Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines




Abstract
:1. Introduction
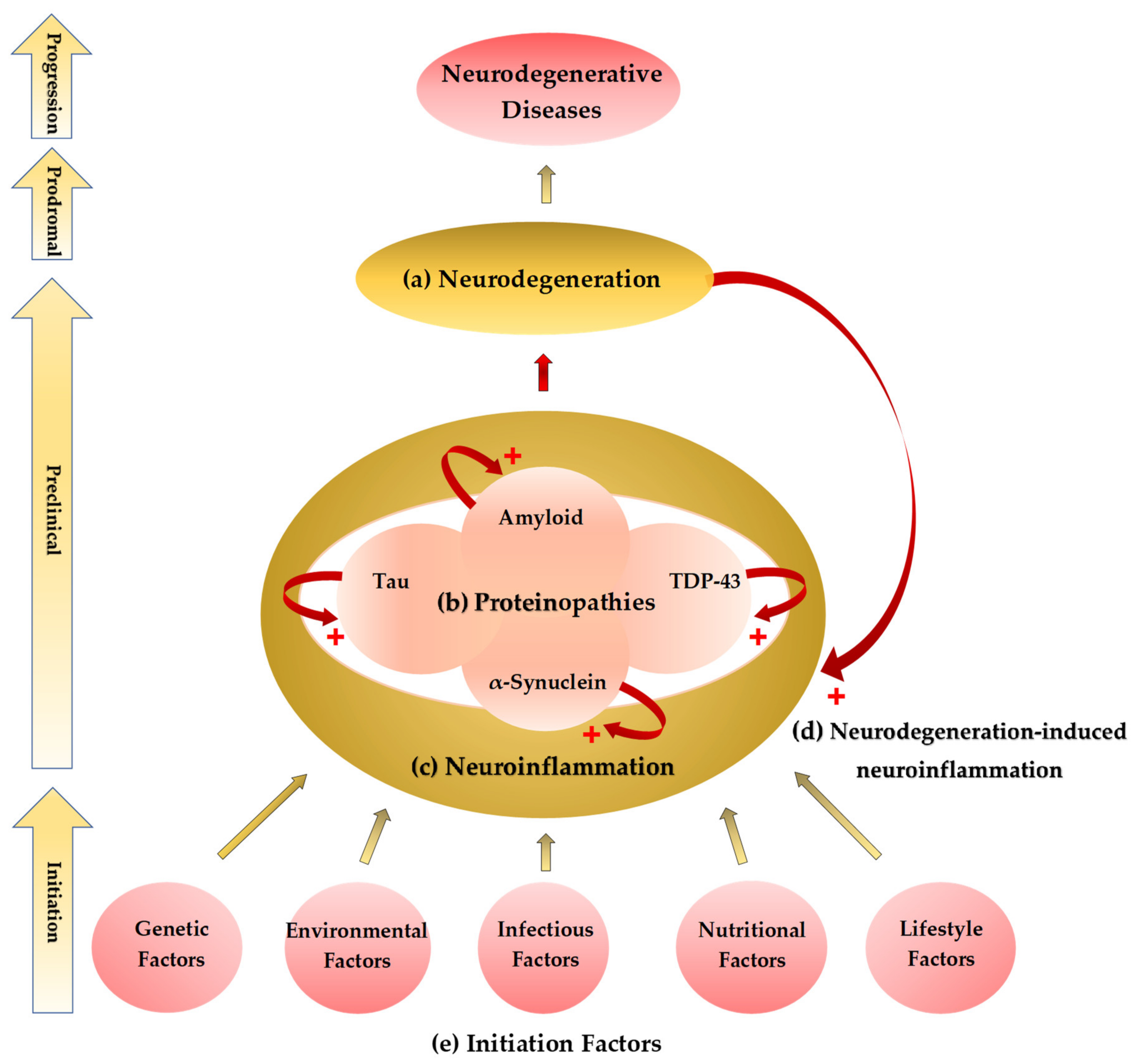
2. The Common Theme: Proteinaceous Deposits, Neurodegeneration, Neuropathies
3. Neuroinflammation: A Common Prelude to Neurodegeneration
3.1. Systematic Reviews on Inflammatory Cytokines in Neurodegenerative Diseases
4. Neurodegeneration-Induced Neuroinflammation, Chronic Inflammation, and Allostatic Loads
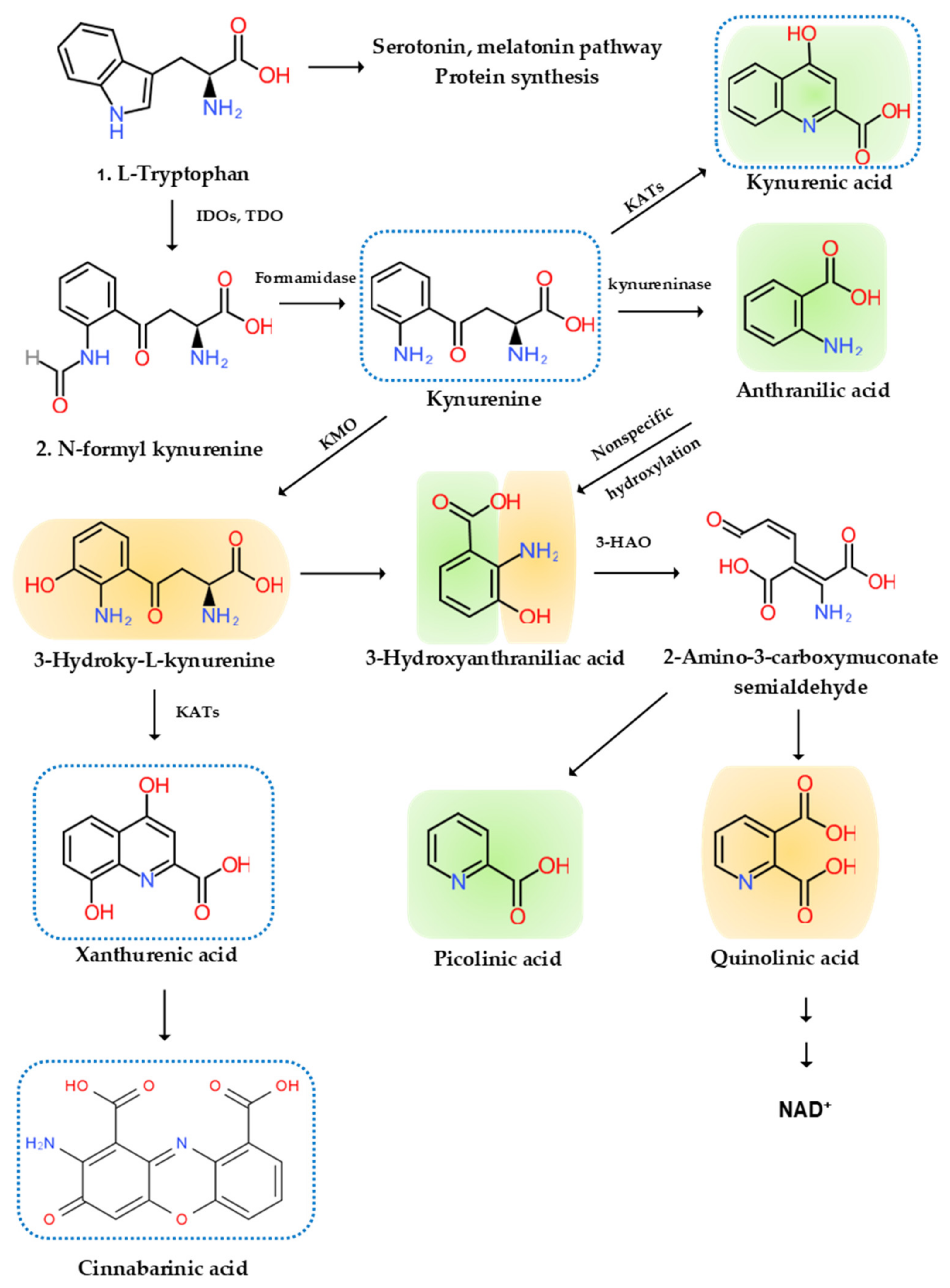
5. The Etiological Links behind Neurodegenerative Diseases: Tryptophan and Bioactive Kynurenines
5.1. Tryptophan
5.2. Bioactive Kynurenines
5.2.1. Neurotoxic Kynurenines
5.2.2. Neuromodulatory Kynurenines
5.2.3. Anti-Inflammatory Kynurenines
5.2.4. Reactive Oxygen Species
5.2.5. Antioxidant
5.2.6. Immune Kynurenines
5.3. Kynurenine Pathway Enzyme Activities
5.4. Systematic Reviews on Kynurenines in Major Neurodegenerative Diseases
5.5. Synthesis of Inflammatory Cytokines and Bioactive Kynurenines: Signs and Symptoms
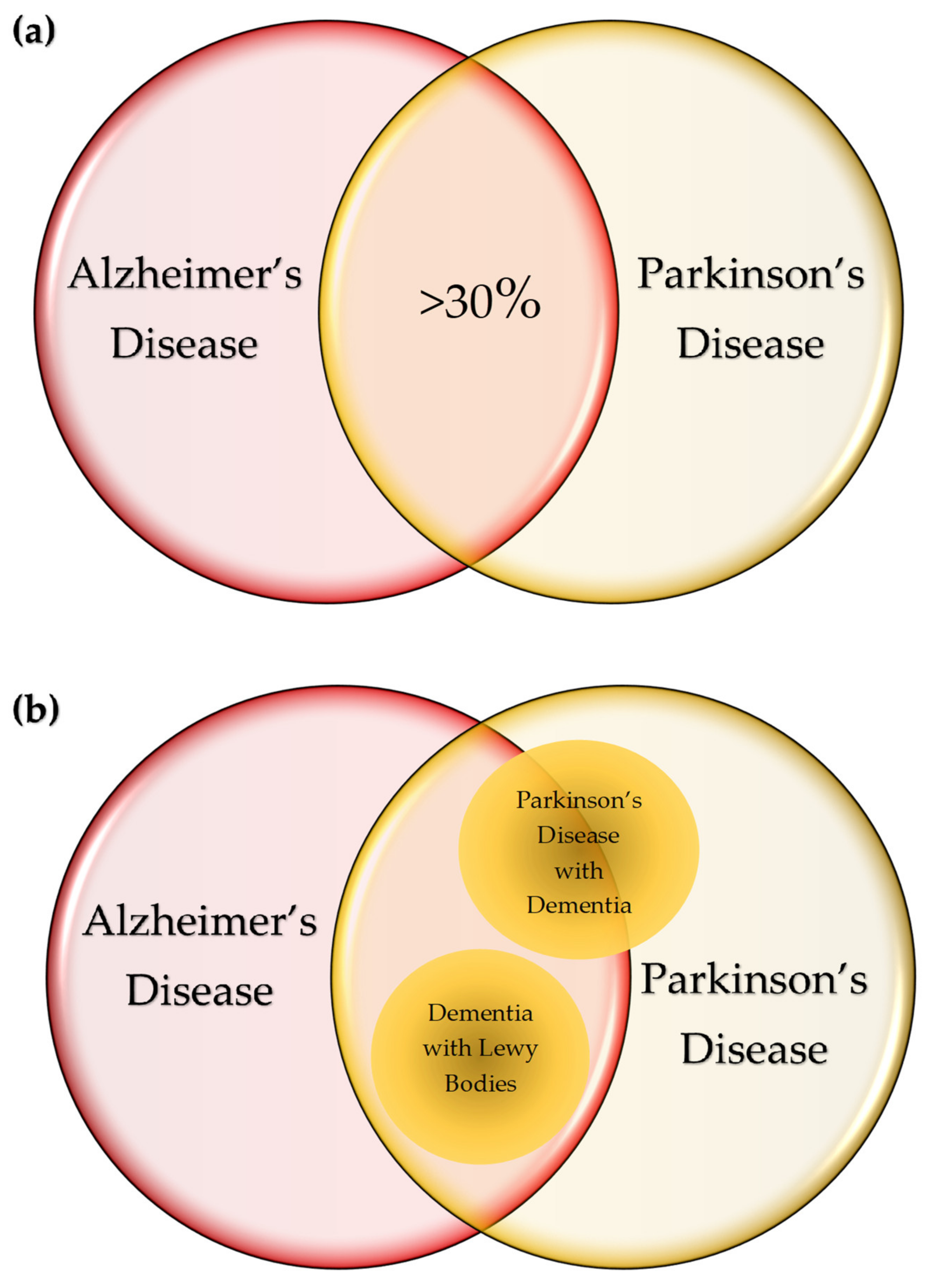
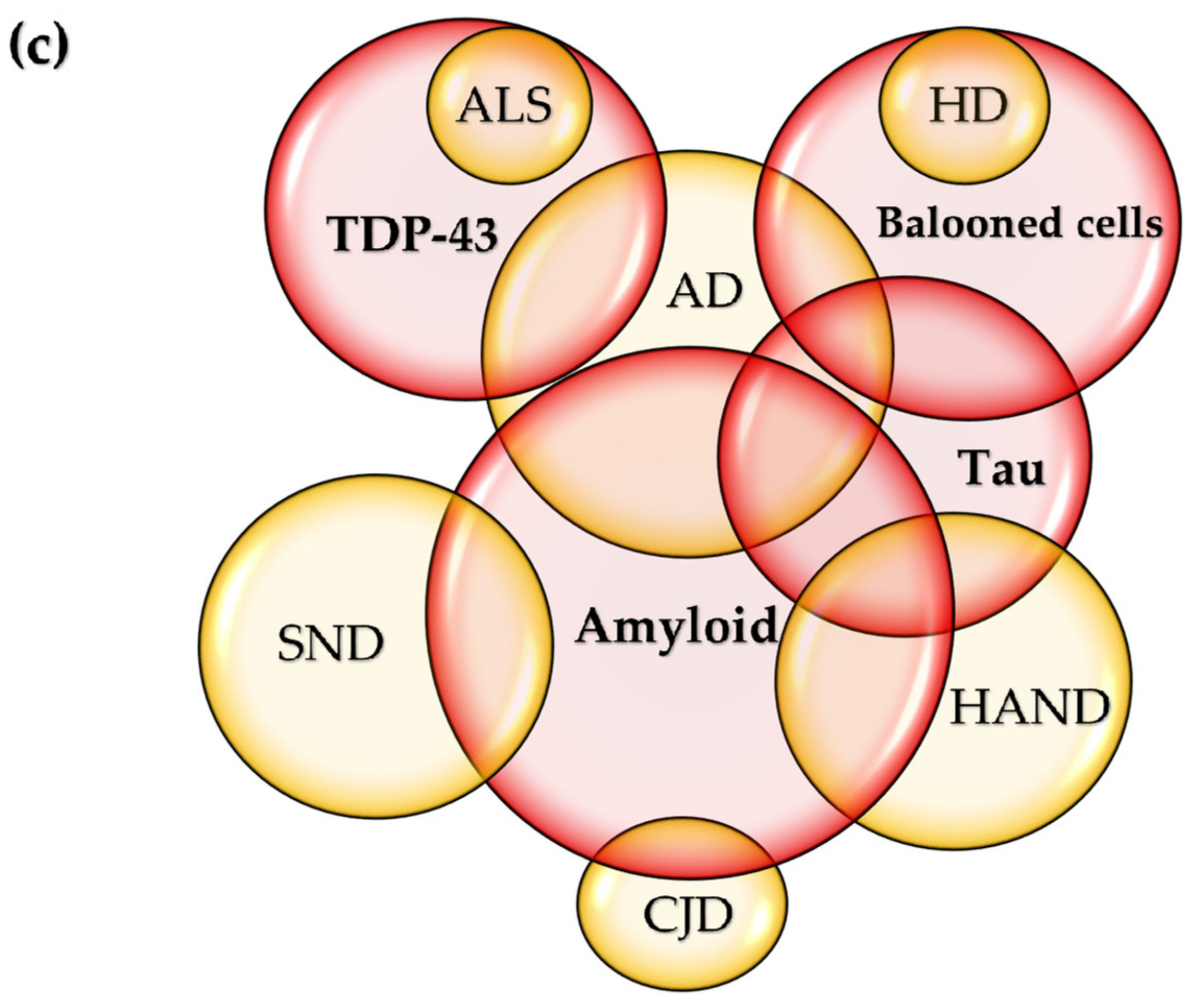
6. Variations on the Theme? Commonalities in Symptomatology and Histopathology
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | anthranilic acid |
| Aβ | amyloid-β |
| AD | Alzheimer’s disease |
| AhR | aryl hydrocarbon receptor |
| AIDS | acquired immune deficiency syndrome |
| ALS | amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APC | antigen-presenting cell |
| ASC | apoptosis-associated speck-like protein containing caspase activation and recruitment domains |
| BBB | blood–brain barrier |
| CA | cinnabarinic acid |
| CD | cluster of differentiation |
| Cdh1 | cadherin 1 |
| CARD | caspase activation and recruitment domains |
| cART | combination antiretroviral therapies |
| CCL5 | C–C motif chemokine ligand 5 |
| CJD | Creutzfeldt–Jakob disease |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DLB | dementia with Lewy bodies |
| GC | glucocorticoid |
| GPR35 | G-protein-coupled receptor 35 |
| 3-HAA | 3-hydroxy-anthralinic acid |
| HAND | HIV-associated neurocognitive disorders |
| HD | Huntington’s disease |
| HIV | human immunodeficiency virus |
| 3-HK | 3-hydroxy-kynurenine |
| IFN | interferon |
| LB | Lewy body |
| IL | interleukin |
| KAT | kynurenine aminotransferase |
| KMO | kynurenine-3-monooxygenase |
| KYN | kynurenine |
| KYNA | kynurenic acid |
| MIP | macrophage inflammatory proteins |
| MS | multiple sclerosis |
| ND | neurodegenerative diseases |
| NMDA | N-methyl-d-aspartate |
| OA | oxidative stress |
| PA | picolinic acid |
| PD | Parkinson’s disease |
| PLP | pyridoxal phosphate |
| PrPC | cellular prion protein |
| PrPSc | scrapie isoform of the prion protein |
| QA | quinolinic acid |
| RANTES | regulated upon activation, normal T cell expressed, and presumably secreted |
| RBC | red blood cell |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SND | stroke-induced secondary neurodegeneration |
| TDO | tryptophan dioxygenase |
| TDP | transactive response DNA-binding protein |
| TGF | transforming growth factor |
| Th | T helper |
| TNF | tumor necrosis factor |
| Tregs | regulatory T cells |
| TRP | tryptophan |
| VEGF | vascular endothelial growth factor |
| XA | xanthurenic acid |
Appendix A


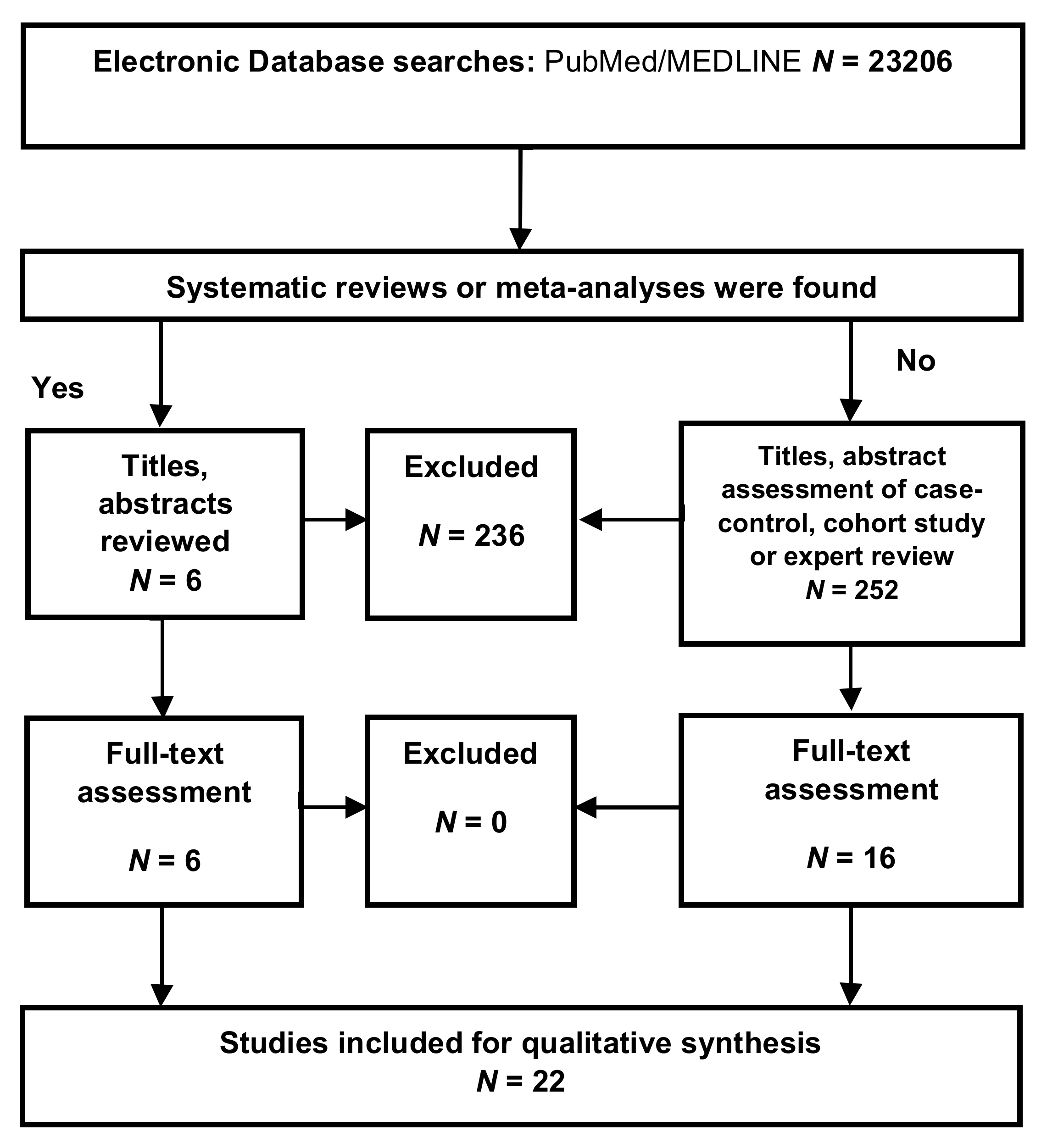
Appendix A.1. Inclusion Criteria
Appendix A.2. Exclusion Criteria
Appendix A.3. Selection Process
Appendix A.4. Data Extraction
Appendix A.5. Assessment of the Methodological Quality

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Study Types | Reference Numbers or Sample Numbers (Disease/Control) | Samples | Risk of Bias |
|---|---|---|---|---|
| Alzheimer’s disease [54,55] | ||||
| Brosseron et al., 2014 [54] | Systematic review | 118 | Plasma, serum, CSF | Low risk |
| Stuart and Baune, 2014 [55] | Systematic review | 96 | Blood, plasma, serum, CSF | |
| Parkinson’s disease [56,57] | ||||
| Qin et al., 2016 [56] | Systematic review | 25 | Blood | Low risk |
| Chen et al., 2018 [57] | Systematic review | 14 | CSF | |
| Multiple sclerosis [58] | ||||
| Wang et al., 2018a [58] | Expert review | - | - | High risk |
| Huntington’s disease [60,61,62] | ||||
| Chang et al., 2015 [60] | Case–control study | 15/16 | Plasma | High risk |
| Bouwens et al., 2015 [61] | Cohort study | 124 | Plasma | |
| Silvestroni et al., 2009 [62] | Case–control study | 6/17 | Post-mortem brain tissue | |
| Amyotrophic lateral sclerosis [57,63] | ||||
| Hu et al., 2017 [63] | Meta-analysis | 25 | Serum | Low risk |
| Chen et al., 2018 [57] | Meta-analysis | 71 | CSF | |
| Creutzfeldt–Jakob disease [64,65,66,67,68] | ||||
| Van Everbroeck et al, 2002 [64] | Case–control study | 19/19 | CSF | Low risk |
