Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance

 ,
, 
Abstract
:1. Introduction
2. Circulating Neutrophils in Glioma Progression and Treatment Resistance
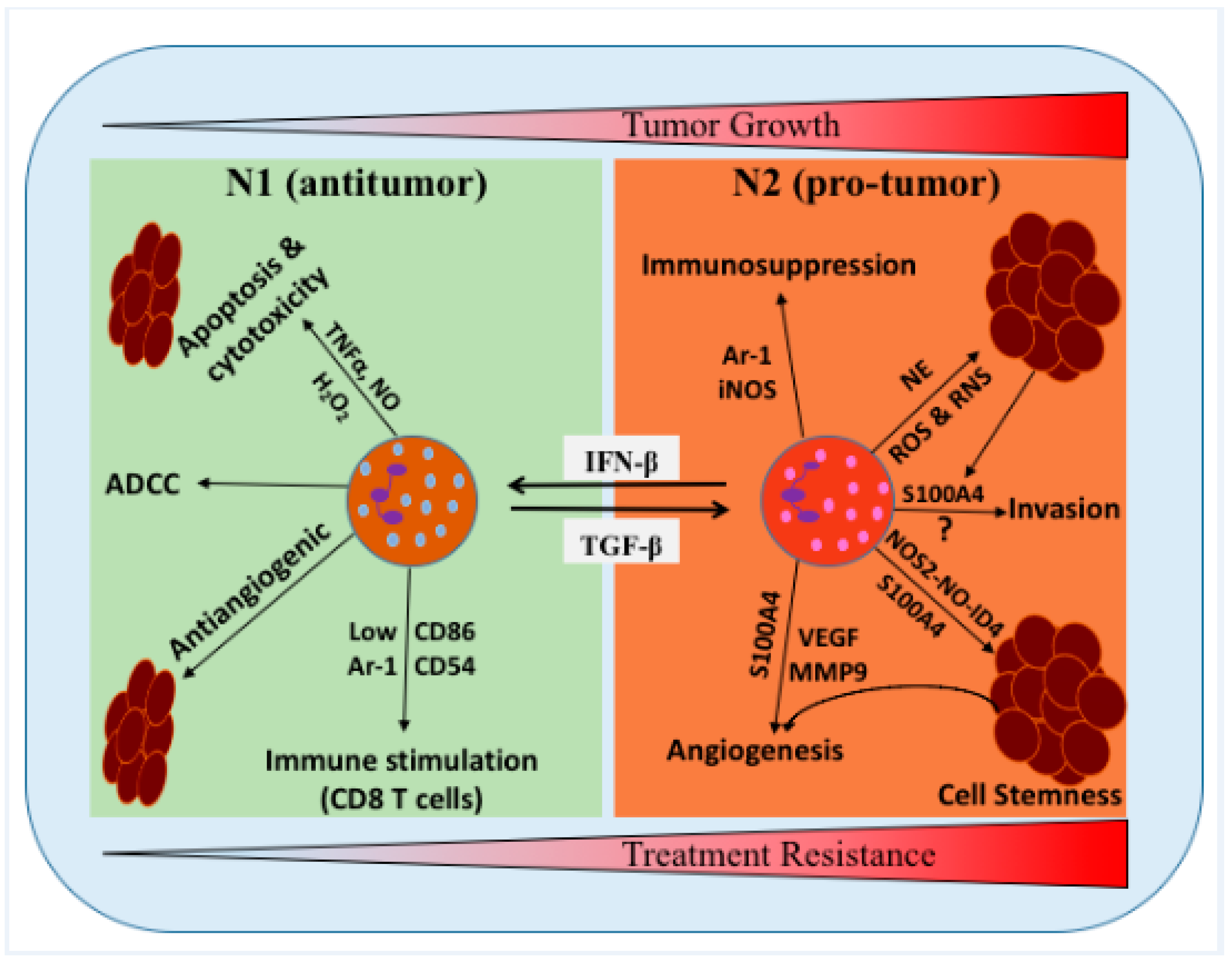
3. TANs in Glioma Progression
4. TANs and Treatment Resistance in Glioma
4.1. Chemotherapy and Anti-VEGF Therapy Resistance
4.2. RT Resistance
4.3. Immunotherapy Resistance
5. MDSCs in Glioma Progression
6. MDSCs and Treatment Resistance in Glioma
6.1. MDSC-Mediated Immunosuppression and Therapy Resistance in Glioma
6.2. MDSCs and RT Resistance
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Arg1 | Arginase-1 |
| GBM | Glioblastoma |
| GM-CSF | Granulocyte macrophage colony stimulating factor |
| GME | Glioma microenvironment |
| GSCs | Glioma stem cells |
| IFN | Interferon |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| MDSCs | Myeloid-derived suppressor cells |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK cells | Natural killer cells |
| NLR | Neutrophil-to-lymphocyte ratio |
| PD-L1 | Programmed death-ligand 1 |
| PMN-MDSCs | Granulocytic polymorphonuclear MDSCs |
| ROS | Reactive oxygen species |
| RT | Radiation therapy |
| STAT-3 | Signal transducer and activator of transcription 3 |
| TANs | Tumor-associated neutrophils |
| TGF-β | Transforming growth factor beta |
| TMZ | Temozolomide |
| VEGF | Vascular endothelial growth factor |
References
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melin, B.S.; Barnholtz-Sloan, J.S.; Wrensch, M.R.; Johansen, C.; Il’yasova, D.; Kinnersley, B.; Ostrom, Q.T.; Labreche, K.; Chen, Y.; Armstrong, G.; et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 2017, 49, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- TCGA Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers (Basel) 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gieryng, A.; Kaminska, B. Myeloid-derived suppressor cells in gliomas. Contemp. Oncol. (Pozn.) 2016, 20, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badie, B.; Schartner, J.M. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 2000, 46, 957–961; discussion 961–962. [Google Scholar]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Verschuere, T.; Toelen, J.; Maes, W.; Poirier, F.; Boon, L.; Tousseyn, T.; Mathivet, T.; Gerhardt, H.; Mathieu, V.; Kiss, R.; et al. Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int. J. Cancer 2014, 134, 873–884. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Ireland, J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology 2011, 13, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Bambury, R.M.; Teo, M.Y.; Power, D.G.; Yusuf, A.; Murray, S.; Battley, J.E.; Drake, C.; O’Dea, P.; Bermingham, N.; Keohane, C.; et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J. Neurooncol. 2013, 114, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Zhou, X.; Niu, X.; Li, J.; Wang, T.; Zhang, H.; Yang, Y.; Liu, Y.; Mao, Q. Neutrophil/Lymphocyte Ratio Is an Independent Prognostic Factor in Elderly Patients with High-Grade Gliomas. World Neurosurg. 2019, 127, e261–e267. [Google Scholar] [CrossRef]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Petersen-Baltussen, H.M.; ter Laan, M.; Wesseling, P.; Adema, G.J. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J. Neuropathol. Exp. Neurol. 2015, 74, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.C.N.A.; Heimberger, A.B. The Role of Myeloid-Derived Suppressor Cells in Immunosuppression in Brain Tumors. In Translational Immunotherapy of Brain Tumors; Sampson, J.H., Ed.; Academic Press: London, UK, 2017; pp. 63–82. [Google Scholar]
