MYC-Associated Factor MAX is a Regulator of the Circadian Clock

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
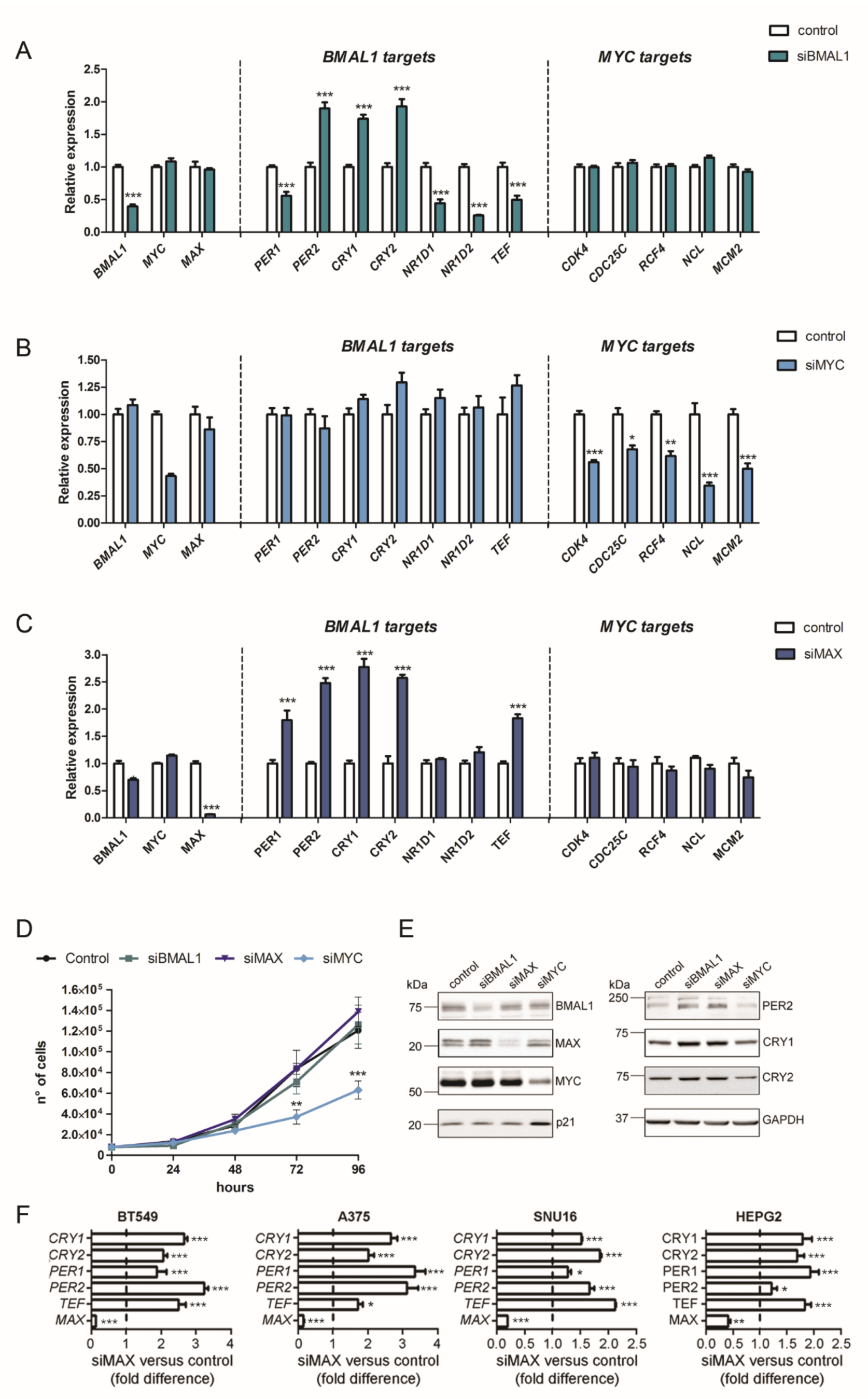
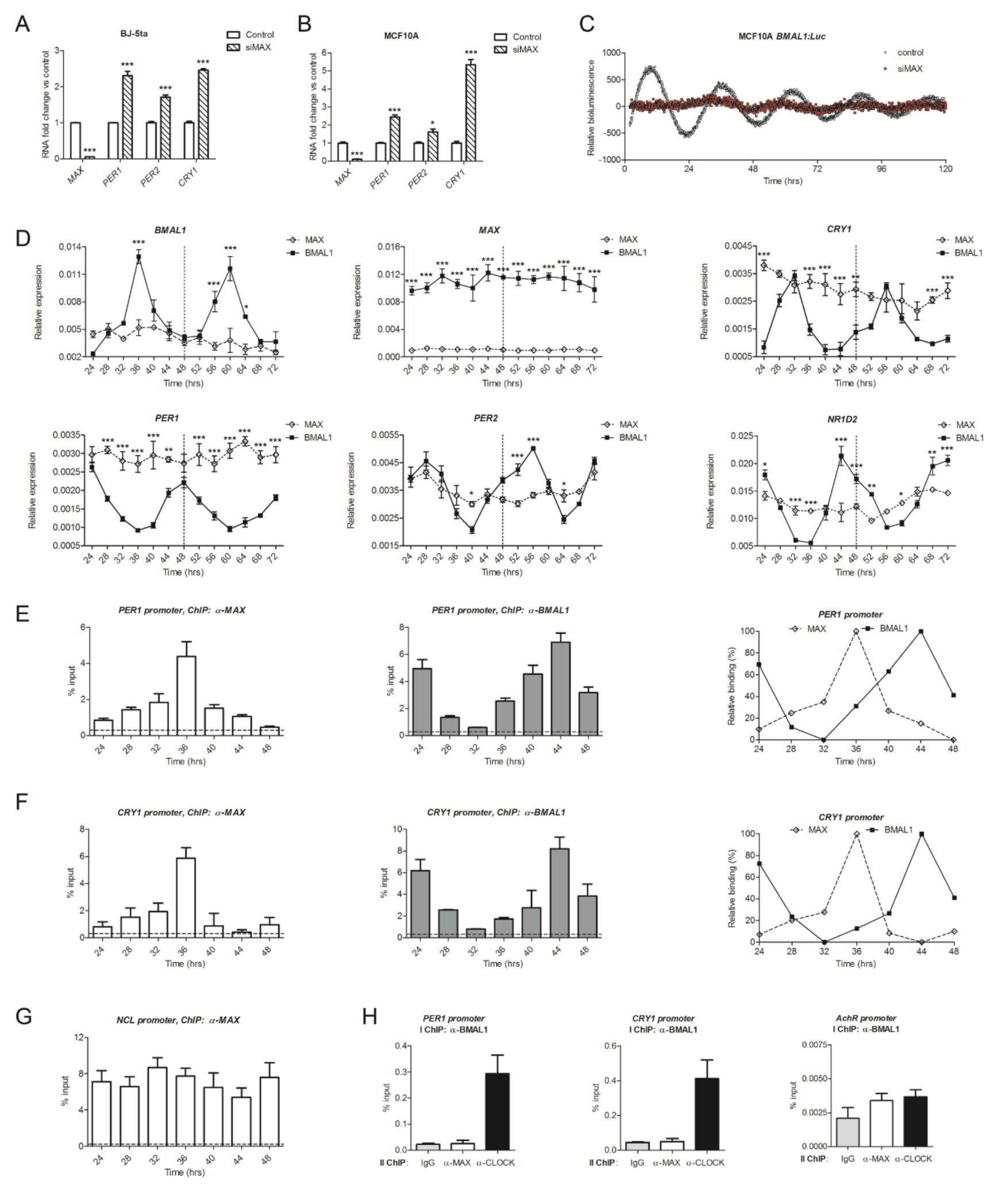
2.1. Knockdown of MAX Represses the Transcription of Core Clock Genes in Cancer Cell Lines
2.2. MAX-Inhibition of the Core Clock Genes is Independent of CRY-Mediated Repression but Requires a Functional E-Box Responsive Element
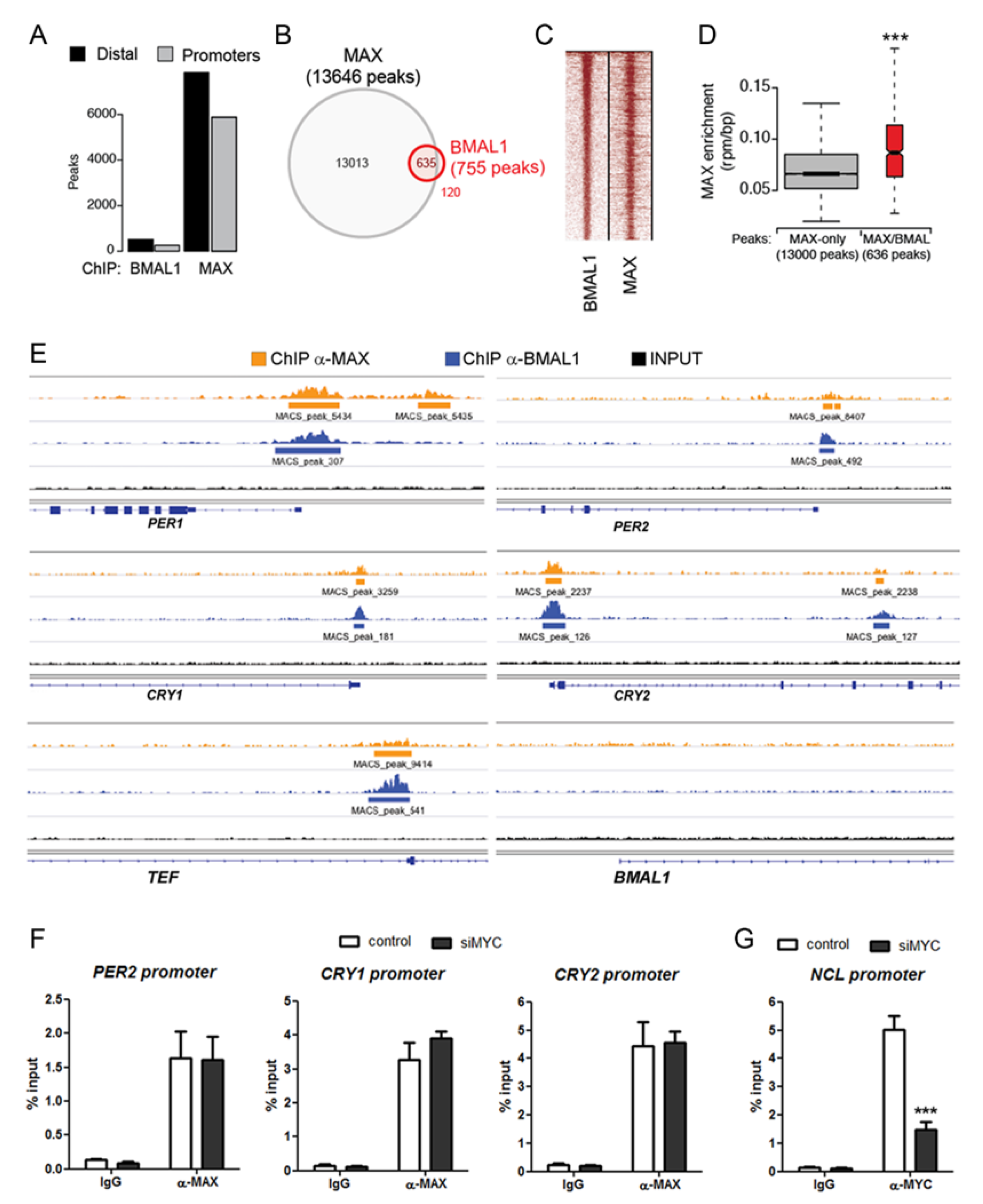
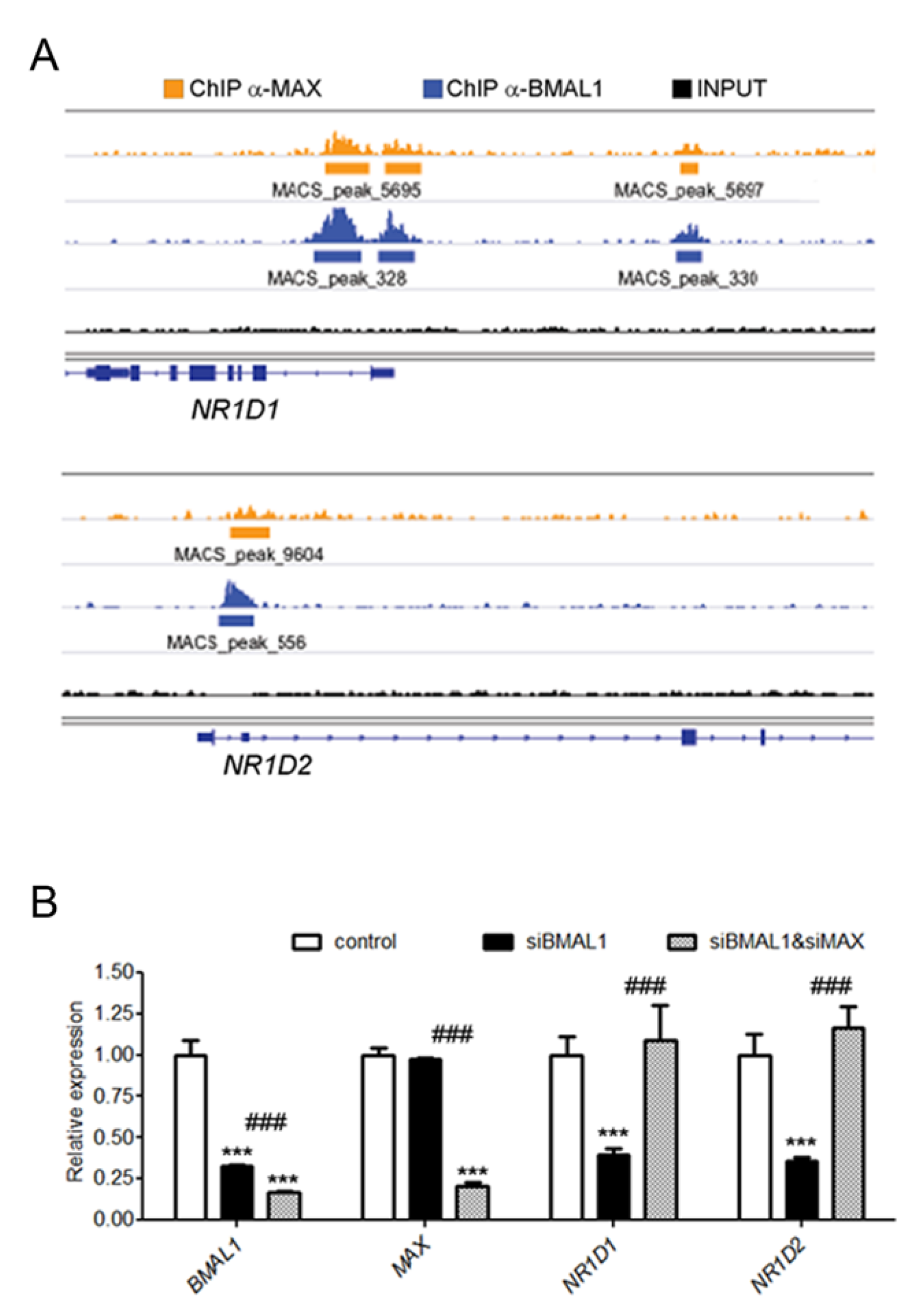
2.3. MAX is Recruited on BMAL1 Bound Genomic Regions in a MYC-Independent Manner
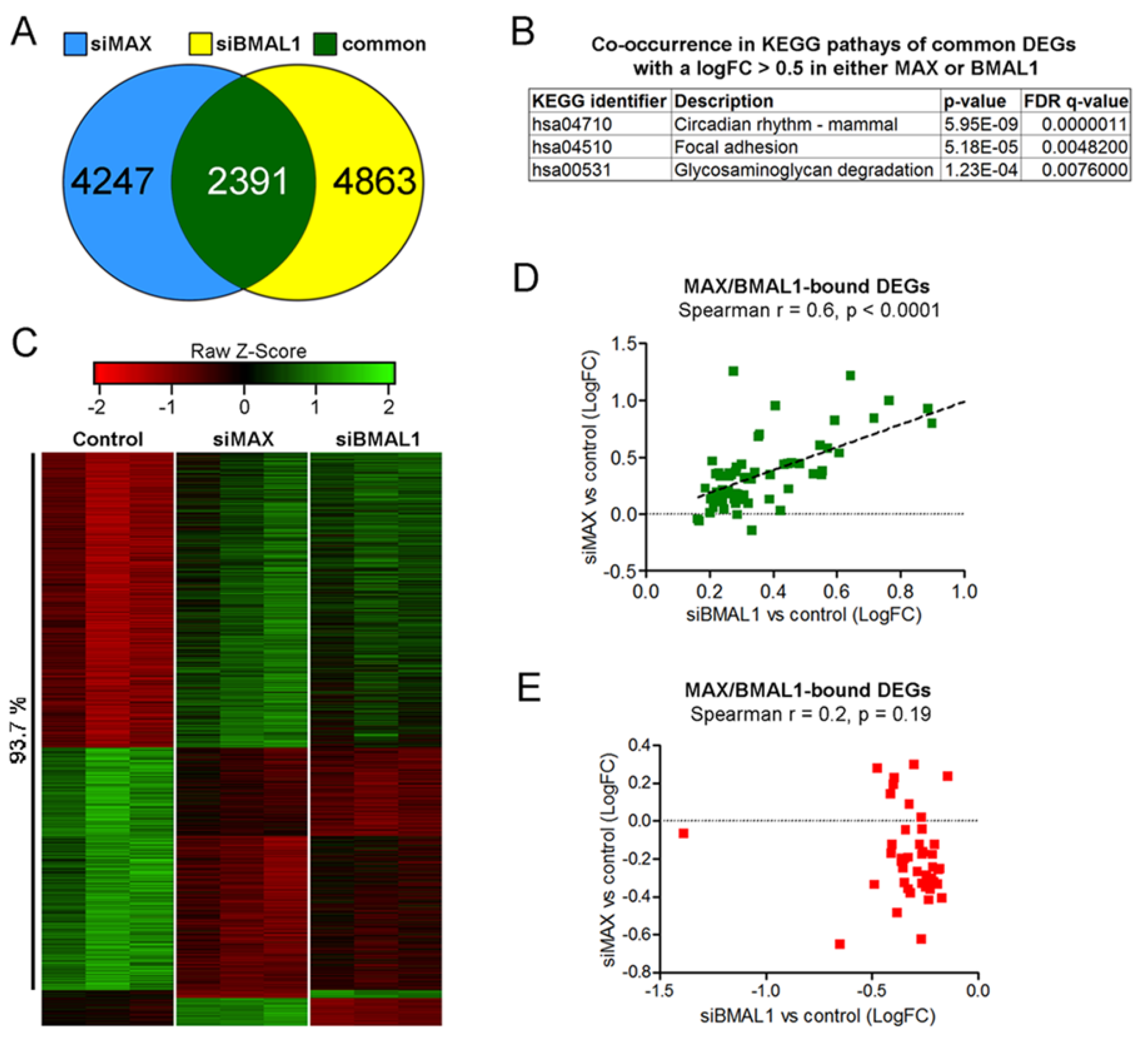
2.4. MAX and BMAL1 Regulate the Expression of Common Transcripts
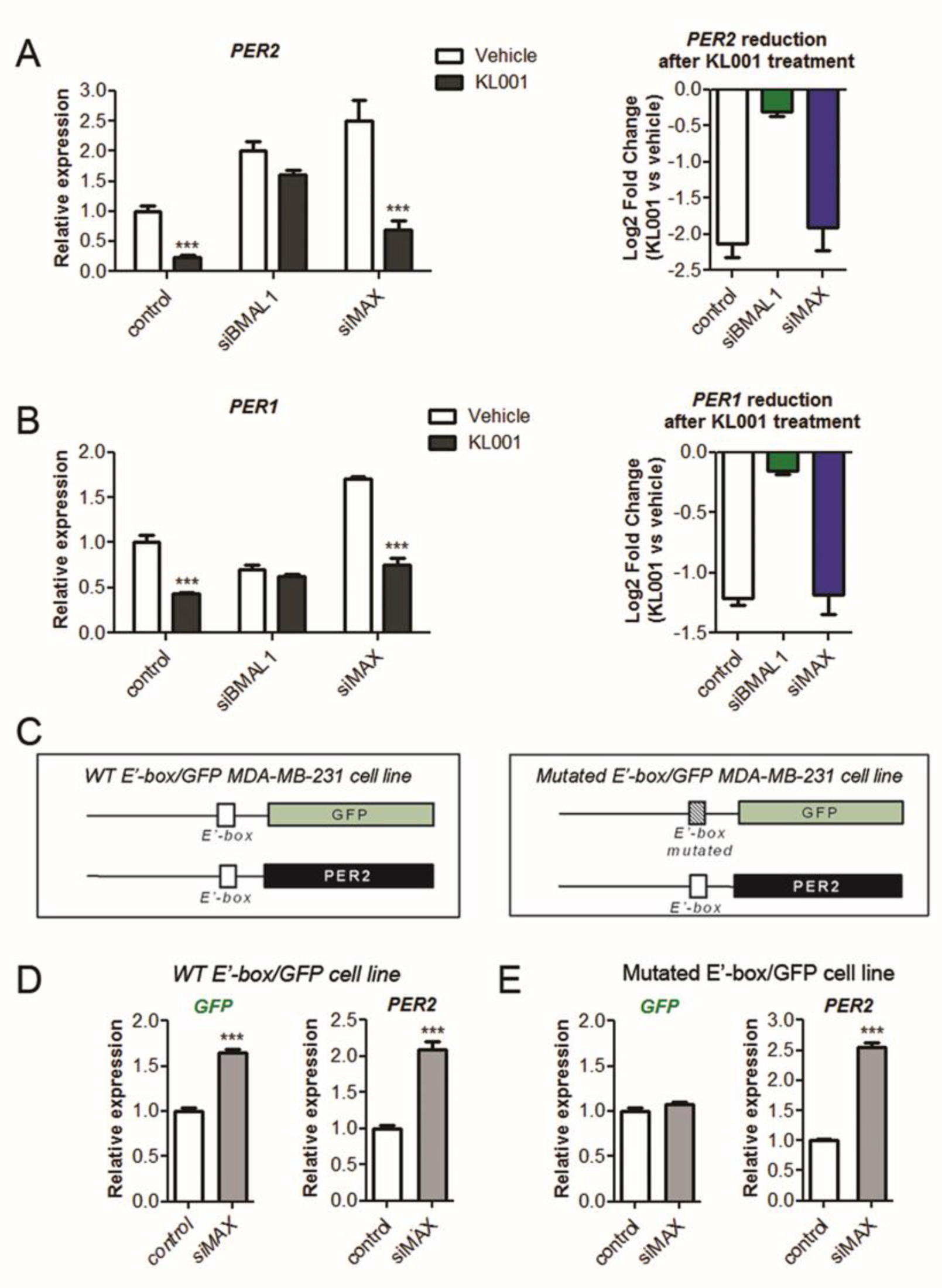
2.5. MAX Regulates Circadian Gene Expression
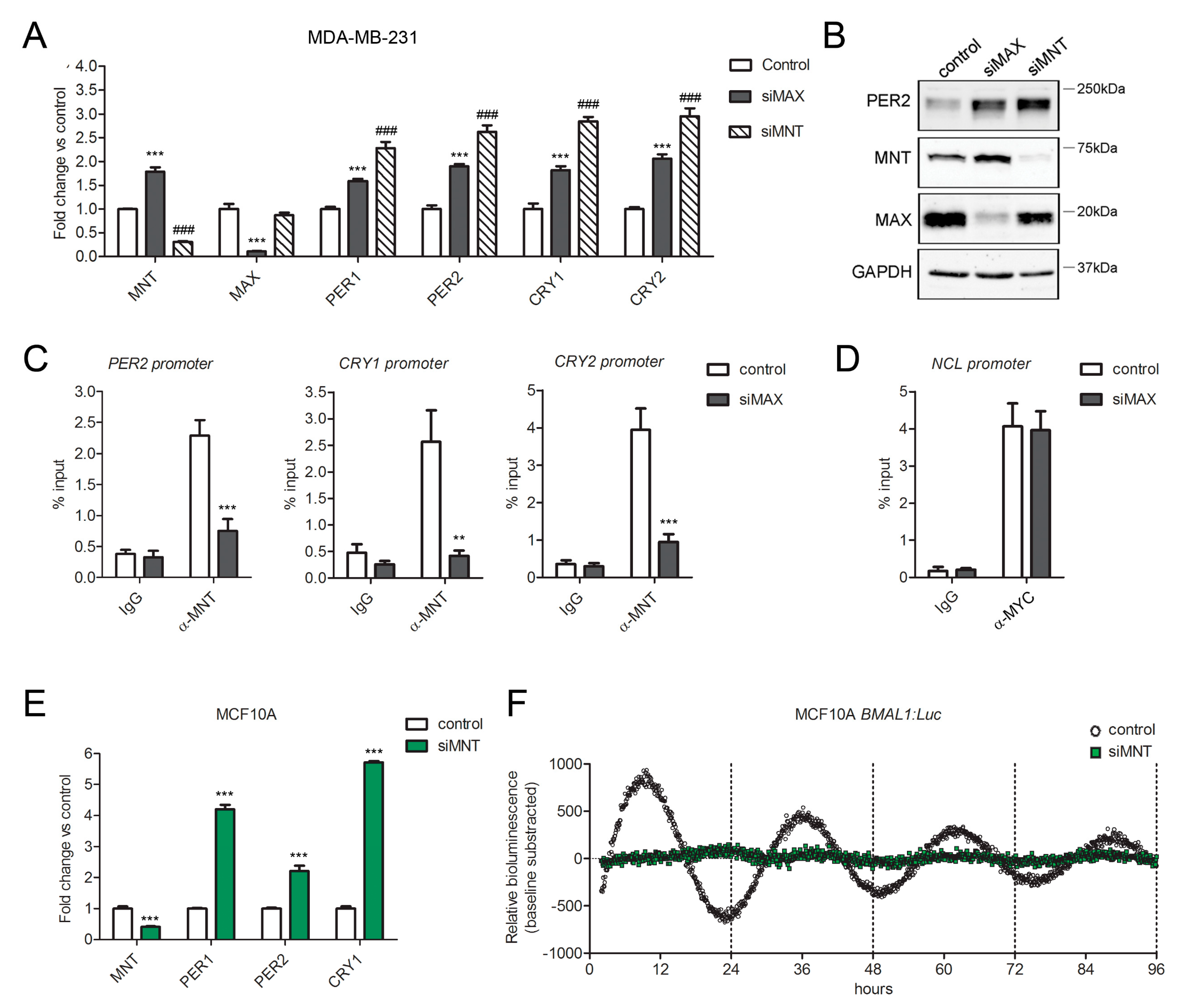
2.6. MAX Repressor Partner, MNT, Cooperates in MAX-Dependent Repression of Clock Genes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. siRNA Transfection
4.3. Cell Proliferation Analysis
4.4. Quantitative RT–PCR
4.5. RNA Sequencing
4.6. Analysis of Differentially Expressed Genes
4.7. Quantitative Chromatin Immunoprecipitation
4.8. Chromatin Immunoprecipitation Sequencing
4.9. ChIPseq Data Analysis
4.10. Immunoblotting
4.11. Generation of PER2 Promoter Reporter Plasmids
4.12. Generation of Reporter Cell Lines
4.13. Real-Time Bioluminescence Monitoring of Circadian Rhythm in Cultured Cells
4.14. Sequencing Data Availability
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMAL1 | Brain and Muscle ARNT-Like 1 |
| MAX | Myc-associated factor X |
| PER1/2 | Period circadian protein homolog 1/2 |
| CRY1/2 | Cryptochrome Circadian Regulator1/2 |
| CLOCK | Circadian Locomotor Output Cycles Kaput |
