The Role of Autophagy and Autophagy Receptor NDP52 in Microbial Infections

Abstract
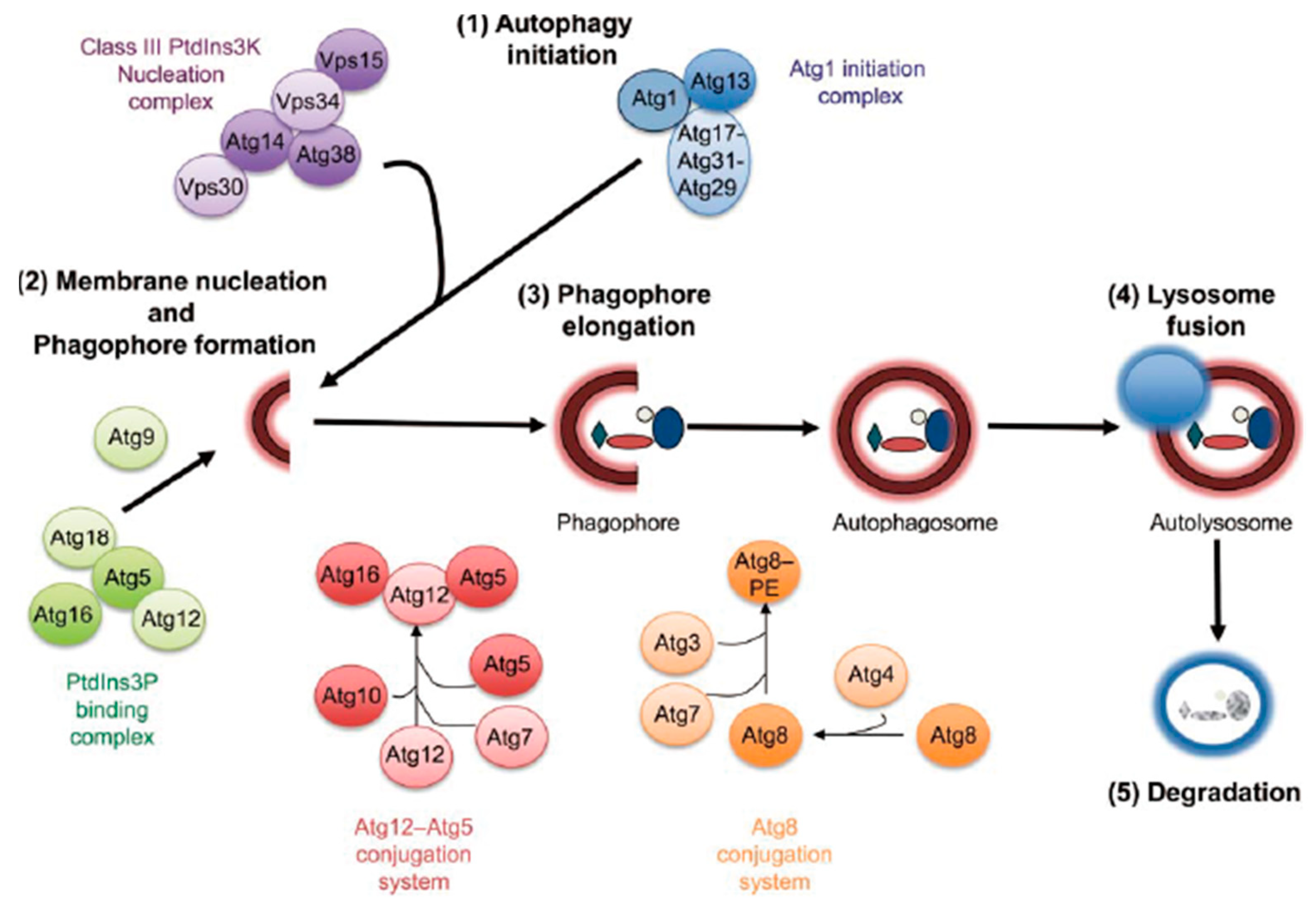
:1. Introduction
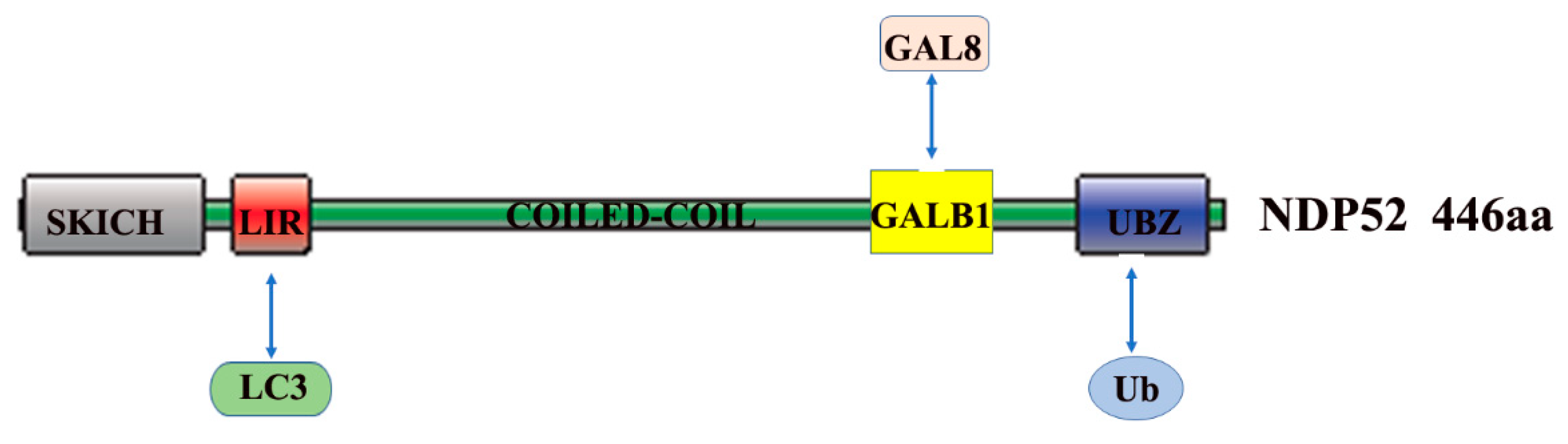
2. The Role and Mechanism of Autophagy and NDP52 in Bacterial Infections
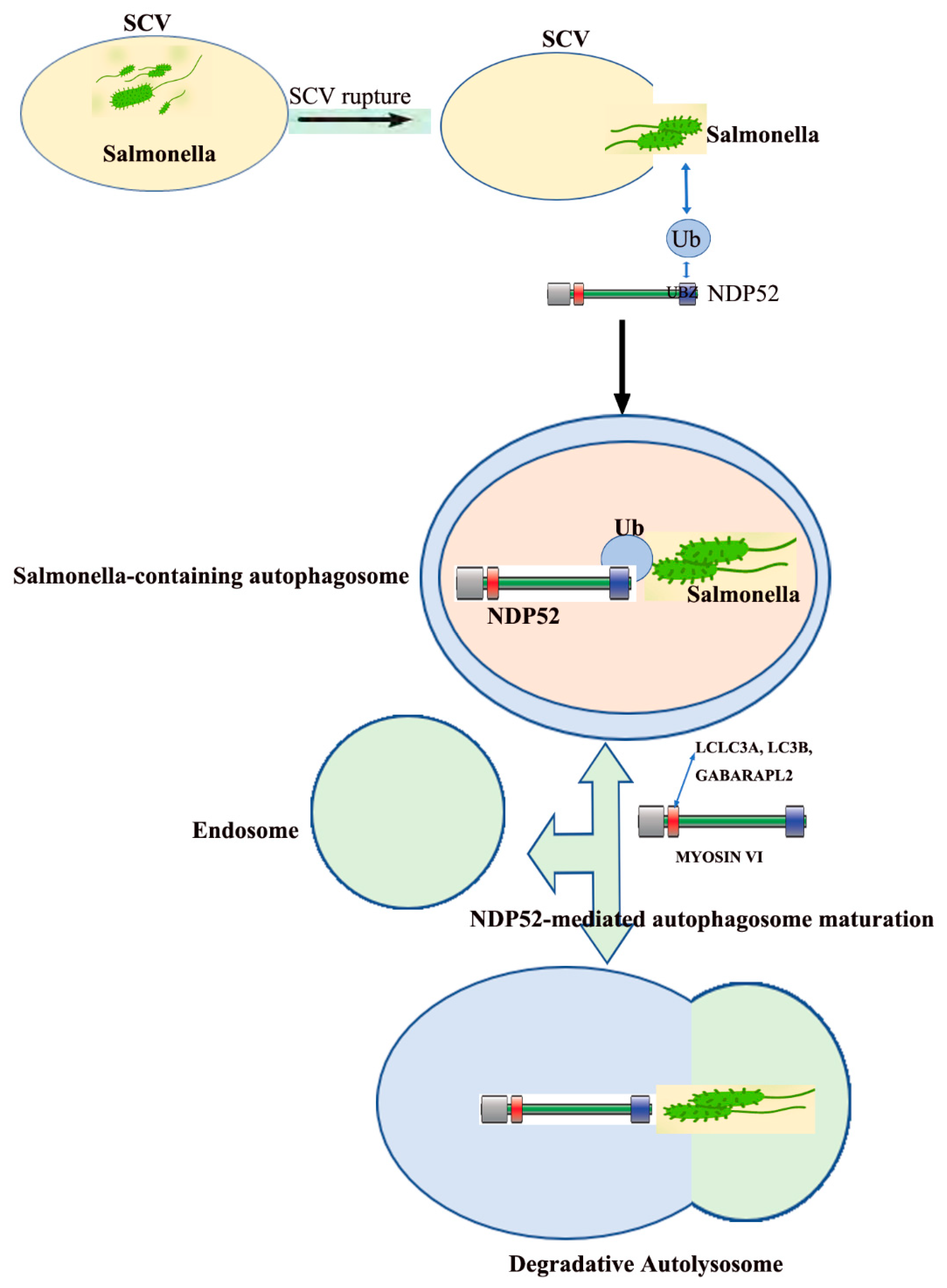
3. The Role of NDP52 in Salmonella Infections
4. The Role of NDP52 in Shigella Infections
5. The Role of NDP52 in Listeria Infections
6. The Role of NDP52 in Streptococcus Infections
7. The Role and Mechanism of NDP52 in Viral Infections
Funding
Conflicts of Interest
References
- Cemma, M.; Brumell, J.H. Interactions of Pathogenic Bacteria with Autophagy Systems. Curr. Biol. 2012, 22, R540–R545. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth Factor Regulation of Autophagy and Cell Survival in the Absence of Apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Ruth Scherz-Shouval, E.S.; Ephraim Fass, H.S., Lidor Gil; Elazar, A.Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Eur. Mol. Biol. Organ. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers atop. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [Green Version]
- Jäättelä, M.H.-H.M. Autophagy An emerging target for cancer therapy. Autophagy 2008, 4, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Gelino, S. Autophagy-An Emerging Anti-Aging Mechanism. J. Clin. Exp. Pathol. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Cossart, S.M.A.P. Bacterial autophagy restriction or promotion of bacterial replication? Trends Cell Biol. 2012, 22, 283–291. [Google Scholar] [CrossRef]
- Ricci, V. Relationship between VacA Toxin and Host Cell Autophagy in Helicobacter pylori Infection of the Human Stomach: A Few Answers, Many Questions. Toxins 2016, 8, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam Lam, R.P.; Gross, C.M.; Riesenberg, L.A.; Vinodkumar, S.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung. Cell Mol. Physiol. 2017, 313, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killian, M.S. Dual role of autophagy in HIV-1 replication and pathogenesis. AIDS Res. Ther. 2012, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingjie Xie, Z.Y.; Liu, Y.; Zheng, M. The role of HBV-Induced autophagy in HBV replication and HBV related-HCC. Life Sci. 2018, 205, 107–112. [Google Scholar] [CrossRef]
