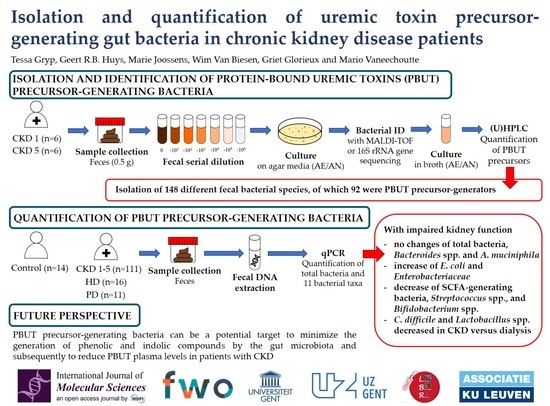
Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
2.1. Culture Media to Isolate Protein-Bound Uremic Toxin Precursor-Generating Bacteria from Fecal Samples
2.2. Isolation and Identification of Protein-Bound Uremic Toxin-Generating Bacteria from Fecal CKD Samples
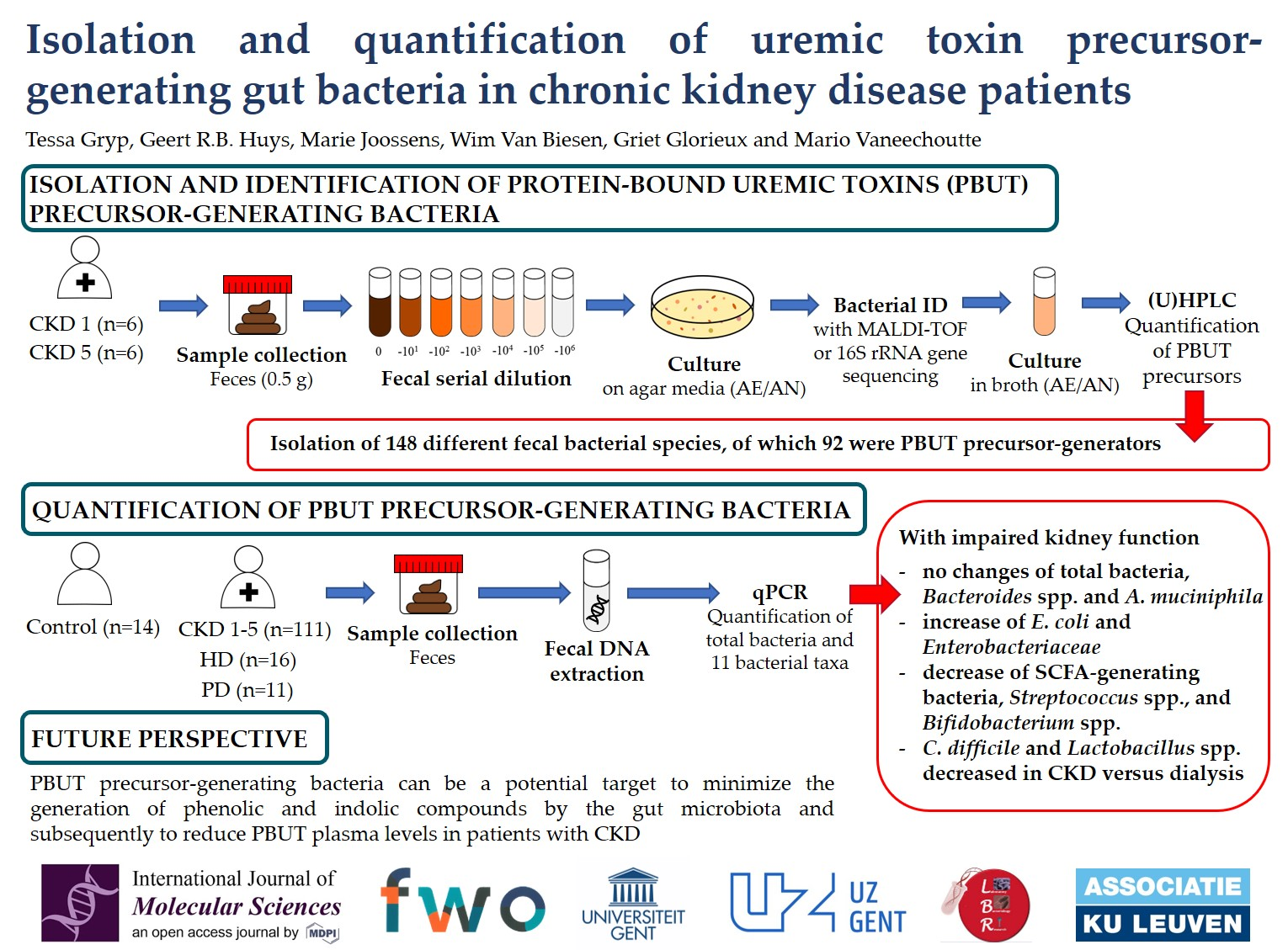
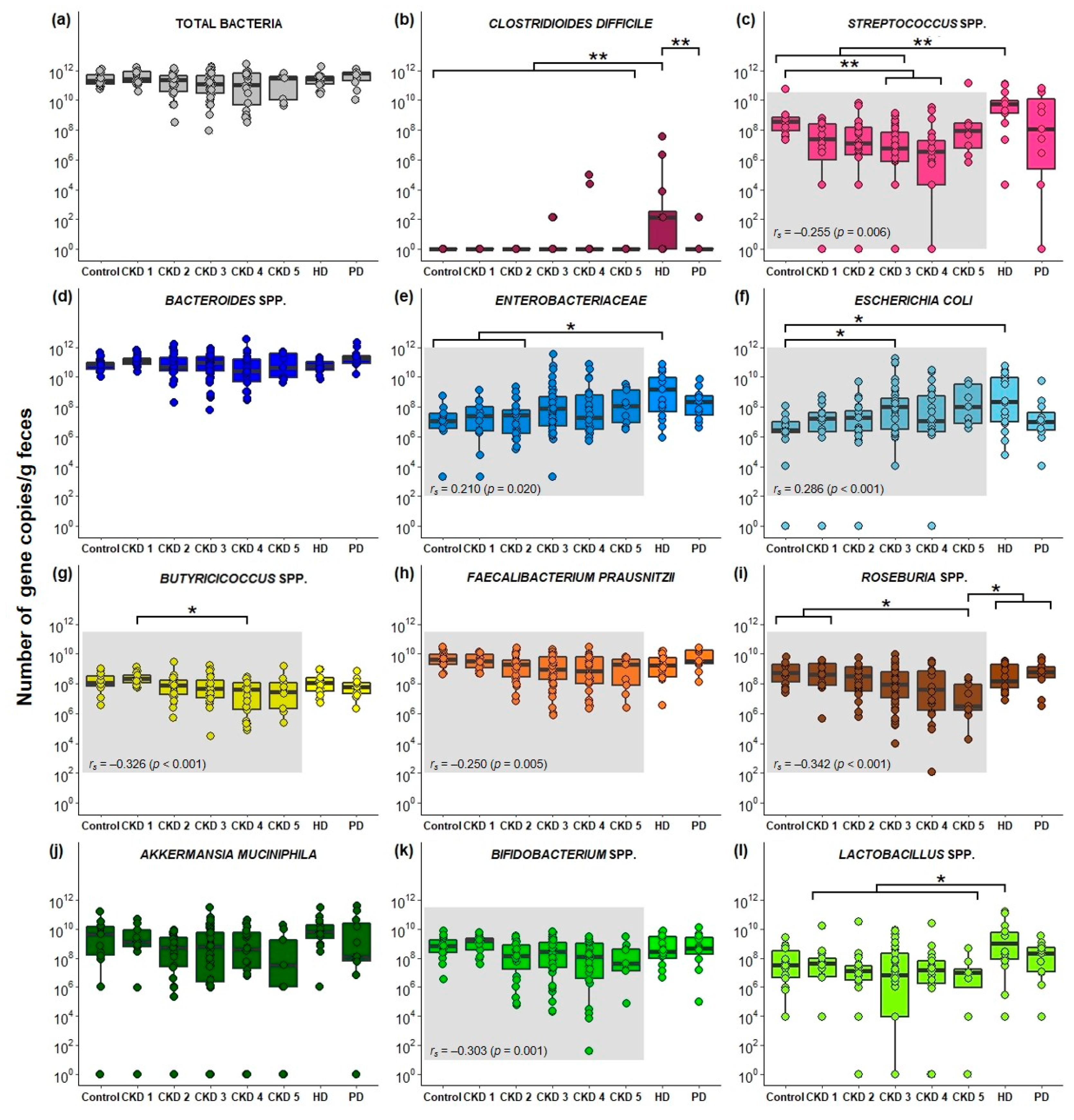
2.3. Quantification of Fecal Bacterial Species in Different Stages Of CKD
3. Discussion
4. Materials and Methods
4.1. Study Population and Sample Collection
4.2. Culture Media
4.3. Isolation of Protein-Bound Uremic Toxin Precursor-Generating Bacteria from CKD Fecal Samples
4.4. Identification of Fecal Protein-Bound Uremic Toxin Precursor-Generating Bacteria
4.5. Quantification of p-Cresol, Phenol, Indole and Indole-3-Acetic Acid in Broth Media
4.6. Quantification of Bacterial Species in Fecal Samples
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | Aromatic amino acids |
| CKD | Chronic kidney disease |
| eGFR | Estimated glomerular filtration rate |
| ESKD | End stage kidney disease |
| FS | Fecal suspension |
| GOS | Galactooligosaccharides |
| HD | Hemodialysis |
| IAA | Indole-3-acetic acid |
| IN | Enriched inulin |
| IxS | Indoxyl sulfate |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| OF | Oligofructose |
| PBUT | Protein-bound uremic toxin |
| pCS | p-Cresyl sulfate |
| pCG | p-Cresyl glucuronide |
| PD | Peritoneal dialysis |
| SCFA | Short chain fatty acid |
| SCH | Schaedler medium |
| YCFAG | Yeast Casitone Fatty Acid Glucose medium |
References
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Glorieux, G.; De Smet, R.; Lameire, N. New insights in uremic toxins. Kidney Int. Suppl. 2003, S6–S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chuang, C.K.; Jayakumar, T.; Liu, H.L.; Pan, C.F.; Wang, T.J.; Chen, H.H.; Wu, C.J. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch. Med. Sci. 2013, 9, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of colon-derived uremic solutes. Clin. J. Am. Soc. Nephrol. 2018, 13, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, S12–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The influence of CKD on colonic microbial metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef]
- De Loor, H.; Bammens, B.; Evenepoel, P.; De Preter, V.; Verbeke, K. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin. Chem. 2005, 51, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Taleb, S. Tryptophan dietary impacts gut barrier and metabolic diseases. Front. Immunol. 2019, 10, 2113. [Google Scholar] [CrossRef]
- Deltombe, O.; Van Biesen, W.; Glorieux, G.; Massy, Z.; Dhondt, A.; Eloot, S. Exploring protein binding of uremic toxins in patients with different stages of chronic kidney disease and during hemodialysis. Toxins 2015, 7, 3933–3946. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of p-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Kuo, C.C.; Chen, Y.M.; Hsu, C.Y.; Lee, W.C.; Tsai, Y.C.; Ng, H.Y.; Kuo, L.C.; Chiou, T.T.; Yang, Y.K.; et al. Factors associated with blood concentrations of indoxyl sulfate and p-cresol in patients undergoing peritoneal dialysis. Perit. Dial. Int. 2010, 30, 456–463. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Wang, F.; Jiang, H.; Liu, H.; Wei, M.; Wang, Z.; Xie, L. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 2014, 59, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Fukuuchi, F.; Hida, M.; Aiba, Y.; Koga, Y.; Endoh, M.; Kurokawa, K.; Sakai, H. Intestinal bacteria-derived putrefactants in chronic renal failure. Clin. Exp. Nephrol. 2002, 6, 99–104. [Google Scholar] [CrossRef]
- Strid, H.; Simren, M.; Stotzer, P.O.; Ringstrom, G.; Abrahamsson, H.; Bjornsson, E.S. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion 2003, 67, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Simenhoff, M.L.; Saukkonen, J.J.; Burke, J.F.; Wesson, L.G., Jr.; Schaedler, R.W.; Gordon, S.J. Bacterial populations of the small intestine in uremia. Nephron 1978, 22, 63–68. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, P.; Jiang, H.; Cheng, S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig. Dis. Sci. 2012, 57, 2856–2862. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.K.; Lai, H.C.; Yu, C.J.; Liang, C.C.; Chang, C.T.; Kuo, H.L.; Yang, Y.F.; Lin, C.C.; Lin, H.H.; Liu, Y.L.; et al. Real-time PCR analysis of the intestinal microbiotas in peritoneal dialysis patients. Appl. Environ. Microbiol. 2012, 78, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [Green Version]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Bourke, E.; Milne, M.D.; Stokes, G.S. Caecal pH and ammonia in experimental uraemia. Gut 1966, 7, 558–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Van der Meulen, R.; Camu, N.; Van Vooren, T.; Heymans, C.; De Vuyst, L. In vitro kinetic analysis of carbohydrate and aromatic amino acid metabolism of different members of the human colon. Int. J. Food Microbiol. 2008, 124, 27–33. [Google Scholar] [CrossRef]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Studies on amine production in the human colon: Enumeration of amine forming bacteria and physiological effects of carbohydrates and pH. Anaerobe 1996, 2, 285–297. [Google Scholar] [CrossRef]
- Elsden, S.R.; Hilton, M.G.; Waller, J.M. The end products of the metabolism of aromatic amino acids by Clostridia. Arch. Microbiol. 1976, 107, 283–288. [Google Scholar] [CrossRef]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.; Campbell, K.L. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): A randomized trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, F.; Taki, K.; Niwa, T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am. J. Kidney Dis. 2003, 41, S142–S145. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2011, 26, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- Guida, B.; Germano, R.; Trio, R.; Russo, D.; Memoli, B.; Grumetto, L.; Barbato, F.; Cataldi, M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: A randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1043–1049. [Google Scholar] [CrossRef]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of increasing dietary fiber on plasma levels of colon-derived solutes in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603–1610. [Google Scholar] [CrossRef] [Green Version]