- Treffers, L.W.; Hiemstra, I.H.; Kuijpers, T.W.; van den Berg, T.K.; Matlung, H.L. Neutrophils in cancer. Immunol. Rev. 2016, 273, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Cancer-related circulating and tumor-associated neutrophils—Subtypes, sources and function. FEBS J. 2018, 285, 4316–4342. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, W.; Yuan, X.; Fu, M.; Qian, H.; Xu, W. Neutrophils in cancer development and progression: Roles, mechanisms, and implications (Review). Int. J. Oncol. 2016, 49, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M.L. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.K.; Waziri, A. Neutrophil degranulation and immunosuppression in patients with GBM: Restoration of cellular immune function by targeting arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, A.; Cederarv, M.; Wolmer-Solberg, N.; Tammik, C.; Stragliotto, G.; Peredo, I.; Fornara, O.; Xu, X.; Dzabic, M.; Taher, C.; et al. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. Oncoimmunology 2015, 5, e1075693. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neurooncol. 2017, 132, 463–471. [Google Scholar] [CrossRef]
- Deng, Q.; He, B.; Liu, X.; Yue, J.; Ying, H.; Pan, Y.; Sun, H.; Chen, J.; Wang, F.; Gao, T.; et al. Prognostic value of pre-operative inflammatory response biomarkers in gastric cancer patients and the construction of a predictive model. J. Transl. Med. 2015, 13, 66. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Sato, K.; Allegretta, M.; Brocke, S.; Lim, M.; Mitchell, D.J.; Steinman, L. Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Res. 1992, 571, 19–25. [Google Scholar] [CrossRef]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [Green Version]
- Schernberg, A.; Nivet, A.; Dhermain, F.; Ammari, S.; Escande, A.; Pallud, J.; Louvel, G.; Deutsch, E. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin. Transl. Radiat. Oncol. 2018, 10, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangere, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef] [PubMed]
- Quillien, V.; Carpentier, A.F.; Gey, A.; Avril, T.; Tartour, E.; Sejalon, F.; Campillo-Gimenez, B.; Vauleon, E. Absolute numbers of regulatory T cells and neutrophils in corticosteroid-free patients are predictive for response to bevacizumab in recurrent glioblastoma patients. Cancer Immunol. Immunother. CII 2019, 68, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hor, W.S.; Huang, W.L.; Lin, Y.S.; Yang, B.C. Cross-talk between tumor cells and neutrophils through the Fas (APO-1, CD95)/FasL system: Human glioma cells enhance cell viability and stimulate cytokine production in neutrophils. J. Leukoc. Biol. 2003, 73, 363–368. [Google Scholar] [CrossRef]
- Fujita, M.; Scheurer, M.E.; Decker, S.A.; McDonald, H.A.; Kohanbash, G.; Kastenhuber, E.R.; Kato, H.; Bondy, M.L.; Ohlfest, J.R.; Okada, H. Role of type 1 IFNs in antiglioma immunosurveillance—Using mouse studies to guide examination of novel prognostic markers in humans. Clin. Cancer Res. 2010, 16, 3409–3419. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, P.; Lombardi, F.; Siragusa, G.; Dehcordi, S.R.; Luzzi, S.; Cimini, A.; Cifone, M.G.; Cinque, B. Involvement of NOS2 Activity on Human Glioma Cell Growth, Clonogenic Potential, and Neurosphere Generation. Int. J. Mol. Sci. 2018, 19, 2801. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.Y.; Ham, S.W.; Kim, J.K.; Jin, X.; Lee, S.Y.; Shin, Y.J.; Choi, C.Y.; Sa, J.K.; Kim, S.H.; Chun, T.; et al. Ly6G(+) inflammatory cells enable the conversion of cancer cells to cancer stem cells in an irradiated glioblastoma model. Cell Death Differ. 2019, 26, 2139–2156. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Awad, R.M.; De Vlaeminck, Y.; Maebe, J.; Goyvaerts, C.; Breckpot, K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front. Immunol. 2018, 9, 1977. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, R.E.; Hill, Q.A.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.E. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: Potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumor Biol. 2017, 39, 1010428317699797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- See, A.P.; Han, J.E.; Phallen, J.; Binder, Z.; Gallia, G.; Pan, F.; Jinasena, D.; Jackson, C.; Belcaid, Z.; Jeong, S.J.; et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J. Neurooncol. 2012, 110, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef] [Green Version]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, T.; Li, G.; Nagpal, S. History and current state of immunotherapy in glioma and brain metastasis. Ther. Adv. Med. Oncol. 2017, 9, 347–368. [Google Scholar] [CrossRef]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [Green Version]
- Iwatsuki, K.; Kumara, E.; Yoshimine, T.; Nakagawa, H.; Sato, M.; Hayakawa, T. Elastase expression by infiltrating neutrophils in gliomas. Neurol. Res. 2000, 22, 465–468. [Google Scholar] [CrossRef]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef]
- Batich, K.A.; Sampson, J.H. Standard of care and future pharmacological treatment options for malignant glioma: An urgent need for screening and identification of novel tumor-specific antigens. Expert Opin. Pharm. 2014, 15, 2047–2061. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, W.; Ren, X.; Wang, Z.; Liu, X.; Li, G.; Han, S.; Jiang, T.; Wu, A. Tumor Purity as an Underlying Key Factor in Glioma. Clin. Cancer Res. 2017, 23, 6279–6291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology (Williston Park) 2015, 29, 720–727. [Google Scholar] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [Green Version]
- Achyut, B.R.; Shankar, A.; Iskander, A.S.; Ara, R.; Angara, K.; Zeng, P.; Knight, R.A.; Scicli, A.G.; Arbab, A.S. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Lett. 2015, 369, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.Y.; Wang, J.X.; Parisini, E.; Dascher, C.C.; Nigrovic, P.A. Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 2013, 94, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Eun, K.; Jeon, H.M.; Kim, S.O.; Choi, S.H.; Lee, S.Y.; Jin, X.; Kim, S.C.; Kim, H. A cell-autonomous positive-signaling circuit associated with the PDGF-NO-ID4-regulatory axis in glioblastoma cells. Biochem. Biophys. Res. Commun. 2017, 486, 564–570. [Google Scholar] [CrossRef]
- Jeon, H.M.; Kim, S.H.; Jin, X.; Park, J.B.; Kim, S.H.; Joshi, K.; Nakano, I.; Kim, H. Crosstalk between glioma-initiating cells and endothelial cells drives tumor progression. Cancer Res. 2014, 74, 4482–4492. [Google Scholar] [CrossRef] [Green Version]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef] [Green Version]
- Kelley, K.; Knisely, J.; Symons, M.; Ruggieri, R. Radioresistance of Brain Tumors. Cancers (Basel) 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Xue, X.; Zhou, H.; Zhang, G. A molecular view of the radioresistance of gliomas. Oncotarget 2017, 8, 100931–100941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Mildenberger, I.; Bunse, L.; Ochs, K.; Platten, M. The promises of immunotherapy in gliomas. Curr. Opin. Neurol. 2017, 30, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Atai, N.A.; Bansal, M.; Lo, C.; Bosman, J.; Tigchelaar, W.; Bosch, K.S.; Jonker, A.; De Witt Hamer, P.C.; Troost, D.; McCulloch, C.A.; et al. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 2011, 132, 39–48. [Google Scholar] [CrossRef]