| Stoeck et al., 2005 [65] | Case–control study | 20/20 | CSF | |
| Stoeck et al., 2006 [66] | Case–control study | 23/111 | CSF | |
| Fujita et al., 2013 [67] | Case–control study | 14/14 | CSF | |
| Stoeck et al., 2014 [68] | Case–control study | 12/12 | CSF | |
| HIV-associated neurocognitive disorders [69,70,71] | ||||
| Correia et al., 2013 [69] | Case–control study | 50/74 | Plasma | High risk |
| Yuan et al., 2013 [70] | Case–control study | 64/43 | Plasma, CSF | |
| Seilhean et al., 1997 [71] | Case–control study | 12/6 | Brain tissue | |
| Stroke-induced secondary neurodegeneration [72,73,74,75] | High risk | |||
| Lambertsen et al., 2012 [72] | Expert review | - | - | |
| Tarkowski et al., 1997 [73] | Case–control study | 30/15 | CSF | |
| Perini et al., 2001 [74] | Case–control study | 42/39 | Serum | |
| Mazzotta et al., 2004 [75] | Case–control study | 18/25 | Plasma | |
| Diseases | Study Types | Reference Numbers or Sample Numbers (Disease/Control) | Samples | Risk of Bias |
|---|---|---|---|---|
| Alzheimer’s disease [172,173,174,175] | ||||
| Guillemin et al., 2005 [172] | Case–control study | 6/4 | Brain tissue | High risk |
| Bonda et al., 2010 [173] | Case–control study | 12/7 | Brain tissue | |
| Gulaj et al., 2010 [174] | Case–control study | 34/18 | Serum | |
| Schwarcz et al., 2013 [175] | Case–control study | 20/19 | Serum | |
| Parkinson’s disease [176,177,178] | ||||
| Hartai et al., 2005 [176] | Case–control study | 19/17 | Plasma, RBC | High risk |
| Lewitt et al., 2013 [177] | Case–control study | 48/57 | CSF | |
| Chang et al., 2018 [178] | Case–control study | 118/37 | Plasma | |
| Multiple sclerosis [132,133,134,135] | ||||
| Lim et al., 2017 [132] | Case–control study cohort study | 139/50 | Plasma, CSF | High risk |
| Aeinehband et al., 2016 [134] | Case–control study cohort study | 30/6 | CSF | |
| Hartai et al., 2005 [135] | Case–control study | 14/13 | CSF, RBC | |
| Huntington’s disease [179,180,181,182] | ||||
| Reynolds and Pearson, 1989 [179] | Case–control study | 12/11 | Postmortem brain tissue | High risk |
| Beal et al., 1992 [180] | Case–control study | 14–30/25–40 | Postmortem brain tissue | |
| Jauch et al., 1995 [181] | Case–control study | 17/17 | Postmortem brain tissue | |
| Stoy et al., 2005 [182] | Case–control study | 15/11 | Postmortem brain tissue | |
| Amyotrophic lateral sclerosis [135,136] | ||||
| Chen et al., 2010 [135] | Case–control study | 35/140 | Serum, CSF, brain tissue | High risk |
| Iłzecka et al., 2003 [136] | Case–control study | 32/30 | Serum, CSF | |
| Creutzfeldt–Jakob disease | ||||
| - | - | - | - | - |
| HIV-associated neurocognitive disorders [137] | ||||
| Baran et al., 2012 [137] | Case–control study | 23/16 | Brain tissue | High risk |
| Stroke-induced secondary neurodegeneration [183,184] | Unclear | |||
| Darlington et al., 2007 [183] | Case–control study | 50/35 | Serum | |
| Yan et al., 2015 [184] | Case–control study | 28/20,11 | Serum, CSF | |
Appendix A.6. Risk of Bias Assessment
| Risk of Bias | Criteria |
|---|---|
| High risk | No meta-analysis or systematic review, fewer than five case–control and/or cohort studies, or presence of only expert review |
| Low risk | Presence of at least one meta-analysis or systematic review, without conflicting results |
| Unclear | Presence of only case–control study or cohort study, meta-analysis with conflicting results, or case–control studies with conflicting results |
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, J.H.; Vora, N. The global burden of neurologic diseases. Neurology 2014, 83, 349–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Sandra, A. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.S.; Rossor, M.N.; Fox, N.C. Alzheimer’s Disease in the 100 Years Since Alzheimer’s Death. Brain 2015, 138(Pt12), 3816–3821. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Le, W. Milestones of Parkinson’s Disease Research: 200 Years of History and Beyond. Neurosci. Bull. 2017, 33, 598–602. [Google Scholar] [CrossRef]
- Taipa, R.; Pinho, J.; Melo-Pires, M. Clinico-Pathological Correlations of the Most Common Neurodegenerative Dementias. Front. Neurol. 2012, 3, 68. [Google Scholar] [CrossRef] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. CSH Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Liyanage, S.M.; Santos, C.; Weavera, D.F. The hidden variables problem in Alzheimer’s disease clinical trial design. Alzheimers Dement. 2018, 4, 628–635. [Google Scholar] [CrossRef]
- Gerez, J.A.; Riek, R. Neurodegenerative diseases distinguished through protein-structure analysis. Nature 2020, 578, 223–224. [Google Scholar]