- Pei, J.; Zhao, M.; Ye, Z.; Gou, H.; Wang, J.; Yi, L.; Dong, X.; Liu, W.; Luo, Y.; Liao, M.; et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy 2013, 10, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Deng, J.; Ye, Z.; Wang, J.; Gou, H.; Liu, W.; Zhao, M.; Liao, M.; Yi, L.; Chen, J. Absence of autophagy promotes apoptosis by modulating the ROS-Dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy 2016, 12, 1738–1758. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Kuo, C.-J.; Hansen, M.; Troemel, E. Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 2018, 14, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Sumpter, R.; Levine, B. Autophagy and innate immunity: Triggering, targeting and tuning. Semin. Cell Dev. Biol. 2010, 21, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.-D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-Like Receptor 4 Is a Sensor for Autophagy Associated with Innate Immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhang, Y.; Liu, L.; Zhang, T.; Han, F.; Cleveland, J.; Wang, F.; McKeehan, W.L.; Li, Y.; Zhang, D. MAP1S Protein Regulates the Phagocytosis of Bacteria and Toll-Like Receptor (TLR) Signaling. J. Biol. Chem. 2016, 291, 1243–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachel Cooney, J.B.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.P.; Barry, J.; Campbell, D.J.A.S. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2009, 16, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, H.; Travassos, L.A.M.C.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; João, G.; Magalhães, L.Y.; Soares, F.; Chea, E.; le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–63. [Google Scholar] [CrossRef]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [Green Version]
- Thomas Sternsdorf, K.J.; Züchner, D.; Will, H. Cellular localization, expression, and structure of the nuclear dot protein 52. J. Cell Biol. 1997, 138, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; Liu, J.; Wang, Y.; Xie, X.; Hu, S.; Pan, L. Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc. Natl. Acad. Sci. USA 2018, 115, E11651–E11660. [Google Scholar] [CrossRef] [Green Version]
- Von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, Á.; Bloor, S.; Rutherford, T.J.; Freund Stefan, M.V.; Komander, D.; Randow, F. LC3C, Bound Selectively by a Noncanonical LIR Motif in NDP52, Is Required for Antibacterial Autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Ivanov, S.; Roy, C.R. NDP52: The missing link between ubiquitinated bacteria and autophagy. Nat. Immunol. 2009, 10, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Do Hoon Kwon, A.H.K.S. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol. Cells 2018, 41, 27–34. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome–Lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2012, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.L.