- De Preter, V.; Ghebretinsae, A.H.; Abrahantes, J.C.; Windey, K.; Rutgeerts, P.; Verbeke, K. Impact of the synbiotic combination of Lactobacillus casei shirota and oligofructose-enriched inulin on the fecal volatile metabolite profile in healthy subjects. Mol. Nutr. Food Res. 2011, 55, 714–722. [Google Scholar] [CrossRef]
- Poesen, R.; Mutsaers, H.A.; Windey, K.; van den Broek, P.H.; Verweij, V.; Augustijns, P.; Kuypers, D.; Jansen, J.; Evenepoel, P.; Verbeke, K.; et al. The influence of dietary protein intake on mammalian tryptophan and phenolic metabolites. PLoS ONE 2015, 10, e0140820. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.; Stewart, C.S.; Flint, H.J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J. Syst Evol. Microbiol. 2002, 52, 2141–2146. [Google Scholar]
- Uniprot. Available online: https://www.uniprot.org/ (accessed on 25 February 2020).
- Aronov, P.A.; Luo, F.J.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.T.; Cummings, J.H.; Allison, C. Protein degradation by human intestinal bacteria. J. Gen. Microbiol. 1986, 132, 1647–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bone, E.; Tamm, A.; Hill, M. The production of urinary phenols by gut bacteria and their possible role in the causation of large bowel cancer. Am. J. Clin. Nutr. 1976, 29, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan metabolism by gut microbiome and gut-brain-axis: An in silico Analysis. Front. Neurosci 2019, 13, 1365. [Google Scholar] [CrossRef] [PubMed]
- Valerio, F.; Lavermicocca, P.; Pascale, M.; Visconti, A. Production of phenyllactic acid by lactic acid bacteria: An approach to the selection of strains contributing to food quality and preservation. FEMS Microbiol. Lett. 2004, 233, 289–295. [Google Scholar] [CrossRef]
- Makras, L.; Triantafyllou, V.; Fayol-Messaoudi, D.; Adriany, T.; Zoumpopoulou, G.; Tsakalidou, E.; Servin, A.; De Vuyst, L. Kinetic analysis of the antibacterial activity of probiotic lactobacilli towards Salmonella enterica serovar Typhimurium reveals a role for lactic acid and other inhibitory compounds. Res. Microbiol. 2006, 157, 241–247. [Google Scholar] [CrossRef]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Joossens, M.; Faust, K.; Gryp, T.; Nguyen, A.T.L.; Wang, J.; Eloot, S.; Schepers, E.; Dhondt, A.; Pletinck, A.; Vieira-Silva, S.; et al. Gut microbiota dynamics and uraemic toxins: One size does not fit all. Gut 2019, 68, 2257–2260. [Google Scholar] [CrossRef]
- Terpstra, M.L.; Sinnige, M.J.; Hugenholtz, F.; Peters-Sengers, H.; Remmerswaal, E.B.; Geerlings, S.E.; Bemelman, F.J. Butyrate production in patients with end-stage renal disease. Int J. Nephrol Renovasc. Dis. 2019, 12, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, M.; Wang, J.; Li, R.; Zhang, Y. Alterations to the gut microbiota and their correlation with inflammatory factors in chronic kidney disease. Front. Cell Infect. Microbiol. 2019, 9, 206. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef]