- Shimizu, S.; Hirose, D.; Hatanaka, H.; Takenoshita, N.; Kaneko, Y.; Ogawa, Y.; Sakurai, H.; Hanyu, H. Role of Neuroimaging as a Biomarker for Neurodegenerative Diseases. Front. Neurol. 2018, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Dias, F.L.; Silva, R.M.; Moraes, E.N.; Caramelli, P. Clinical and autonomic profile of patients with Alzheimer’s disease and mixed dementia patients. Rev. Assoc. Médica Bras. 2013, 59, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.F.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Xu, W.; Li, J.Q.; Wang, J.; Lai, T.J.; et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J. Affect. Dis. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Martinez-Bellver, S.; Giraldo, E.; Teruel-Marti, V.; Lloret, A.; Vina, J. A beta induces excitotoxicity mediated by APC/C-Cdh1 depletion that can be prevented by glutaminase inhibition promoting neuronal survival. Sci. Rep. 2016, 6, 31158. [Google Scholar] [CrossRef] [Green Version]
- Greenamyre, J.T.; Penney, J.B.; Young, A.B.; D’Amato, C.J.; Hicks, S.P.; Shoulson, I. Alterations in L-glutamate binding in Alzheimer’s and Huntington’s diseases. Science 1985, 227, 1496–1499. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182. [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkaoui, M.A.; Geoffroy, P.A.; Roze, E.; Degos, B.; Garcin, B. Functional Motor Symptoms in Parkinson’s Disease and Functional Parkinsonism: A Systematic Review. J. Neuropsychi. Clin. 2020, 32, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papagno, C.; Trojano, L. Cognitive and Behavioral Disorders in Parkinson’s Disease: An Update. I: Cognitive Impairments. Neurol. Sci. 2018, 39, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Broen, M.P.G.; Narayen, N.E.; Kuijf, M.L.; Dissanayaka, N.N.W.; Leentjens, A.F.G. Prevalence of Anxiety in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2016, 31, 1125–1133. [Google Scholar] [CrossRef]
- Kale, N.; Magana, S.; Agaoglu, J.; Tanik, O. Assessment of autonomic nervous system dysfunction in multiple sclerosis and association with clinical disability. Neurol. Int. 2009, 1, e5. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Boeschoten, R.E.; Braamse, A.M.J.; Beekman, A.T.F.; Cuijpers, P.; van Oppen, P.; Dekker, J.; Uitdehaag, B.M.J. Prevalence of Depression and Anxiety in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Sci. 2017, 372, 331–341. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [Green Version]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W. Clinical and Pathological Insights into the Dynamic Nature of the White Matter Multiple Sclerosis Plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Folstein, S. Huntington’s Disease a Disorder of Families; Johns Hopkins University Press: Baltimore, MD, USA, 1989. [Google Scholar]
- Dale, M.; van Duijn, E. Anxiety in Huntington’s Disease. J. Neuropsychiatry Clin. Neurosci. 2015, 27, 262–271. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neurophatology of huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [PubMed] [Green Version]
- Mueller, K.A.; Glajch, K.E.; Huizenga, M.N.; Wilson, R.A.; Granucci, E.J.; Dios, A.M.; Tousley, A.R.; Iuliano, M.; Weisman, E.; LaQuaglia, M.J.; et al. Hippo Signaling Pathway Dysregulation in Human Huntington’s Disease Brain and Neuronal Stem Cells. Sci. Rep. 2018, 8, 11355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbay, S.; Kahana, E.; Zilber, N.; Cooper, G.; Pintov, S.; Leibowitz, Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol. 1985, 232, 295–300. [Google Scholar] [CrossRef]
- Piccione, E.; Sletten, D.; Staff, N.; Low, P. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve 2015, 51, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, T.; Jozsa, F.; Al-Chalabi, A. Nonmotor Symptoms in Amyotrophic Lateral Sclerosis: A Systematic Review. Int. Rev. Neurobio. 2017, 134, 1409–1441. [Google Scholar]
- Carvalho, T.L.; de Almeida, L.M.; Lorega, C.M.; Barata, M.F.O.; Ferreira, M.L.B.; de Brito-Marques, P.R.; da Cunha Correia, C. Depression and anxiety in individuals with amyotrophic lateral sclerosis: A systematic review. Trends Psychiatry Psychother. 2016, 38, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Krasnianski, A.; Bohling, G.T.; Harden, M.; Zerr, I. Psychiatric Symptoms in Patients with Sporadic Creutzfeldt-Jakob Disease in Germany. J. Clin. Psychiatry 2015, 76, 1209–1215. [Google Scholar] [CrossRef]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein Front. Mol. Biosci. 2017, 4, 19. [Google Scholar]