| NR1D1 | Nuclear receptor subfamily 1, group D, member 1 |
| NR1D2 | Nuclear receptor subfamily 1, group D, member 2 |
| CCG | Clock Controlled Genes |
| CDK4 | Cyclin-dependent kinase 4 |
| CDC25C | Cell Division Cycle 25C |
| RCF4 | Replication complex factor4 |
| NCL | Nucleolin |
| MCM2 | Minichromosome Maintenance Complex Component 2 |
| CDKN1A | Cyclin Dependent Kinase Inhibitor 1A |
| ChIP-seq | Chromatin Immuno-Precipitation sequencing |
| NGS | next generation sequencing |
| DEGs | differentially expressed genes |
| BHLHE41 | Basic Helix-Loop-Helix Family Member E41 |
| HIF1A | hypoxia-inducible factor-1 alpha |
| mTOR | mammalian target of rapamycin |
| ATCC | American Type Culture Collection |
References
- Dibner, C.; Schibler, U.; Albrecht, U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schibler, U.; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Reinke, H.; Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Boil. 2019, 20, 227–241. [Google Scholar] [CrossRef]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet. 2006, 15, R271–R277. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2016, 18, 164–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercolani, L.; Ferrari, A.; De Mei, C.; Parodi, C.; Wade, M.; Grimaldi, B. Circadian clock: Time for novel anticancer strategies? Pharmacol. Res. 2015, 100, 288–295. [Google Scholar] [CrossRef]
- Nagoshi, E.; Saini, C.; Bauer, C.; Laroche, T.; Naef, F.; Schibler, U. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 2004, 119, 693–705. [Google Scholar] [CrossRef]
- Lananna, B.V.; Nadarajah, C.J.; Izumo, M.; Cedeño, M.R.; Xiong, D.D.; Dimitry, J.; Tso, C.F.; McKee, C.A.; Griffin, P.; Sheehan, P.W.; et al. Cell-Autonomous Regulation of Astrocyte Activation by the Circadian Clock Protein BMAL1. Cell Rep. 2018, 25, 1–9.e5. [Google Scholar] [CrossRef] [Green Version]