- Stadlbauer, V.; Horvath, A.; Ribitsch, W.; Schmerbock, B.; Schilcher, G.; Lemesch, S.; Stiegler, P.; Rosenkranz, A.R.; Fickert, P.; Leber, B. Structural and functional differences in gut microbiome composition in patients undergoing haemodialysis or peritoneal dialysis. Sci. Rep. 2017, 7, 15601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.K.; Wu, Y.Y.; Yang, Y.F.; Ting, I.W.; Lin, C.C.; Yen, T.H.; Chen, J.H.; Wang, C.H.; Huang, C.C.; Lin, H.C. The effect of probiotics on serum levels of cytokine and endotoxin in peritoneal dialysis patients: A randomised, double-blind, placebo-controlled trial. Benef. Microbes 2015, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaide, M.A.I.; Singh, R.; Datta, P.; Rewers-Felkins, K.A.; Salguero, M.V.; Al-Obaidi, I.; Kottapalli, K.R.; Vasylyeva, T.L. Gut microbiota-dependent trimethylamine-N-oxide and serum biomarkers in patients with T2DM and advanced CKD. J. Clin. Med. 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facklam, R. What happened to the streptococci: Overview of taxonomic and nomenclature changes. Clin. Microbiol. Rev. 2002, 15, 613–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J. Probiotics and prebiotics: A brief overview. J. Ren. Nutr. 2002, 12, 76–86. [Google Scholar] [CrossRef] [PubMed]
- van Passel, M.W.; Kant, R.; Zoetendal, E.G.; Plugge, C.M.; Derrien, M.; Malfatti, S.A.; Chain, P.S.; Woyke, T.; Palva, A.; de Vos, W.M.; et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PLoS ONE 2011, 6, e16876. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.E. Colonic bacterial metabolites and human health. Curr. Opin. Microbiol 2013, 16, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Eeckhaut, V.; Machiels, K.; Perrier, C.; Romero, C.; Maes, S.; Flahou, B.; Steppe, M.; Haesebrouck, F.; Sas, B.; Ducatelle, R.; et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 2013, 62, 1745–1752. [Google Scholar] [CrossRef]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie van Leeuwenhoek 2016, 109, 1389–1396. [Google Scholar] [CrossRef]
- Pflughoeft, K.J.; Versalovic, J. Human microbiome in health and disease. Ann. Rev. Pathol 2012, 7, 99–122. [Google Scholar] [CrossRef]
- Ramesh, M.S.; Yee, J. Clostridioides difficile infection in chronickidney disease/end-stage renal disease. Adv. Chronic Kidney Dis 2019, 26, 30–34. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Lesaffer, G. p-Cresol: A toxin revealing many neglected but relevant aspects of uraemic toxicity. Nephrol Dial. Transplant. 1999, 14, 2813–2815. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; De Smet, R.; Waterloos, M.A.; Van Landschoot, N.; Vogeleere, P.; Hoste, E.; Ringoir, S. Mechanisms of uremic inhibition of phagocyte reactive species production: Characterization of the role of p-cresol. Kidney Int. 1995, 47, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Cerini, C.; Dou, L.; Anfosso, F.; Sabatier, F.; Moal, V.; Glorieux, G.; De Smet, R.; Vanholder, R.; Dignat-George, F.; Sampol, J.; et al. P-cresol, a uremic retention solute, alters the endothelial barrier function in vitro. Thromb. Haemost. 2004, 92, 140–150. [Google Scholar]