- Sarnataro, D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navia, B.; Jordan, B.; Price, R. The AIDS dementia complex: I. Clinical features. Ann. Neurol. 1986, 19, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Korpelainen, J.T.; Sotaniemi, K.A.; Mäkikallio, A.; Huikuri, H.V.; Myllylä, V.V. Dynamic Behavior of Heart Rate in Ischemic Stroke. Stroke 1999, 30, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Fure, B.; Wyller, T.; Engedal, K.; Thommessen, B. Emotional symptoms in acute ischemic stroke. Clin. Auton. Res. 2006, 21, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Walker, F.R.; Nilsson, M.; Jones, K. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 2013, 14, 1262–1276. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Jiang, C. Inflammation: The Common Pathway of Stress-Related Diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef]
- Allison, D.J.; Ditor, D.S. The common inflammatory etiology of depression and cognitive impairment: A therapeutic target. J. Neuroinflammation 2014, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Schiffrin, E.L. Inflammation, immunity and development of essential hypertension. J. Hypertens. 2014, 32, 228–229. [Google Scholar] [CrossRef]
- Brew, B.J.; Crowe, S.M.; Landay, A.; Cysique, L.A.; Guillemin, G. Neurodegeneration and ageing in the HAART era. J. Neuroimmune Pharmacol. 2009, 4, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wolkowitz, O.M.; Epel, E.S.; Reus, V.I.; Mellon, S.H. Depression gets old fast: Do stress and depression accelerate cell aging? Depress. Anxiety 2010, 27, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Comparative Overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, M.J.; Baune, B.T. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: A systematic review of biomarker studies. Neurosci. Biobehav. Rev. 2014, 42, 93–115. [Google Scholar] [CrossRef]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Song, F.; Fernandez-Escobar, A.; Luo, G.; Wang, J.H.; Sun, Y. The properties of cytokines in multiple sclerosis: Pros and cons. Am. J. Med. Sci. 2018, 356, 552–560. [Google Scholar] [CrossRef]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef]
- Chang, K.H.; Wu, Y.R.; Chen, Y.C.; Chen, C.M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef]
- Bouwens, J.A.; van Duijn, E.; Cobbaert, C.M.; Roos, R.A.C.; van der Mast, R.C.; Giltay, E.J. Affiliations expand. Disease stage and plasma levels of cytokines in Huntington’s disease: A 2-year follow-up study. Mov. Disord. 2017, 32, 1103–1104. [Google Scholar] [CrossRef]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Möller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cao, C.; Qin, X.Y.; Yu, Y.; Yuan, J.; Zhao, Y.; Cheng, Y. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: A meta-analysis study. Sci. Rep. 2017, 7, 9094. [Google Scholar] [CrossRef] [PubMed]
- Van Everbroeck, B.; Dewulf, E.; Pals, P.; Lübke, U.; Martin, J.J.; Cras, P. The Role of Cytokines, Astrocytes, Microglia and Apoptosis in Creutzfeldt-Jakob Disease. Neurobiol. Aging 2002, 23, 59–64. [Google Scholar] [CrossRef]
- Stoeck, K.; Bodemer, M.; Ciesielczyk, B.; Meissner, B.; Bartl, M.; Heinemann, U.; Zerr, I. Interleukin 4 and Interleukin 10 Levels Are Elevated in the Cerebrospinal Fluid of Patients With Creutzfeldt-Jakob Disease. Arch. Neurol. 2005, 62, 1591–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- And Anti-Inflammatory Cytokines in the CSF of Patients With Creutzfeldt-Jakob Disease. J. Neuroimmunol. 2006, 172, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Matsui, N.; Takahashi, Y.; Iwasaki, Y.; Yoshida, M.; Yuasa, T.; Izumi, Y.; Kaji, R. Increased interleukin-17 in the Cerebrospinal Fluid in Sporadic Creutzfeldt-Jakob Disease: A Case-Control Study of Rapidly Progressive Dementia. J. Neuroinflammation 2013, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Stoeck, K.; Schmitz, M.; Ebert, E.; Schmidt, C.; Zerr, I. Immune Responses in Rapidly Progressive Dementia: A Comparative Study of Neuroinflammatory Markers in Creutzfeldt-Jakob Disease, Alzheimer’s Disease and Multiple Sclerosis. J. Neuroinflammation 2014, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.; Cohen, R.; Gongvatana, A.; Ross, S.; Olchowski, J.; Devlin, K.; Tashima, K.; Navia, B.; Delamonte, S. Relationship of plasma cytokines and clinical biomarkers to memory performance in HIV. J. Neuroimmunol. 2013, 265, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Qiao, L.; Wei, F.; Yin, J.; Liu, L.; Ji, Y.; Smith, D.; Li, N.; Chen, D. Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. J. Neurovirol. 2013, 19, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Seilhean, D.; Kobayashi, K.; He, Y.; Rosenblum, O.; Katlama, C.; Bricaire, F.; Duyckaerts, C.; Hauw, J.J. Tumor necrosis factor-alpha, microglia and astrocytes in AIDS dementia complex. Acta Neuropathol. 1997, 93, 508–517. [Google Scholar] [CrossRef]