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-Linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Viret, C.; Faure, M. Dual function of CALCOCO2/NDP52 during xenophagy. Autophagy 2015, 11, 965–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, T.L.M.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-Coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Morriswood, B.; Ryzhakov, G.; Puri, C.; Arden, S.D.; Roberts, R.; Dendrou, C.; Kendrick-Jones, J.; Buss, F. T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J. Cell Sci. 2007, 120, 2574–2585. [Google Scholar] [CrossRef] [Green Version]
- Verlhac, P.; Grégoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy Receptor NDP52 Regulates Pathogen-Containing Autophagosome Maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Shintani, T. Autophagy in Health and Disease A Double Edged Sword. Science 2004, 306, 990–995. [Google Scholar]
- Viret, C.; Rozières, A.; Faure, M. Novel Insights into NDP52 Autophagy Receptor Functioning. Trends Cell Biol. 2018, 28, 255–257. [Google Scholar] [CrossRef]
- Evans, R.J.; Sundaramurthy, V.; Frickel, E.-M. The Interplay of Host Autophagy and Eukaryotic Pathogens. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Sudhakar, P.; Jacomin, A.-C.; Hautefort, I.; Samavedam, S.; Fatemian, K.; Ari, E.; Gul, L.; Demeter, A.; Jones, E.; Korcsmaros, T.; et al. Targeted interplay between bacterial pathogens and host autophagy. Autophagy 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Sorbara, M.T.; Philpott, D.J.; Girardin, S.E. Bacterial autophagy. Autophagy 2014, 8, 1848–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. p62 and NDP52 Proteins Target IntracytosolicShigellaandListeriato Different Autophagy Pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Muhlinen, N.; Thurston, T.; Ryzhako, G.; Bloor, S.; Randow, F. NDP52, a novel autophagy receptor for ubiquitin-Decorated cytosolic bacteria. Autophagy 2014, 6, 288–289. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.A.; Neilson, A.A. Typhoid and paratyphoid fever in travellers. Aust. Fam. Physician 2010, 39, 847–851. [Google Scholar]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; O′Brien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M. The Global Burden of NontyphoidalSalmonellaGastroenteritis. Clin. Infect. Dis. 2010, 50, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Arjyal, A.; Basnyat, B.; Koirala, S.; Karkey, A.; Dongol, S.; Agrawaal, K.K.; Shakya, N.; Shrestha, K.; Sharma, M.; Lama, S.; et al. Gatifloxacin versus chloramphenicol for uncomplicated enteric fever: An open-Label, randomised, controlled trial. Lancet Infect. Dis. 2011, 11, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Chitnis, C.E.; de Jong, H.K.; Parry, C.M.; van der Poll, T.; Wiersinga, W.J. Host–Pathogen Interaction in Invasive Salmonellosis. PLoS Pathog. 2012, 8, e1002933. [Google Scholar] [CrossRef]