- Chang, C.Y.; Tai, J.A.; Li, S.; Nishikawa, T.; Kaneda, Y. Virus-stimulated neutrophils in the tumor microenvironment enhance T cell-mediated anti-tumor immunity. Oncotarget 2016, 7, 42195–42207. [Google Scholar] [CrossRef]
- Youn, J.I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425, author reply 426. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Moses, K.; Trellakis, S.; Lang, S.; Brandau, S. Neutrophils and granulocytic myeloid-derived suppressor cells: Immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol. Immunother. 2012, 61, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol. Immunother 2002, 51, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandruzzato, S.; Brandau, S.; Britten, C.M.; Bronte, V.; Damuzzo, V.; Gouttefangeas, C.; Maurer, D.; Ottensmeier, C.; van der Burg, S.H.; Welters, M.J.; et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: Results from an interim study. Cancer Immunol. Immunother. 2016, 65, 161–169. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.C.; Roggerson, K.M.; Lee, H.C.; Golden-Mason, L.; Rosen, H.R.; Hahn, Y.S. Hepatitis C Virus-Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-gamma Production by Altering Cellular Metabolism via Arginase-1. J. Immunol. 2016, 196, 2283–2292. [Google Scholar] [CrossRef] [Green Version]
- Tumino, N.; Turchi, F.; Meschi, S.; Lalle, E.; Bordoni, V.; Casetti, R.; Agrati, C.; Cimini, E.; Montesano, C.; Colizzi, V.; et al. In HIV-positive patients, myeloid-derived suppressor cells induce T-cell anergy by suppressing CD3zeta expression through ELF-1 inhibition. Aids 2015, 29, 2397–2407. [Google Scholar] [CrossRef]
- Schrijver, I.T.; Theroude, C.; Roger, T. Myeloid-Derived Suppressor Cells in Sepsis. Front. Immunol. 2019, 10, 327. [Google Scholar] [CrossRef] [Green Version]
- Barnie, P.A.; Zhang, P.; Lv, H.; Wang, D.; Su, X.; Su, Z.; Xu, H. Myeloid-derived suppressor cells and myeloid regulatory cells in cancer and autoimmune disorders. Exp. Ther. Med. 2017, 13, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Yong, V.W.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Dubinski, D.; Wolfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 2016, 18, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.F.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008, 83, 1136–1144. [Google Scholar] [CrossRef]
- Kohanbash, G.; McKaveney, K.; Sakaki, M.; Ueda, R.; Mintz, A.H.; Amankulor, N.; Fujita, M.; Ohlfest, J.R.; Okada, H. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013, 73, 6413–6423. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011, 71, 2664–2674. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.-Y.; Ma, J.-W.; Dong, J. Myeloid-derived suppressor cells and nonresolving inflammatory cells in glioma microenvironment: Molecular mechanisms and therapeutic strategies. Glioma 2018, 1, 2–8. [Google Scholar]
- Ding, A.S.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef] [Green Version]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef] [Green Version]
- Cripps, J.G.; Gorham, J.D. MDSC in autoimmunity. Int. Immunopharmacol. 2011, 11, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Perng, P.; Lim, M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front. Oncol. 2015, 5, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G.; et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Angelini, G.; Gardella, S.; Ardy, M.; Ciriolo, M.R.; Filomeni, G.; Di Trapani, G.; Clarke, F.; Sitia, R.; Rubartelli, A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc. Natl. Acad. Sci. USA 2002, 99, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Bunt, S.K.; Clements, V.K.; Hanson, E.M.; Sinha, P.; Ostrand-Rosenberg, S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 2009, 85, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in Radiotherapy for Glioblastoma. Front. Neurol. 