- Lambertsen, K.; Biber, K.; Finsen, B. Inflammatory cytokines in experimental and human stroke. J. Cerebr. Blood F. Met. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarkowski, E.; Rosengren, L.; Blomstrand, C.; Wikkelsö, C.; Jensen, C.; Ekholm, S.; Tarkowski, A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin. Exp. Immunol. 1997, 110, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Morra, M.; Alecci, M.; Galloni, E.; Marchi, M.; Toso, V. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci. 2001, 22, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, G.; Sarchielli, P.; Caso, V.; Paciaroni, M.; Floridi, A.; Floridi, A.; Gallai, V. Different cytokine levels in thrombolysis patients as predictors for clinical outcome. Eur. J. Neurol. 2004, 11, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Kashyap, R.; Kabra, D.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann. Indian Acad. Neurol. 2012, 15, 181–185. [Google Scholar]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia Contributes to Plaque Growth by Cell Death Due to Uptake of tau β in the Brain of Alzheimer’s Disease Mouse Model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Perea, J.R.; Llorens-Martín, M.; Ávila, J.; Bolós, M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front. Cell Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef] [Green Version]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau Activates NLRP3-ASC Inflammasome Exacerbating Exogenously Seeded and Non-Exogenously Seeded Tau Pathology in Vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Vázquez Rodríguez, P.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early Microglial Activation and Peripheral Inflammation in Dementia with Lewy Bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef]
- Chung, H.K.; Ho, H.A.; Pérez-Acuña, D.; Lee, S.J. Modeling α-Synuclein Propagation with Preformed Fibril Injections. J. Mov. Disord. 2019, 12, 139–151. [Google Scholar] [CrossRef]
- Duffy, M.F.; Collier, T.J.; Patterson, J.R.; Kemp, C.J.; Luk, K.C.; Tansey, M.G.; Paumier, K.L.; Kanaan, N.M.; Fischer, D.L.; Polinski, N.K.; et al. Lewy Body-Like Alpha-Synuclein Inclusions Trigger Reactive Microgliosis Prior to Nigral Degeneration. J. Neuroinflammation 2018, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Thammisetty, S.S.; Pedragosa, J.; Weng, Y.C.; Calon, F.; Planas, A.; Krizcorresponding, J. Age-related deregulation of TDP-43 after stroke enhances NF-κB-mediated inflammation and neuronal damage. J. Neuroinflammation 2018, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; Khoury, J.E. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszekcorresponding, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Miranda-Saksena, M.; Saksena, N.K. Viruses and neurodegeneration. Virol. J. 2013, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking Cellular Stress Responses to Systemic Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef]
- Selye, H. The Stress of Life, Rev. ed.; McGraw-Hill: New York, NY, USA, 1976. [Google Scholar]
- McEwen, B.; Stellar, E. Stress and the individual. Mechanisms leading to disease. Arch. Intern. Med. 1993, 153, 2093–2101. [Google Scholar] [CrossRef]
- Sterling, P.; Eyer, J. Allostasis: A new paradigm to explain arousal pathology. In Handbook of Life Stress, Cognition, and Health; Fisher, S., Reason, J.T., Eds.; Wiley: Chichester, NY, USA, 1988. [Google Scholar]
- Van der Goot, A.T.; Nollen, E.A.A. Tryptophan Metabolism: Entering the Field of Aging and Age-Related Pathologies. Trends Mol. Med. 2013, 19, 336–344. [Google Scholar] [CrossRef]
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, T.A.; Nguyen, J.C.D.; Polglaze, K.E.; Bertrand, P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int. J. Tryptophan Res. 2018, 11, 1178646918802282. [Google Scholar] [CrossRef] [Green Version]
- Lieben, C.K.; Blokland, A.; Deutz, N.E.; Jansen, W.; Han, G.; Hupperts, R.M. Intake of tryptophan-enriched whey protein acutely enhances recall of positive loaded words in patients with multiple sclerosis. Clin. Nutr. 2018, 37, 321–328. [Google Scholar] [CrossRef]
- Herédi, J.; Berkó, A.M.; Jankovics, F.; Iwamori, T.; Iwamori, N.; Ono, E.; Horváth, S.; Kis, Z.; Toldi, J.; Vécsei, L.; et al. Astrocytic and neuronal localization of kynurenine aminotransferase-2 in the adult mouse brain. Brain Struct. Funct. 2017, 222, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef]
- Rózsa, E.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural. Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [Green Version]