- Shearman, L.P. Interacting Molecular Loops in the Mammalian Circadian Clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef]
- Zheng, B.; Albrecht, U.; Kaasik, K.; Sage, M.; Lu, W.; Vaishnav, S.; Li, Q.; Sun, Z.; Eichele, G.; Bradley, A.; et al. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell 2001, 105, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Van Der Horst, G.; Muijtjens, M.; Kobayashi, K.; Takano, R.; Kanno, S.-I.; Takao, M.; De Wit, J.; Verkerk, A.; Eker, A.P.M.; Van Leenen, D.; et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 1999, 398, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Vitaterna, M.H.; Selby, C.P.; Todo, T.; Niwa, H.; Thompson, C.; Fruechte, E.M.; Hitomi, K.; Thresher, R.J.; Ishikawa, T.; Miyazaki, J.; et al. Differential regulation of mammalian Period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. USA 1999, 96, 12114–12119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.-W.; DiTacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Buhr, E.D.; Takahashi, J.S. Molecular Components of the Mammalian Circadian Clock in Circadian Clocks; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–27. [Google Scholar]
- Miller, B.H.; McDearmon, E.L.; Panda, S.; Hayes, K.R.; Zhang, J.; Andrews, J.L.; Antoch, M.P.; Walker, J.R.; Esser, K.A.; HogenEsch, J.B.; et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 3342–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; HogenEsch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Preitner, N.; Damiola, F.; Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The Orphan Nuclear Receptor REV-ERBα Controls Circadian