- Yan, Z.; Zhong, H.M.; Maher, N.; Torres, R.; Leo, G.C.; Caldwell, G.W.; Huebert, N. Bioactivation of 4-methylphenol (p-cresol) via cytochrome P450-mediated aromatic oxidation in human liver microsomes. Drug Metab. Dispos. 2005, 33, 1867–1876. [Google Scholar]
- Dou, L.; Cerini, C.; Brunet, P.; Guilianelli, C.; Moal, V.; Grau, G.; De Smet, R.; Vanholder, R.; Sampol, J.; Berland, Y. P-cresol, a uremic toxin, decreases endothelial cell response to inflammatory cytokines. Kidney Int. 2002, 62, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef] [Green Version]
- De Preter, V.; Vanhoutte, T.; Huys, G.; Swings, J.; De Vuyst, L.; Rutgeerts, P.; Verbeke, K. Effects of Lactobacillus casei Shirota, Bifidobacterium breve, and oligofructose-enriched inulin on colonic nitrogen-protein metabolism in healthy humans. Am. J. Physiol. 2007, 292, G358–G368. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, R.; Pechenyak, B.; Vyas, U.; Ranganathan, P.; Weinberg, A.; Liang, P.; Mallappallil, M.C.; Norin, A.J.; Friedman, E.A.; Saggi, S.J. Randomized controlled trial of strain-specific probiotic formulation (Renadyl) in dialysis patients. BioMed Res. Int. 2014, 2014, 568571. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Wei, G.; Baird, B.C.; Murtaugh, M.; Chonchol, M.B.; Raphael, K.L.; Greene, T.; Beddhu, S. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 2012, 81, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Barrios, C.; Beaumont, M.; Pallister, T.; Villar, J.; Goodrich, J.K.; Clark, A.; Pascual, J.; Ley, R.E.; Spector, T.D.; Bell, J.T.; et al. Gut-microbiota-metabolite axis in early renal function decline. PLoS ONE 2015, 10, e0134311. [Google Scholar] [CrossRef] [Green Version]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. MicrobiolyOpen 2019, 8, e00678. [Google Scholar] [CrossRef]
- Snauwaert, E.; Holvoet, E.; Van Biesen, W.; Raes, A.; Glorieux, G.; Vande Walle, J.; Roels, S.; Vanholder, R.; Askiti, V.; Azukaitis, K.; et al. Uremic toxin concentrations are related to residual kidney function in the pediatric hemodialysis population. Toxins 2019, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Gryp, T.; Glorieux, G.; Joossens, M.; Vaneechoutte, M. Comparison of five assays for DNA extraction from bacterial cells in human faecal samples. J. Appl. Microbiol. 2020. [Google Scholar] [CrossRef]
- Bergstrom, A.; Licht, T.R.; Wilcks, A.; Andersen, J.B.; Schmidt, L.R.; Gronlund, H.A.; Vigsnaes, L.K.; Michaelsen, K.F.; Bahl, M.I. Introducing GUt low-density array (GULDA): A validated approach for qPCR-based intestinal microbial community analysis. FEMS Microbiol. Lett. 2012, 337, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.C.; Derrien, M.; Isolauri, E.; de Vos, W.M.; Salminen, S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol. 2007, 73, 7767–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.; McKay, L.; Williams, D.; Garrett, V.; Gentry, R.; Sayler, G. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl. Environ. Microbiol. 2006, 72, 4214–4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauther, K.A.; Cobaugh, K.L.; Jantz, L.M.; Sparer, T.E.; DeBruyn, J.M. Estimating time since death from postmortem human gut microbial communities. J. Forensic Sci. 2015, 60, 1234–1240. [Google Scholar] [CrossRef]