- Robotka, H.; Toldi, J.; Vecsei, L. L-Kynurenine: Metabolism and mechanism of neuroprotection. Future Neurol. 2008, 3, 169–188. [Google Scholar] [CrossRef]
- Grant, R.S.; Coggan, S.E.; Smythe, G.A. The Physiological Action of Picolinic Acid in the Human Brain. Int. J. Tryptophan Res. 2009, 2, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Török, N.; Majláth, Z.; Fülöp, F.; Toldi, J.; Vécsei, L. Brain Aging and Disorders of the Central Nervous System: Kynurenines and Drug Metabolism. Curr. Drug Metab. 2016, 17, 412–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A. Hypothesis kynurenic and quinolinic acids: The main players of the kynurenine pathway and opponents in inflammatory disease. Med. Hypotheses 2018, 118, 129–138. [Google Scholar] [CrossRef]
- Bala, S.; Kambo, J.S.; Saini, V.; Prasad, D.N. Anti-inflammatory, analgesic evaluation and molecular docking studies of N-phenyl anthranilic acid-based 1,3,4-oxadiazole analogues. J. Chem. 2013, 2013, 412053. [Google Scholar] [CrossRef] [Green Version]
- Dehhaghi, M.; Panahi, H.K.S.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12, 1178646919852996. [Google Scholar] [CrossRef] [Green Version]
- Boros, F.A.; Bohár, Z.; Vécsei, L. Genetic alterations affecting the genes encoding the enzymes of the kynurenine pathway and their association with human diseases. Mutat. Res. 2018, 776, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Boros, F.A.; Vécsei, L. Immunomodulatory Effects of Genetic Alterations Affecting the Kynurenine Pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef]
- Romero, R.; Chaemsaithong, P.; Yeo, L.; Docheva, N.; Chaiyasit, N. Fetal and Maternal Responses to Intraamniotic Infection. In Fetal and Neonatal Physiology, 5th ed.; Barichello, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 1, pp. 144–159. [Google Scholar]
- Ciorba, M.A. Kynurenine pathway metabolites: Relevant to vitamin B-6 deficiency and beyond. Am. J. Clin. Nutr. 2013, 98, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Pescosolido, N.; Barbato, A.; Giannotti, R.; Komaiha, C.; Lenarduzzi, F. Age-related changes in the kinetics of human lenses: Prevention of the cataract. Int. J. Ophthalmol. 2016, 9, 1506–1517. [Google Scholar]
- Majláth, Z.; Toldi, J.; Vécsei, L. The potential role of kynurenines in Alzheimer’s disease: Pathomechanism and therapeutic possibilities by influencing the glutamate receptors. J. Neural Transm. 2014, 121, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Huitróna, R.; Blanco-Ayalaa, T.; Ugalde-Muñiza, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S. Targeting the Broadly Pathogenic Kynurenine Pathway; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Fucíková, J.; Jelinkova, L.P.; Bartunkova, J.; Radek, S. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Routy, B.; Graziani, G.M.; Vikram, M. The Kynurenine Pathway Is a Double-Edged Sword in Immune-Privileged Sites and in Cancer: Implications for Immunotherapy. Int. J. Tryptophan Res. 2016, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pour, S.R.; Morikawa, H.; Kiani, N.A.; Yang, M.; Azimi, A.; Shafi, G.; Shang, M.; Baumgartner, R.; Ketelhuth, D.F.J.; Kamleh, M.A.; et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci. Rep. 2019, 9, 12150. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Tullius, S.G.; Biefer, H.R.; Li, S.; Trachtenberg, A.J.; Edtinger, K.; Quante, M.; Krenzien, F.; Uehara, H.; Yang, X.; Kissick, H.T.; et al. NAD protects against EAE by regulating CD4 T-cell differentiation. Nat. Commun. 2014, 5, 5101. [Google Scholar] [CrossRef]
- Recalcati, S.; Gammella, E.; Cairo, G. Ironing out Macrophage Immunometabolism. Pharmaceuticals (Basel) 2019, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Heng, B.; Lim, C.K.; Lovejoy, D.B.; Bessede, A.; Gluch, L.; Guillemin, G.J. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget 2016, 7, 6506–6520. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.; Hoey, L.; Hughes, C.F.; Ward, M.; McNulty, H. Causes, Consequences and Public Health Implications of Low B-Vitamin Status in Ageing. Nutrients 2016, 8, 725. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.F.; Ward, M.; Tracey, F.; Hoey, L.; Molloy, A.M.; Pentieva, K.; McNulty, H. B-Vitamin Intake and Biomarker Status in Relation to Cognitive Decline in Healthy Older Adults in a 4-Year Follow-Up Study. Nutrients 2017, 9, 53. [Google Scholar] [CrossRef]
- Rossi, F.; Miggiano, R.; Ferraris, D.M.; Rizzi, M. The Synthesis of Kynurenic Acid in Mammals: An Updated Kynurenine Aminotransferase Structural KATalogue. Front. Mol. Biosci. 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Salvo, M.L.; Safo, M.K.; Contestabile, R. Biomedical aspects of pyridoxal 5’-phosphate availability. Front. Biosci. 2012, 4, 897–913. [Google Scholar]