2017, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.F.; Maity, A. Radiotherapy and the tumor microenvironment: Mutual influence and clinical implications. Adv. Exp. Med. Biol. 2014, 772, 147–165. [Google Scholar]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Klein, B.; Loven, D.; Lurie, H.; Rakowsky, E.; Nyska, A.; Levin, I.; Klein, T. The effect of irradiation on expression of HLA class I antigens in human brain tumors in culture. J. Neurosurg. 1994, 80, 1074–1077. [Google Scholar] [CrossRef] [Green Version]
- Garnett, C.T.; Palena, C.; Chakraborty, M.; Tsang, K.Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef]
- Olschowka, J.A.; Kyrkanides, S.; Harvey, B.K.; O’Banion, M.K.; Williams, J.P.; Rubin, P.; Hansen, J.T. ICAM-1 induction in the mouse CNS following irradiation. Brain Behav. Immun. 1997, 11, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Nishioka, A.; Hamada, N.; Terashima, M.; Inomata, T.; Yoshida, S.; Seguchi, H.; Kishimoto, S. Expression of fas (CD95/APO-1) antigen induced by radiation therapy for diffuse B-cell lymphoma: Immunohistochemical study. Clin. Cancer Res. 1997, 3 (12 Pt 1), 2211–2216. [Google Scholar]
- Newcomb, E.W.; Demaria, S.; Lukyanov, Y.; Shao, Y.; Schnee, T.; Kawashima, N.; Lan, L.; Dewyngaert, J.K.; Zagzag, D.; McBride, W.H.; et al. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin. Cancer Res. 2006, 12, 4730–4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.; Su, M. Nanoparticle location and material dependent dose enhancement in X-ray radiation therapy. J. Phys. Chem. C Nanomater. Interfaces 2012, 116, 23047–23052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.K.; Chen, W.C.; Lai, S.F.; Liu, C.J.; Wang, C.L.; Wang, C.H.; Chen, H.H.; Hua, T.E.; Cheng, Y.Y.; Wu, M.K.; et al. Enhancement of irradiation effects on cancer cells by cross-linked dextran-coated iron oxide (CLIO) nanoparticles. Phys. Med. Biol. 2010, 55, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Hainfeld, J.F.; Slatkin, D.N.; Smilowitz, H.M. The use of gold nanoparticles to enhance radiotherapy in mice. Phys. Med. Biol. 2004, 49, N309–N315. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Muroski, M.E.; Miska, J.; Lee-Chang, C.; Shen, Y.; Rashidi, A.; Zhang, P.; Xiao, T.; Han, Y.; Lopez-Rosas, A.; et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomedicine 2019, 16, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.; Jaffray, D.; Milosevic, M. Radiation effects on the tumor microenvironment: Implications for nanomedicine delivery. Adv. Drug Deliv. Rev. 2017, 109, 119–130. [Google Scholar] [CrossRef]
- Chiblak, S.; Tang, Z.; Lemke, D.; Knoll, M.; Dokic, I.; Warta, R.; Moustafa, M.; Mier, W.; Brons, S.; Rapp, C.; et al. Carbon irradiation overcomes glioma radioresistance by eradicating stem cells and forming an antiangiogenic and immunopermissive niche. JCI Insight 2019, 4, 1–15. [Google Scholar] [CrossRef]
- Monzen, S.; Yoshino, H.; Kasai-Eguchi, K.; Kashiwakura, I. Characteristics of myeloid differentiation and maturation pathway derived from human hematopoietic stem cells exposed to different linear energy transfer radiation types. PLoS ONE 2013, 8, e59385. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Montero, C.M.; Finke, J.; Montero, A.J. Myeloid-derived suppressor cells in cancer: Therapeutic, predictive, and prognostic implications. Semin. Oncol. 2014, 41, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Schuster, J.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; Desjardins, A.; Ashby, L.S.; et al. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J. Clin. Oncol. 2015, 33 (Suppl. S15), 2009. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; Fu, B.D.; Hsu, F.P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4(+) T-lymphocyte counts. CNS Oncol. 2018, 7, Cns22. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Mechanism/Inference | Test Systems | Specific Cells Used | References |