- Kubota, H.; Sakai, T.; Gawad, A.; Makino, H.; Akiyama, T.; Ishikawa, E.; Oishi, K. Development of TaqMan-based quantitative PCR for sensitive and selective detection of toxigenic Clostridium difficile in human stools. PLoS ONE 2014, 9, e111684. [Google Scholar] [CrossRef]
- Bartosch, S.; Fite, A.; Macfarlane, G.T.; McMurdo, M.E. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl. Environ. Microbiol. 2004, 70, 3575–3581. [Google Scholar] [CrossRef] [Green Version]
- Chern, E.C.; Siefring, S.; Paar, J.; Doolittle, M.; Haugland, R.A. Comparison of quantitative PCR assays for Escherichia coli targeting ribosomal RNA and single copy genes. Lett. Appl. Microbiol. 2011, 52, 298–306. [Google Scholar] [CrossRef]
- Ramirez-Farias, C.; Slezak, K.; Fuller, Z.; Duncan, A.; Holtrop, G.; Louis, P. Effect of inulin on the human gut microbiota: Stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br. J. Nutr. 2009, 101, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.F.; Cao, W.W.; Cerniglia, C.E. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl. Environ. Microbiol. 1996, 62, 1242–1247. [Google Scholar] [CrossRef] [Green Version]
- Rinttila, T.; Kassinen, A.; Malinen, E.; Krogius, L.; Palva, A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl. Microbiol. 2004, 97, 1166–1177. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sorensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Collado, M.C.; Delgado, S.; Maldonado, A.; Rodriguez, J.M. Assessment of the bacterial diversity of breast milk of healthy women by quantitative real-time PCR. Lett. Appl. Microbiol. 2009, 48, 523–528. [Google Scholar] [CrossRef]
- Forootan, A.; Sjoback, R.; Bjorkman, J.; Sjogreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | CKD 1 (n = 6) | CKD 5 (n = 6) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Neg | pC | Phe | Ind | IAA | Neg | pC | Phe | Ind | IAA | |
| ACTINOBACTERIA | ||||||||||
| Actinomycetaceae | ||||||||||
| Actinomyces sp. | O4 | O4 | ||||||||
| Bifidobacteriaceae | ||||||||||
| Bifidobacterium adolescentis | N9 | N1 | ||||||||
| Bifidobacterium bifidum c | N1 | N2 | ||||||||
| Bifidobacterium catenulatum/ pseudocatenulatum a | N9 | N2 | ||||||||
| Bifidobacterium dentium | N1 | N1 | ||||||||
| Bifidobacterium longum c | N6 | N5 | ||||||||
| Parascardovia denticolens | NC | |||||||||
| Scardovia wiggsiae | N1 | |||||||||
| Brevibacteriaceae | ||||||||||
| Brevibacterium ravenspurgense | O1 | |||||||||
| Coriobacteriaceae | ||||||||||
| Collinsella aerofaciens | O1 | N4 | ||||||||
| Corynebacteriaceae | ||||||||||
| Corynebacterium amycolatum | O1 | O1 | ||||||||
| Corynebacterium aurimucosum | NC | NC | ||||||||
| Corynebacterium coyleae | O3 | |||||||||
| Eggerthellaceae | ||||||||||
| Eggerthella lenta | N3 | |||||||||