- Parra, M.; Stahl, S.; Hellmann, H. Vitamin B6 and Its Role in Cell Metabolism and Physiology. Cells 2018, 7, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majláth, Z.; Török, N.; Toldi, J.; Vécsei, L. Memantine and Kynurenic Acid: Current Neuropharmacological Aspects. Curr. Neuropharmacol. 2016, 14, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Markey, S.P.; Heyes, M.P. Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience 1992, 51, 25–39. [Google Scholar] [CrossRef]
- Guillemin, G.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef]
- Chen, Y.; Stankovic, R.; Cullen, K.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef]
- Iłzecka, I.; Kocki, J.; Stelmasiak, T.; Turski, Z. WA Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Hainfellner, J.; Kepplinger, B.; Mazal, P.R.; Schmid, H.; Budka, H. Kynurenic acid metabolism in the brain of HIV-1 infected patients. J. Neural Transm. 2000, 107, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Jennekens, F.G. A short history of the notion of neurodegenerative disease. J. Hist. Neurosci. 2014, 23, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Khurshid Baig, M.H.; Mushtaq, G.; Kamal, M.A.; Greig, N.H.; Choi, I. Commonalities in Biological Pathways, Genetics, and Cellular Mechanism between Alzheimer Disease and Other Neurodegenerative Diseases: An In Silico-Updated Overview. Curr. Alzheimer Res. 2017, 14, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.J.; Babulal, G.M.; Monsell, S.E.; Cairns, N.J.; Roe, C.M.; Morris, J.C. Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol. 2015, 72, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Kragh-Sorensen, P. Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch. Neurol. 2003, 60, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Gomperts, S.N. Lewy Body Dementias: Dementia with Lewy Bodies and Parkinson Disease Dementia. Lifelong Learn. Neurol. 2016, 22, 435–463. [Google Scholar] [CrossRef]
- Berlyand, Y.; Weintraub, D.; Xie, S.X.; Mellis, I.A.; Doshi, J.; Rick, J.; McBride, J.; Davatzikos, C.; Shaw, L.M.; Hurtig, H.; et al. An Alzheimer’s Disease-Derived Biomarker Signature Identifies Parkinson’s Disease Patients with Dementia. PLoS ONE 2016, 11, e0147319. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A.; Korczyn, A.D. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef] [Green Version]
- Giguère, N.; Nanni, S.B.; Trudeau, L.E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Parisi, J.E.; Petrucelli, L.; Jack, C.R.; Petersen, R.C.; Dickson, D.W. Staging TDP43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014, 127, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Vignos, M.; Dudman, J.; Chang, A.; Fisher, E.; Staugaitis, S.M.; Battapady, H.; Mork, S.; Ontaneda, D.; Jones, S.E.; et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: A retrospective study. Lancet Neurol. 2018, 17, 870–884. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.L.; Nopoulos, P.C.; Gonzalez-Alegre, P. Human Immunodeficiency Virus Infection in Huntington’s Disease is Associated with an Earlier Age of Symptom Onset. J. Huntingt. Dis. 2018, 7, 163–166. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. β-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. CSH Perspect. Med. 2017, 8, a023507. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-Like Properties of Amyloid-β Assemblies: Implications for Alzheimer’s Disease. CSH Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [Green Version]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Maryam, S.; Naini, A.; Mandelkow, E.M.; Mandelkow, E.; Buée, L.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef]
- Milanini, B.; Valcour, V. Differentiating HIV-Associated Neurocognitive Disorders from Alzheimer’s Disease: An Emerging Issue in Geriatric NeuroHIV. Curr. HIV/AIDS Rep. 2017, 14, 123–132. [Google Scholar] [CrossRef]
- Vijayan, M.; Reddy, P.H. Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J. Alzheimers Dis. 2016, 54, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, J.; Tsuji, S. Genomic Aspects of Sporadic Neurodegenerative Diseases. Biochem. Biophys. Res. Commun. 2014, 452, 221–225. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
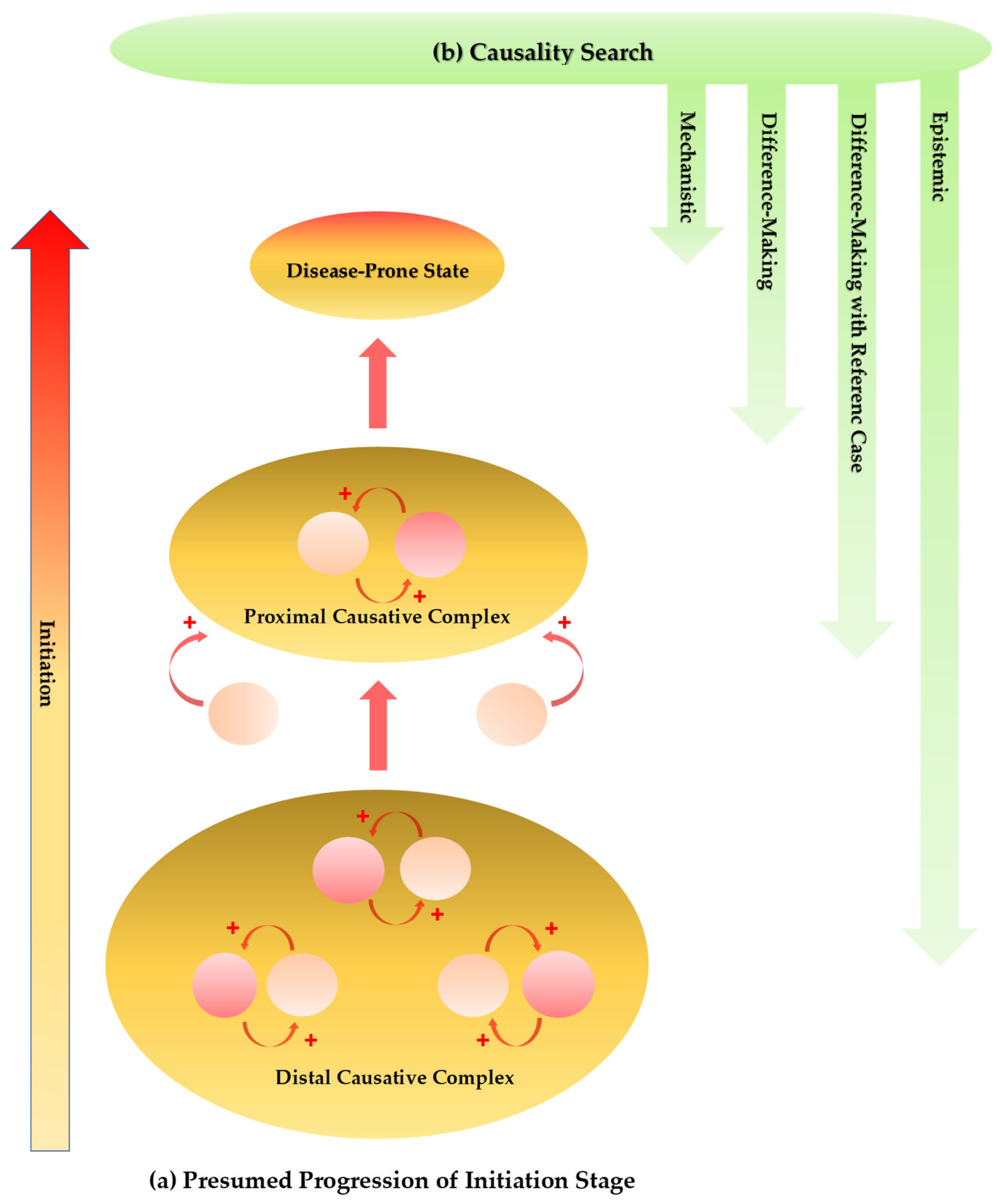