- Radtke, A.L.; O′Riordan, M.X.D. Homeostatic maintenance of pathogen-Containing vacuoles requires TBK1-dependent regulation ofaquaporin-1. Cell. Microbiol. 2008, 10, 2197–2207. [Google Scholar] [CrossRef]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy Controls Salmonella Infection in Response to Damage to theSalmonella-Containing Vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef] [Green Version]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2014, 9, 1256–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Autophagy Proteins in Viral Exocytosis and Anti-Viral Immune Responses. Viruses 2017, 9, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320.e326–329.e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.; Sansonetti, P.J.; Marteyn, B.S. Shigella Diversity and Changing Landscape: Insights for the Twenty-First Century. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.M.; Girardin, S.; Philpott, D.J. Nod proteins link bacterial sensing and autophagy. Autophagy 2014, 6, 409–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martineza, J.; Johann, A.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartnerb, M.O.; Green, D. 1. Microtubule-Associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Pro. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-Associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Krokowski, S.; Mostowy, S. Bacterial cell division is recognized by the septin cytoskeleton for restriction by autophagy. Autophagy 2019, 15, 937–939. [Google Scholar] [CrossRef]
- Krokowski, S.; Lobato-Márquez, D.; Mostowy, S. Mitochondria promote septin assembly into cages that entrap Shigella for autophagy. Autophagy 2017, 14, 913–914. [Google Scholar] [CrossRef] [Green Version]
- Campbell-Valois, F.-X.; Sachse, M.; Sansonetti, P.J.; Parsot, C.; Casadevall, A. Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. mBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Chakravortty, D.; Baxt, L.A.; Goldberg, M.B. Host and Bacterial Proteins That Repress Recruitment of LC3 to Shigella Early during Infection. PLoS ONE 2014, 9, e94653. [Google Scholar] [CrossRef] [Green Version]