Transcription within the Positive Limb of the Mammalian Circadian Oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Pett, J.P.; Korenčič, A.; Wesener, F.; Kramer, A.; Herzel, H. Feedback Loops of the Mammalian Circadian Clock Constitute Repressilator. PLoS Comput. Boil. 2016, 12, e1005266. [Google Scholar] [CrossRef]
- Kim, J.K.; Forger, D. A mechanism for robust circadian timekeeping via stoichiometric balance. Mol. Syst. Boil. 2012, 8, 630. [Google Scholar] [CrossRef]
- Lee, Y.; Chen, R.; Lee, H.-M.; Lee, C. Stoichiometric Relationship among Clock Proteins Determines Robustness of Circadian Rhythms*. J. Boil. Chem. 2011, 286, 7033–7042. [Google Scholar] [CrossRef] [Green Version]
- Roenneberg, T.; Merrow, M. The Circadian Clock and Human Health. Curr. Boil. 2016, 26, R432–R443. [Google Scholar] [CrossRef]
- Altman, B.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.M.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shostak, A.; Ruppert, B.; Ha, N.; Bruns, P.; Toprak, U.H.; Lawerenz, C.; Eils, R.; Schlesner, M.; Diernfellner, A.; Brunner, M. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat. Commun. 2016, 7, 11807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.; Hsieh, A.L.; Gouw, A.M.; Dang, C.V. Correspondence: Oncogenic MYC persistently upregulates the molecular clock component REV-ERBα. Nat. Commun. 2017, 8, 14862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shostak, A.; Ruppert, B.; Diernfellner, A.; Brunner, M. Correspondence: Reply to ‘Oncogenic MYC persistently upregulates the molecular clock component REV-ERBα’. Nat. Commun. 2017, 8, 14918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderton, G.K. Transcription: The transcriptional effects of MYC. Nat. Rev. Cancer 2014, 14, 513. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hänel, F.; Eilers, M. An alternative pathway for gene regulation by Myc. EMBO J. 1997, 16, 5672–5686. [Google Scholar] [CrossRef]