|---|---|---|---|
| Glioma-derived factors affect circulating neutrophils and influence their infiltration into the tumors | In vivo human | Blood neutrophils and tumor sections | [36] |
| Neutrophils enhance proliferation of GSCs and promote glioma progression and resistance against anti-vascular endothelial growth factor (VEGF) therapy via upregulation of S100A4 | Mixed (in vitro and in vivo in both human and mouse) | Tumor tissue microarray, GSCs and mouse xenografts | [18] |
| Neutrophil degranulation is associated with elevated levels of circulating Arg1, which promotes tumor growth and immunosuppression | In vitro and in vivo human | Blood neutrophils and tumor sections | [37] |
| Increased neutrophil activation levels indicate early signs of tumor progression and provide prognostic value in glioblastoma (GBM) | In vivo human | Blood neutrophils and serum | [38] |
| Immunosuppression within the tumor is driven by the overexpression and production of G-CSF and S100A4 | Mixed (in vitro and in vivo in both human and mouse) | Glioma cells, GSCs and blood samples | [18,41] |
| IL-6 and IL-8 partially mediated by glioma cells have a protective effect on blood neutrophils | In vitro human | Blood neutrophils and glioma cells | [46] |
| Depletion of neutrophils via monoclonal antibody against Ly6G prolongs the survival of mice with developing gliomas | Mixed (in vitro and in vivo in mouse, and in vitro human) | Transgenic mice and patients’ blood | [47] |
| TANs are associated with tumor aggressiveness in mutant-IDH1 glioma | Mixed (in vivo mouse and human) | Transgenic mice, patients tumor tissue and blood cells/RNA | [21] |
| Primary glioma cells sustaining NOS2 activity promote proliferation, migration, and neurosphere generation and represent a prognostic factor for glioma malignancy and recurrence | Human in vitro | Glioma cell lines and primary culture | [48] |
| Radiation-induced infiltrating Ly6G+ neutrophils support the conversion of GBM tumor cells to GSCs via the regulation of nitrosative stress and dedifferentiation (NOS2-NO-ID4) signaling in newly diagnosed/recurrent GBM patients, and this is negatively associated with survival and radiation therapy outcomes | Mixed (in vitro and in vivo in both human and mouse) | Human glioma cell lines, tumor single cells, and glioma mouse models | [49] |
| In a CIBERSORT comparative analysis of immune cell fractions, mesenchymal subtypes of GBM have higher levels of TANs than other subtypes | Human in vitro and in vivo | GSCs and GBM tumor tissue | [50] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061954
Khan S, Mittal S, McGee K, Alfaro-Munoz KD, Majd N, Balasubramaniyan V, de Groot JF. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. International Journal of Molecular Sciences. 2020; 21(6):1954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061954
Chicago/Turabian StyleKhan, Sabbir, Sandeep Mittal, Kain McGee, Kristin D. Alfaro-Munoz, Nazanin Majd, Veerakumar Balasubramaniyan, and John F. de Groot. 2020. "Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance" International Journal of Molecular Sciences 21, no. 6: 1954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061954