- Krokowski, S.; Mostowy, S. Interactions between Shigella flexneri and the Autophagy Machinery. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.-K.; Yuk, J.-M.; Shin, D.-M.; Sasakawa, C. Roles of Autophagy in Elimination of Intracellular Bacterial Pathogens. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association Between Variants of PRDM1 and NDP52 and Crohn’s Disease, Based on Exome Sequencing and Functional Studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Thurston, T.L.M.; Boyle, K.B.; Allen, M.; Ravenhill, B.J.; Karpiyevich, M.; Bloor, S.; Kaul, A.; Noad, J.; Foeglein, A.; Matthews, S.A.; et al. Recruitment of TBK1 to cytosol-Invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J. 2016, 35, 1779–1792. [Google Scholar] [CrossRef]
- Boyle, K.B.; Ravenhill, B.J.; Randow, F. CALCOCO2/NDP52 initiates selective autophagy through recruitment of ULK and TBK1 kinase complexes. Autophagy 2019, 15, 1655–1656. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, M.d.S.; Ribeiro, R.d.M.; Travassos, L.H. Autophagy and Its Interaction With Intracellular Bacterial Pathogens. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nat. Microbiol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Braga, V.; Vázquez, S.; Vico, V.; Pastorino, V.; Mota, M.I.; Legnani, M.; Schelotto, F.; Lancibidad, G.; Varela, G. Prevalence and serotype distribution of Listeria monocytogenes isolated from foods in Montevideo-Uruguay. Braz. J. Microbiol. 2017, 48, 689–694. [Google Scholar] [CrossRef]
- Vazquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Dominguez-Bernal, G.; Goebel, W.; Gonzalez-Zorn, B.; Wehland, J.; Kreft, J. Listeria Pathogenesis and Molecular Virulence Determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2017, 16, 32–46. [Google Scholar] [CrossRef]
- Cossart, P.; Vicente, F.; Mengaud, J.; Baquero, F.; Perez-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes direct evidence obtained by gene complementation. Infect. Immun. 1989. [Google Scholar] [CrossRef] [Green Version]
- Birmingham, C.L.; Higgins, D.E.; Brumell, J.H. Avoiding death by autophagy: Interactions ofListeria monocytogeneswith the macrophage autophagy system. Autophagy 2014, 4, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Freitag, N.E.; Cemma, M.; Lam, G.Y.; Stöckli, M.; Higgins, D.E.; Brumell, J.H. Strain-Specific Interactions of Listeria monocytogenes with the Autophagy System in Host Cells. PLoS ONE 2015, 10, e0125856. [Google Scholar] [CrossRef] [Green Version]