- Gebhardt, A.; Frye, M.; Herold, S.; Benitah, S.A.; Braun, K.; Samans, B.; Watt, F.E.; Elsässer, H.-P.; Eilers, M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Boil. 2006, 172, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Wiese, K.E.; Walz, S.; Von Eyss, B.; Wolf, E.; Athineos, D.; Sansom, O.J.; Eilers, M. The Role of MIZ-1 in MYC-Dependent Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014290. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Kondratov, R.; Kanagal-Shamanna, R.; Kondratova, A.A.; Gorbacheva, V.Y.; Antoch, M.P. Dual role of the CLOCK/BMAL1 circadian complex in transcriptional regulation. FASEB J. 2006, 20, 530–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, F.; Matsubara, C.; Myung, J.; Yoritaka, T.; Kamimura, N.; Tsutsumi, S.; Kanai, A.; Suzuki, Y.; Sassone-Corsi, P.; Aburatani, H.; et al. Genome-Wide Profiling of the Core Clock Protein BMAL1 Targets Reveals a Strict Relationship with Metabolism. Mol. Cell. Boil. 2010, 30, 5636–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ukai-Tadenuma, M.; Yamada, R.G.; Xu, H.; Ripperger, J.A.; Liu, A.C.; Ueda, H.R. Delay in Feedback Repression by Cryptochrome 1 Is Required for Circadian Clock Function. Cell 2011, 144, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, A.J.; Menet, J.S. Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genet. 2018, 14, e1007156. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Hirota, T.; Lee, J.W.; John, P.C.S.; Sawa, M.; Iwaisako, K.; Noguchi, T.; Pongsawakul, P.Y.; Sonntag, T.; Welsh, D.K.; Brenner, D.A.; et al. Identification of Small Molecule Activators of Cryptochrome. Science 2012, 337, 1094–1097. [Google Scholar] [CrossRef] [Green Version]