| Microbacteriaceae | ||||||||||
| Microbacterium sp. | O2 | |||||||||
| Micrococcaceae | ||||||||||
| Kocuria sp. | O1 | |||||||||
| Pseudoglutamicibacter albus | O1 | |||||||||
| Propionibacteriaceae | ||||||||||
| Cutibacterium acnes | N2 | N2 | N1 | N1 | ||||||
| Propionibacterium freudenreichii | N1 | |||||||||
| BACTEROIDETES | ||||||||||
| Bacteroidaceae | ||||||||||
| Bacteroides sp. | N1 | N1 | N1 | N1 | N1 | |||||
| Bacteroides caccae | N1 | N1 | N1 | N1 | N1 | N1 | ||||
| Bacteroides cellulosilyticus | N1 | N1 | ||||||||
| Bacteroides coprocola | N1 | N1 | ||||||||
| Bacteroides eggerthii | N1 | N1 | N1 | N1 | N1 | N1 | ||||
| Bacteroides faecis | N1 | N1 | ||||||||
| Bacteroides finegoldii | N1 | N1 | N1 | |||||||
| Bacteroides fragilis | N1 | N1 | N1 | O1 | O1 | O1 | ||||
| Bacteroides intestinalis | NC | |||||||||
| Bacteroides massiliensis | N1 | N1 | ||||||||
| Bacteroides ovatus | N1 | N1 | N1 | N1 | N1 | |||||
| Bacteroides pyogenes | N1 | N1 | ||||||||
| Bacteroides salyersiae | N1 | N1 | ||||||||
| Bacteroides stercoris | NC | |||||||||
| Bacteroides thetaiotaomicron | N2 | N2 | ||||||||
| Bacteroides uniformis | N1 | N1 | N1 | N1 | N2 | N2 | N4 | N3 | ||
| Bacteroides vulgatus | N2 | N1 | N2 | N1 | N1 | |||||
| Odoribacteriaceae | ||||||||||
| Odoribacter splanchnicus | N3 | N3 | N3 | N3 | ||||||
| Prevotellaceae | ||||||||||
| Prevotella copri | N1 | N1 | ||||||||
| Rikenellaceae | ||||||||||
| Alistipes putredinis | N1 | N1 | ||||||||
| Alistipes shahii | N2 | N2 | ||||||||
| Tannerellaceae | ||||||||||
| Parabacteroides distasonis | N2 | N2 | ||||||||
| Parabacteroides goldsteinii | N1 | N1 | ||||||||
| Parabacteroides johnsonii | N1 | N1 | ||||||||
| Parabacteroides merdae | N1 | N2 | N3 | |||||||
| FUSOBACTERIA | ||||||||||
| Fusobacteriaceae | ||||||||||
| Fusobacterium sp. | N1 | N1 | N1 | |||||||
| FIRMICUTES | ||||||||||
| Acidaminococcaceae | ||||||||||
| Acidaminococcus intestine | N1 | |||||||||
| Bacillaceae | ||||||||||
| Bacillus amyloliquefaciens | N1 | N1 | N1 | |||||||
| Bacillus cereus | O5 | |||||||||
| Bacillus licheniformis | O1 | |||||||||
| Bacillus pumilus | O1 | |||||||||
| Bacillus sonorensis | O1 | |||||||||
| Bacillus subtilis | O1 | O1 | ||||||||
| Clostridiaceae | ||||||||||
| Clostridium perfringens | N1 | N1 | ||||||||
| Clostridium tertium | O1 | O1 | ||||||||
| Enterococcaceae | ||||||||||
| Enterococcus avium | O1 | O1 | ||||||||
| Enterococcus casseliflavus | O1 | O1 | O1 | |||||||
| Enterococcus durans d | O2 | |||||||||
| Enterococcus faecalis d | O2 | O2 | ||||||||
| Enterococcus faecium d | O9 | N1 | N1 | O3 | N2 | |||||
| Erysipelotrichaceae | ||||||||||
| Coprobacillus cateniformis | NC | |||||||||
| Catenibacterium mitsuokai | N1 | |||||||||
| Erysipelatoclostridium ramosum c | N2 | |||||||||
| Lachnospiraceae | ||||||||||
| Lachnoclostridium scindens | N1 | N1 | ||||||||
| Lachnoclostridium symbiosum c | N1 | N1 | N1 | |||||||
| Lactobacillaceae | ||||||||||
| Lactobacillus buchneri | NC | |||||||||
| Lactobacillus delbrueckii c | N1 | |||||||||
| Lactobacillus paracasei | NC | NC | ||||||||