- Py, B.F.; Lipinski, M.M.; Yuan, J. Autophagy LimitsListeria monocytogenesIntracellular Growth in the Early Phase of Primary Infection. Autophagy 2014, 3, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A.; Roy, C.R. Avoidance of Autophagy Mediated by PlcA or ActA Is Required for Listeria monocytogenes Growth in Macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Ogawa, M.; Hain, T.; Yoshida, M.; Fukumatsu, M.; Kim, M.; Mimuro, H.; Nakagawa, I.; Yanagawa, T.; Ishii, T.; et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 2009, 11, 1233–1240. [Google Scholar] [CrossRef]
- Thompson, K.M.; Sterkel, A.K.; McBride, J.A.; Corliss, R.F. The Shock of Strep: Rapid Deaths Due to Group a Streptococcus. Acad. Forensic Pathol. 2018, 8, 136–149. [Google Scholar] [CrossRef]
- Ralph, A.P.; Carapetis, J.R. Group A Streptococcal Diseases and Their Global Burden. Curr. Top. Microbiol. Immunol. 2012, 368, 1–27. [Google Scholar] [CrossRef]
- Nakagawa, I. Autophagy Defends Cells Against Invading Group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Minowa-Nozawa, A.; Nozawa, T.; Okamoto-Furuta, K.; Kohda, H.; Nakagawa, I. Rab35 GTPase recruits NDP52 to autophagy targets. EMBO J. 2017, 36, 2790–2807. [Google Scholar] [CrossRef]
- Barnett, T.C.; Liebl, D.; Seymour, L.M.; Gillen, C.M.; Lim, J.Y.; LaRock, C.N.; Davies, M.R.; Schulz, B.L.; Nizet, V.; Teasdale, R.D.; et al. The Globally Disseminated M1T1 Clone of Group A Streptococcus Evades Autophagy for Intracellular Replication. Cell Host Microbe 2013, 14, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Takada, N.; Shizukuishi, S.; Tomokiyo, M.; Chang, B.; Yoshida, M.; Kakuta, S.; Tanida, I.; Ryo, A.; Guan, J.-L.; et al. Streptococcus pneumoniae triggers hierarchical autophagy through reprogramming of LAPosome-Like vesicles via NDP52-Delocalization. Commun. Biol. 2020, 3. [Google Scholar] [CrossRef] [Green Version]
- Korioth, F.; Gieffers, C.; Maul, G.G.; Frey, J. Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. J. Cell Biol. 1995. [Google Scholar] [CrossRef]
- Conenello, G.M.; Palese, P. Influenza A Virus PB1-F2: A Small Protein with a Big Punch. Cell Host Microbe 2007, 2, 207–209. [Google Scholar] [CrossRef] [Green Version]
- Leymarie, O.; Meyer, L.; Tafforeau, L.; Lotteau, V.; Costa, B.D.; Delmas, B.; Chevalier, C.; Le Goffic, R. Influenza virus protein PB1-F2 interacts with CALCOCO2 (NDP52) to modulate innate immune response. J. Gen. Virol. 2017, 98, 1196–1208. [Google Scholar] [CrossRef]
- Furuse, Y.; Oshitani, H. Global Transmission Dynamics of Measles in the Measles Elimination Era. Viruses 2017, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Joubert, P.-E.; Meiffren, G.; Grégoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.-O.; Vidal, M.; Lotteau, V.; et al. Autophagy Induction by the Pathogen Receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef]