- Hardin, P. Transcription Regulation within the Circadian Clock: The E-box and Beyond. J. Boil. Rhythm. 2004, 19, 348–360. [Google Scholar] [CrossRef]
- Lüscher, B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene 2001, 277, 1–14. [Google Scholar] [CrossRef]
- Honma, S.; Kawamoto, T.; Takagi, Y.; Fujimoto, K.; Sato, F.; Noshiro, M.; Kato, Y.; Honma, K.-I. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002, 419, 841–844. [Google Scholar] [CrossRef]
- Ramanathan, C.; Khan, S.K.; Kathale, N.D.; Xu, H.; Liu, A.C. Monitoring Cell-autonomous Circadian Clock Rhythms of Gene Expression Using Luciferase Bioluminescence Reporters. J. Vis. Exp. 2012, 19, 4234. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Monreal, M.A.; Trevino, V.; Moreno-Cuevas, J.E.; Scott, S.-P. Identification of circadian-related gene expression profiles in entrained breast cancer cell lines. Chronobiol. Int. 2016, 33, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Corlazzoli, F.; Gregorski, A.; Azmi, N.H.A.; Sacchi, N. Identification of an estrogen-regulated circadian mechanism necessary for breast acinar morphogenesis. Cell Cycle 2012, 11, 3691–3700. [Google Scholar] [CrossRef] [Green Version]
- Don, S.S.L.; Lin, H.-H.; Furtado, J.J.; Qraitem, M.; Taylor, S.R.; Farkas, M.E. Circadian oscillations persist in low malignancy breast cancer cells. Cell Cycle 2019, 18, 2447–2453. [Google Scholar]
