regeneration in adult white matter. J. Neurosci. 2004, 24, 9282–9290. [Google Scholar] [CrossRef] [Green Version]
- Györfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68, 8–27. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. IJMS 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors-insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J. Targeting TGF-β Signaling for Therapeutic Gain. Cold Spring Harb. Perspect Biol. 2017, 9, a022301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccines Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Invest. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- Jahangirian, H.; Kalantari, K.; Izadiyan, Z.; Rafiee-Moghaddam, R.; Shameli, K.; Webster, T.J. A review of small molecules and drug delivery applications using gold and iron nanoparticles. Int. J. Nanomed. 2019, 14, 1633–1657. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Kandasamy, M.; Lehner, B.; Kraus, S.; Sander, P.R.; Marschallinger, J.; Rivera, F.J.; Trümbach, D.; Ueberham, U.; Reitsamer, H.A.; Strauss, O.; et al. TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J. Cell. Mol. Med. 2014, 18, 1444–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11, eaaw8283. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bösl, M.; Bosserhoff, A.; Köstler, J.; Wagner, R.; Tamm, E.R.; et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; Ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. 2012, 6, 107–114. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2018, 23, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Küspert, S.; Wirkert, E.; Heydn, R.; Jurek, B.; Johannesen, S.; Hsam, O.; Korte, S.; Ludwig, F.; Mecklenburg, L.; et al. Modulating the neurogenic niche to recondition the central nervous system; Nature Publishing Group: London, UK, 2020. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specific and X-Reactive Candidates | SUM | X05177 and Derivatives | |||
|---|---|---|---|---|---|
| Human and NHP | Human, NHP and Rodent | Human, NHP and Rodent | |||
| Number of Candidates | Number of Candidates | ||||
| 17mer | 13 | 9 | 22 | 17mer | 1 |
| 16mer | 19 | 7 | 26 | 16mer | 2 |
| 15mer | 15 | 3** | 18 | 15mer | - |
| 14mer | 7 | 9 | 16 | 14mer | 2** |
| 13mer | 17 | 4 | 21 | 13mer | - |
| 12mer | 9 | 1 | 10 | 12mer | - |
| Σ | 80 | 33 | 113 | Σ | 5 |
| ID | Length | Position | 5′-3′ Sequence |
|---|---|---|---|
| X05134 | 16 | 2064 | GbsTbsAbsdGsdTsdGsdTsdTsdTsdAsdGsdGsdGsAbsGbsCb |
| X07080 | 16 | 2064 | GbsTbsAbsGbsdTsdGsdTsdTsdTsdAsdGsdGsdGsAbsGbsCb |
| X07083 | 16 | 2064 | GbsTbsAbsdGsdTsdGsdTsdTsdTsdAsdGsdGsdGsdAsGbsCb |
| X07081 | 16 | 2064 | GbsTbsAbsdGsdTsdGsdTsdTsdTsdAsdGsdGsGbsAbsGbsCb |
| X07084 | 16 | 2064 | GbsTbsdAsdGsdTsdGsdTsdTsdTsdAsdGsdGsdGsAbsGbsCb |
| X07082 | 16 | 2064 | GbsTbsAbsGbsdTsdGsdTsdTsdTsdAsdGsdGsGbsAbsGbsCb |
| X05135 | 16 | 2072 | GbsCbsTbsdAsdTsdTsdTsdGsdGsdTsdAsdGsdTsGbsTbsTb |
| X07085 | 16 | 2072 | GbsCbsTbsAbsdTsdTsdTsdGsdGsdTsdAsdGsdTsGbsTbsTb |
| X07088 | 16 | 2072 | GbsCbsTbsdAsdTsdTsdTsdGsdGsdTsdAsdGsdTsdGsTbsTb |