- Deretic, V.; Richetta, C.; Grégoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained Autophagy Contributes to Measles Virus Infectivity. PLoS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef]
- Petkova, D.; Verlhac, P.; Rozières, A.; Baguet, J.; Claviere, M.; Kretz-Remy, C.; Mahieux, R.; Viret, C.; Faure, M. Distinct Contributions of Autophagy Receptors in Measles Virus Replication. Viruses 2017, 9, 123. [Google Scholar] [CrossRef]
- Inomata, M.; Niida, S.; Shibata, K.-I.; Into, T. Regulation of Toll-Like receptor signaling by NDP52-Mediated selective autophagy is normally inactivated by A20. Cell. Mol. Life Sci. 2011, 69, 963–979. [Google Scholar] [CrossRef] [Green Version]
- Baillet, N.; Krieger, S.; Journeaux, A.; Caro, V.; Tangy, F.; Vidalain, P.-O.; Baize, S. Autophagy Promotes Infectious Particle Production of Mopeia and Lassa Viruses. Viruses 2019, 11, 293. [Google Scholar] [CrossRef] [Green Version]
- Mohamud, Y.; Qu, J.; Xue, Y.C.; Liu, H.; Deng, H.; Luo, H. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2018, 26, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prévost, M.-C.; Pierre, P.; et al. Species-Specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol. Cell 2017, 68, 308.e304–322.e304. [Google Scholar] [CrossRef] [Green Version]
- Becher, P.; Avalos Ramirez, R.; Orlich, M.; Cedillo Rosales, S.; König, M.; Schweizer, M.; Stalder, H.; Schirrmeier, H.; Thiel, H.-J. Genetic and antigenic characterization of novel pestivirus genotypes: Implications for classification. Virology 2003, 311, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Yuan, J.; Deng, S.; Chen, Y.; Xie, B.; Wu, K.; Zhu, M.; Xu, H.; Huang, Y.; Yang, J.; et al. Activation of Interleukin-1β Release by the Classical Swine Fever Virus Is Dependent on the NLRP3 Inflammasome, Which Affects Virus Growth in Monocytes. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Bensaude, E. Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apoptosis and interferon synthesis during the establishment of long-Term infection of porcine vascular endothelial cells. J. Gen. Virol. 2004, 85, 1029–1037. [Google Scholar] [CrossRef]
- Johns, H.L.; Bensaude, E.; La Rocca, S.A.; Seago, J.; Charleston, B.; Steinbach, F.; Drew, T.W.; Crooke, H.; Everett, H. Classical swine fever virus infection protects aortic endothelial cells from pIpC-Mediated apoptosis. J. Gen. Virol. 2009, 91, 1038–1046. [Google Scholar] [CrossRef]