- Bach, J.F. Causality in medicine. Comptes Rendus Biol. 2019, 342, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutch, D.M.; Zulyniak, M.A.; Rudkowska, I.; Tejero, M.E. Lifestyle Genomics: Addressing the Multifactorial Nature of Personalized Health. Lifestyle Genom. 2018, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.E.; Herrera, P.F.; Laura, R. Effect of nutrition on neurodegenerative diseases. A systematic review. Nutr. Neurosci. 2019, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Corpas, R.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Sanfeliu, C.; Pallàsa, M. Understanding Epigenetics in the Neurodegeneration of Alzheimer’s Disease: SAMP8 Mouse Model. J. Alzheimers Dis. 2018, 62, 943–963. [Google Scholar] [CrossRef] [Green Version]
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Invest. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Mackie, J.L. The Cement of the Universe. A Study of Causation; Oxford University Press: Oxford, UK, 1976. [Google Scholar]
- Egger, G.; Dixon, J. Beyond Obesity and Lifestyle: A Review of 21st Century Chronic Disease Determinants. BioMed Res. Int. 2014, 2014, 731685. [Google Scholar] [CrossRef] [Green Version]
- Fedak, K.M.; Bernal, A.; Capshaw, Z.A.; Gross, S. Applying the Bradford Hill criteria in the 21st century: How data integration has changed causal inference in molecular epidemiology. Emerg. Themes Epidemiol. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, L.F.S.C.; Dalgalarrondo, P.; Banzato, C.E.M. On the notion of causality in medicine: Addressing Austin Bradford Hill and John L. Mackie. Rev. Psiquiatr. Clín. 2014, 41, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Konopka, T.; Smedley, D. Incremental data integration for tracking genotype-disease associations. PLoS Comput. Biol. 2020, 16, e1007586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, F.; Williamson, J. Epistemic Causality and Evidence-Based Medicine. Hist. Phil. Life Sci. 2011, 33, 563–581. [Google Scholar]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 hippocampus dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Bonda, D.; Mailankot, M.; Stone, J.G.; Garrett, M.R.; Staniszewska, M.; Castellani, R.J.; Siedlak, S.L.; Zhu, X.; Lee, H.; Perry, G.; et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010, 15, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Schwarcz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345. [Google Scholar] [CrossRef]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. Arizona Parkinson’s Disease Consortium. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Cheng, M.L.; Tang, H.Y.; Huang, C.Y.; Wu, Y.R.; Chen, C.M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Pearson, S.J. Increased brain 3-hydroxykynurenine in Huntington’s disease. Lancet 1989, 2, 979–980. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Jauch, D.; Urbańska, E.M.; Guidetti, P.; Bird, E.D.; Vonsattel, J.P.; Whetsell, W.O., Jr.; Schwarcz, R. Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: Focus on kynurenine aminotransferases. J. Neurol. Sci. 1995, 130, 39–47. [Google Scholar] [CrossRef]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef]
- Yan, E.B.; Frugier, T.; Lim, C.K.; Heng, B.; Sundaram, G.; Tan, M.; Rosenfeld, J.V.; Walker, D.W.; Guillemin, G.J.; Morganti-Kossmann, M.C. Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflamm. 2015, 30, 110. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.P.; Green, S. Cochrane Handbook for Systematic Reviews of Interventions, version 5.1.0.; The Cochrane Collaboration: London, UK, 2011; Available online: https://handbook-5-1.cochrane.org/front_page.htm (accessed on 8 December 2019).






| Diseases | Pro-Inflammatory Cytokines | Anti-Inflammatory Cytokines |
|---|---|---|
| Alzheimer’s disease | ↑ | ↑ |
| Parkinson’s disease | ↑ | ↑ |
| Multiple sclerosis | ↑ | ↓ |
| Huntington’s disease | ↑ | ↑ |
| Amyotrophic lateral sclerosis | ↑ | - |
| Creutzfeldt–Jakob disease | ↑ | ↑ |
| HIV-associated neurocognitive disorders | ↑ | ↑ |
| Stroke-induced secondary neurodegeneration | ↑ | ↓ |
| Diseases | Neurotoxic Kynurenines | Neuromodulatory Kynurenines |
|---|---|---|
| Alzheimer’s disease | ↑ | ↓ |
| Parkinson’s disease | ↑ | ↓ |
| Multiple sclerosis | ↑ | ↑ ↓ |
| Huntington’s disease | ↑ | ↓ |
| Amyotrophic lateral sclerosis | ↑ | ↑ ↓ |
| Creutzfeldt–Jakob disease | ? | ? |
| HIV-associated neurocognitive disorders | ↑ | ↑ |
| Stroke-induced secondary neurodegeneration | ↑ | ↑ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072431
Tanaka M, Toldi J, Vécsei L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. International Journal of Molecular Sciences. 2020; 21(7):2431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072431
Chicago/Turabian StyleTanaka, Masaru, József Toldi, and László Vécsei. 2020. "Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines" International Journal of Molecular Sciences 21, no. 7: 2431. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072431