| X05137 | 16 | 4217 | CbsAbsTbsdGsdAsdAsdTsdGsdGsdAsdCsdCsdAsGbsTbsAb |
| X07091 = NVP-13 | 16 | 4217 | CbsAbsTbsdGsdAsdAsdTsdGsdGsdAsdCsdCsAbsGbsTbsAb |
| X05099 | 15 | 429 | CbsGbsAbsdTsdAsdCsdGsdCsdGsdTsdCsdCsAbsCbsAb |
| X07070 | 15 | 429 | CbsGbsAbsTbsdAsdCsdGsdCsdGsdTsdCsdCsAbsCbsAb |
| X05160 | 17 | 2355 | CbsAbsGbsGbsdCsdAsdTsdTsdAsdAsdTsdAsdAsAbsGbsTbsGb |
| X07095 | 17 | 2355 | CbsAbsGbsdGsdCsdAsdTsdTsdAsdAsdTsdAsdAsdAsGbsTbsGb |
| X05082 | 14 | 355 | CbsTbsCbsdGsdTsdCsdAsdTsdAsdGsdAsCbsCbsGb |
| X07065 | 14 | 355 | CbsTbsdCsdGsdTsdCsdAsdTsdAsdGsdAsCbsCbsGb |
| X07064 | 14 | 355 | CbsTbsCbsdGsdTsdCsdAsdTsdAsdGsdAsdCsCbsGb |
| Molecular Formula (Sodium Salt) | C164H183O83N64S15P15Na15 |
|---|---|
| Molecular weight | 5365.3 Da (free acid) / 5693.94 Da (sodium salt) |
| Description | White to off-white powder, odourless |
| Stereochemistry | NVP-13 is a mixture of 215 stereoisomers |
| Hygroscopicity | The drug substance is hygroscopic |
| Crystalline form | Amorphous |
| pH | The pH value of a 5 mg/mL solution of NVP-13 in - aCSF is approximately 7.1 - water for injection (WfI) is approximately 7.2, - isotonic sterile saline (0.9% NaCl) is approx. 6.5. |
| Solubility | The compound is a sodium salt that is soluble in aqueous solution. The solubility is ≥ 30 mg/mL |
| A549 cells | |||
|---|---|---|---|
| Primary Antibody | Dilution | Company | Order Number |
| FN (rabbit) | 1:50 | Proteintech (St. Leon-Rot, Germany) | 15613-1-AP |
| pSmad2 (rabbit) | 1:50 | Cell Signaling (Danvers, MA, USA) | cs3104s |
| TGF-βRII (rabbit) | 1:50 | Millipore (Darmstadt, Germany) | 06-227 |
| Secondary Antibody | Dilution | Company | Order Number |
| Cy3 goat-anti-rabbit | 1:1000 | Life Technologies (Darmstadt, Germany) | A10520 |
| Alexa Fluor 488 | 1:1500 | Life Technologies (Darmstadt, Germany) | A21441 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuespert, S.; Heydn, R.; Peters, S.; Wirkert, E.; Meyer, A.-L.; Siebörger, M.; Johannesen, S.; Aigner, L.; Bogdahn, U.; Bruun, T.-H. Antisense Oligonucleotide in LNA-Gapmer Design Targeting TGFBR2—A Key Single Gene Target for Safe and Effective Inhibition of TGFβ Signaling. Int. J. Mol. Sci. 2020, 21, 1952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061952
Kuespert S, Heydn R, Peters S, Wirkert E, Meyer A-L, Siebörger M, Johannesen S, Aigner L, Bogdahn U, Bruun T-H. Antisense Oligonucleotide in LNA-Gapmer Design Targeting TGFBR2—A Key Single Gene Target for Safe and Effective Inhibition of TGFβ Signaling. International Journal of Molecular Sciences. 2020; 21(6):1952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061952
Chicago/Turabian StyleKuespert, Sabrina, Rosmarie Heydn, Sebastian Peters, Eva Wirkert, Anne-Louise Meyer, Mareile Siebörger, Siw Johannesen, Ludwig Aigner, Ulrich Bogdahn, and Tim-Henrik Bruun. 2020. "Antisense Oligonucleotide in LNA-Gapmer Design Targeting TGFBR2—A Key Single Gene Target for Safe and Effective Inhibition of TGFβ Signaling" International Journal of Molecular Sciences 21, no. 6: 1952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061952