- Fan, S.; Wu, K.; Luo, C.; Li, X.; Zhao, M.; Song, D.; Ma, S.; Zhu, E.; Chen, Y.; Ding, H.; et al. Dual NDP52 Function in Persistent CSFV Infection. Front. Microbiol. 2020, 10. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function of Autophagy and NDP52 in Bacterial Infections | ||||
|---|---|---|---|---|
| Bacteria | Induction/Inhibition Autophagy | Autophagy in Bacterial Infections | NDP52 in Bacterial Infections | Reference |
| Salmonella | Induction | Inhibition | NDP52 binds Salmonella. | [37] |
| Shigella | Induction | Inhibition | NDP52 targets Shigella. | [42,43] |
| Listeria | Induction | Inhibition | NDP52 targets the Listeria ActA mutant. | [43] |
| Streptococcus | Inhibition (M protein) | Inhibition | NDP52 binds the Salmonella ubiquitin coat. | [44] |
| Function of Autophagy and NDP52 in Virus Infections | ||||
|---|---|---|---|---|
| Virus | Induction/Inhibition Autophagy | Autophagy in Virus Infections | NDP52 in Virus Infections | Reference |
| CSFV | Induction | Promotes CSFV replication | NDP52 interacts with E2 protein | [100] |
| MeV | Induction | Promotes Mev replication | NDP52 interacts with MeV-C and MeV-V protein | [89] |
| CVB3 | Induction | Promotes CVB3 replication | NDP52 interacts withVP1, promoting the ubiquitination of VP1 protein | [93] |
| CHIV | Induction | Promotes CHIV replication | NDP52 interacts withVP1, reducing CHIV infection | [94] |
| Influenza Virus | Induction | Promotes Influenza Virus replication | PB1-F2 interacts with NDP52 | [86] |
| HSV1 | Inhibition | Restricts HSV1 replication | HSV1 removes NDP52 from the nucleus | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, S.; Wu, K.; Zhao, M.; Zhu, E.; Ma, S.; Chen, Y.; Ding, H.; Yi, L.; Zhao, M.; Chen, J. The Role of Autophagy and Autophagy Receptor NDP52 in Microbial Infections. Int. J. Mol. Sci. 2020, 21, 2008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062008
Fan S, Wu K, Zhao M, Zhu E, Ma S, Chen Y, Ding H, Yi L, Zhao M, Chen J. The Role of Autophagy and Autophagy Receptor NDP52 in Microbial Infections. International Journal of Molecular Sciences. 2020; 21(6):2008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062008
Chicago/Turabian StyleFan, Shuangqi, Keke Wu, Mengpo Zhao, Erpeng Zhu, Shengming Ma, Yuming Chen, Hongxing Ding, Lin Yi, Mingqiu Zhao, and Jinding Chen. 2020. "The Role of Autophagy and Autophagy Receptor NDP52 in Microbial Infections" International Journal of Molecular Sciences 21, no. 6: 2008. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062008