- Rey, G.; Cesbron, F.; Rougemont, J.; Reinke, H.; Brunner, M.; Naef, F. Genome-Wide and Phase-Specific DNA-Binding Rhythms of BMAL1 Control Circadian Output Functions in Mouse Liver. PLoS Boil. 2011, 9, e1000595. [Google Scholar] [CrossRef] [Green Version]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and Max proteins possess distinct transcriptional activities. Nature 1992, 359, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Terragni, J.; Nayak, G.; Banerjee, S.; Medrano, J.-L.; Graham, J.R.; James, F.; Sepulveda, S.; Cooper, G.M. The E-Box binding factors Max/Mnt, MITF and USF1 Act coordinately with FoxO to regulate expression of Pro-apoptotic and cell cycle control genes by phosphatidylinositol 3-kinase/Akt/GSK3 signaling. J. Biol. Chem. 2011, 286, 36215–36227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlin, P.J.; Huang, J. The MAX-interacting transcription factor network. Semin. Cancer Boil. 2006, 16, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Boil. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83. [Google Scholar] [CrossRef]
- Yu, C.; Fang, X.; Huang, T.; Yang, S.; Jiang, J.; Sun, C. Hypoxia disrupts the expression levels of circadian rhythm genes in hepatocellular carcinoma. Mol. Med. Rep. 2015, 11, 4002–4008. [Google Scholar] [CrossRef]
- Jaspers, T.; Morrell, M.; Simonds, A.; Adcock, I.M.; Durham, A. The role of hypoxia and the circadian rhythm in sleep apnoea. Eur. Respir. J. 2015, 46, OA298. [Google Scholar]
- Mortola, J.P. Hypoxia and circadian patterns. Respir. Physiol. Neurobiol. 2007, 158, 274–279. [Google Scholar] [CrossRef]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Tang, D.; Liu, N.; Xiong, W.; Huang, H.; Li, Y.; Ma, Z.; Zhao, H.; Chen, P.; Qi, X.; et al. Reciprocal Regulation between the Circadian Clock and Hypoxia Signaling at the Genome Level in Mammals. Cell Metab. 2017, 25, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, I.-M.; Wang, Y.; Liang, C.