| Lactobacillus rhamnosus | O1/N1 | O1/N1 | ||||||||
| Lactobacillus ruminis | NC | |||||||||
| Lactobacillus sakei | NC | |||||||||
| Pediococcus acidilactici c | O1 | O1 | ||||||||
| Oscillospiraceae | ||||||||||
| Oscillibacter sp. | N1 | N1 | N1 | N1 | ||||||
| Ruminococcaceae | ||||||||||
| Flavonifractor plautii | NC | |||||||||
| Staphylococcaceae | ||||||||||
| Staphylococcus capitis | N1 | N1 | ||||||||
| Staphylococcus epidermidis | N1 | N1 | ||||||||
| Staphylococcus haemolyticus | O2 | O2 | O2 | |||||||
| Staphylococcus hominis | NC | |||||||||
| Streptococcaceae | ||||||||||
| Lactococcus lactis | NC | |||||||||
| Streptococcus agalactiae | O1 | |||||||||
| Streptococcus anginosus | NC | |||||||||
| Streptococcus gordonii | O1 | O1 | ||||||||
| Streptococcus lutetiensis | O1/N2 | |||||||||
| Streptococcus oralis/mitis a | O3 | |||||||||
| Streptococcus parasanguinis | NC | |||||||||
| Streptococcus sanguinis/ australis b | N1 | N1 | ||||||||
| Streptococcus vestibularis/ salivarius a | O1 | O1 | O3 | |||||||
| Veillonellaceae | ||||||||||
| Megasphaera elsdenii | NC | NC | ||||||||
| Veillonella atypica | NC | |||||||||
| Veillonella dispar | NC | |||||||||
| PROTEOBACTERIA | ||||||||||
| Caulobacteraceae | ||||||||||
| Brevundimonas diminuta | O1 | |||||||||
| Desulfovibrionaceae | ||||||||||
| Bilophila wadsworthia | N1 | N1 | ||||||||
| Enterobacteriaceae | ||||||||||
| Citrobacter freundii | O1 | O1 | ||||||||
| Enterobacter cloacae/kobei/ludwigii /xiangfangensis/hormaechei a | O6/N1 | |||||||||
| Escherichia coli * | O7/N1 | O7/N1 | O15/N4 | O15/N4 | ||||||
| Klebsiella variicola c | O1 | O1 | ||||||||
| Pseudomonadaceae | ||||||||||
| Pseudomonas aeruginosa | O1 | O1 | ||||||||
| Pseudomonas stutzeri | O1 | |||||||||
| Sutterellaceae | ||||||||||
| Sutterella wadsworthensis | NC | |||||||||
| Xanthomonodaceae | ||||||||||
| Stenotrophomonas maltophilia | NC | |||||||||
| NO IDENTIFICATION° | ||||||||||
| MB 208_49 | N1 | N1 | N1 | N1 | ||||||
| MB 210_6 | N1 | |||||||||
| MB 210_38 | N1 | N1 | ||||||||
| MB 242_16 | N1 | |||||||||
| MB 256_36 | N1 | |||||||||
| MB 256_39 | N1 | N1 | ||||||||
| MB 256_61 | N1 | N1 | N1 | |||||||
| MB 263_32 | N1 | |||||||||
| MB 275_15 | N1 | N1 | ||||||||
| MB 275_16 | N1 | |||||||||
| MB 300_21 | N1 | N1 | ||||||||
| MB 314_01 | O1 | O1 | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2020, 21, 1986. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061986
Gryp T, Huys GRB, Joossens M, Van Biesen W, Glorieux G, Vaneechoutte M. Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients. International Journal of Molecular Sciences. 2020; 21(6):1986. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061986
Chicago/Turabian StyleGryp, Tessa, Geert R.B. Huys, Marie Joossens, Wim Van Biesen, Griet Glorieux, and Mario Vaneechoutte. 2020. "Isolation and Quantification of Uremic Toxin Precursor-Generating Gut Bacteria in Chronic Kidney Disease Patients" International Journal of Molecular Sciences 21, no. 6: 1986. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061986