-W.; Bahri, N.; Quattrone, A.; Doyle, L.; Mariño-Enríquez, A.; Lauria, A.; Zhu, M.; Debiec-Rychter, M.; et al. MAX inactivation is an early event in GIST development that regulates p16 and cell proliferation. Nat. Commun. 2017, 8, 14674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Boil. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, V.; Ceol, A.; Ogier, A.G.E.; De Pretis, S.; Galeota, E.; Kishore, K.; Bora, P.; Croci, O.; Campaner, S.; Amati, B.; et al. Integrated Systems for NGS Data Management and Analysis: Open Issues and Available Solutions. Front. Genet. 2016, 7, 960. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- De Mei, C.; Ercolani, L.; Parodi, C.; Veronesi, M.; Vecchio, C.L.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R.; et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 2014, 34, 2597–2608. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaževitš, O.; Bolshette, N.; Vecchio, D.; Guijarro, A.; Croci, O.; Campaner, S.; Grimaldi, B. MYC-Associated Factor MAX is a Regulator of the Circadian Clock. Int. J. Mol. Sci. 2020, 21, 2294. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072294
Blaževitš O, Bolshette N, Vecchio D, Guijarro A, Croci O, Campaner S, Grimaldi B. MYC-Associated Factor MAX is a Regulator of the Circadian Clock. International Journal of Molecular Sciences. 2020; 21(7):2294. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072294
Chicago/Turabian StyleBlaževitš, Olga, Nityanand Bolshette, Donatella Vecchio, Ana Guijarro, Ottavio Croci, Stefano Campaner, and Benedetto Grimaldi. 2020. "MYC-Associated Factor MAX is a Regulator of the Circadian Clock" International Journal of Molecular Sciences 21, no. 7: 2294. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072294