Impact of Type 2 Diabetes Mellitus on Human Bone Marrow Stromal Cell Number and Phenotypic Characteristics

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
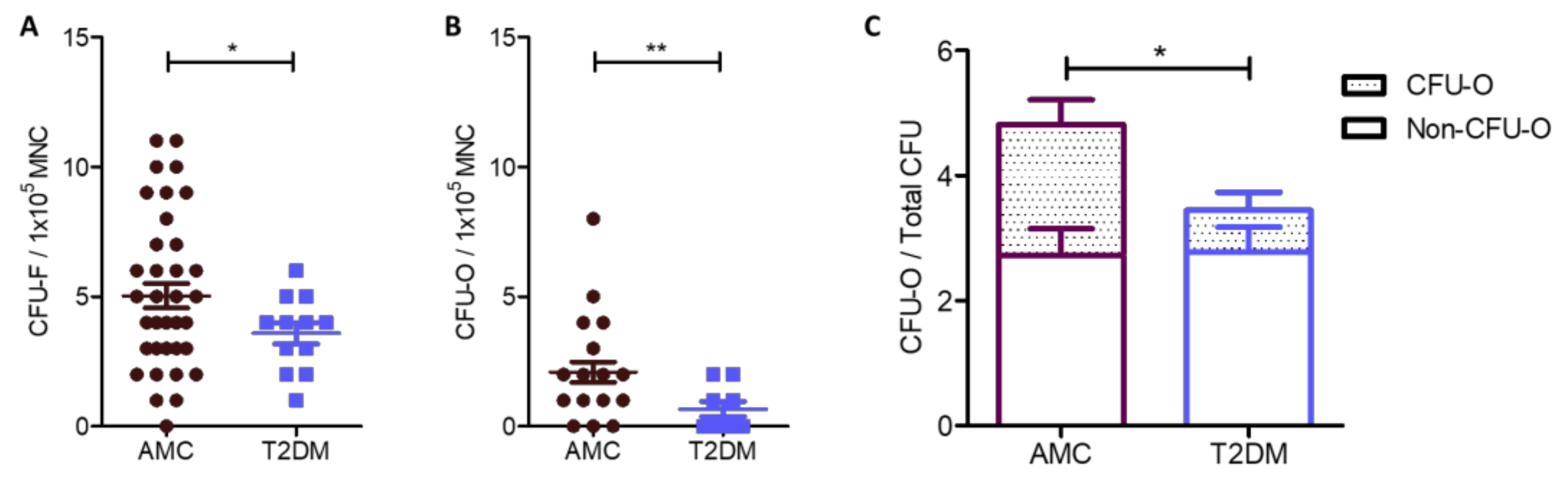
2.1. Reduced Numbers of MSCs in the Bone Marrow of People Living With T2DM Compared to AMC, Due to a Specific Decrease in the Size of the Osteoblast-Committed Subpopulation
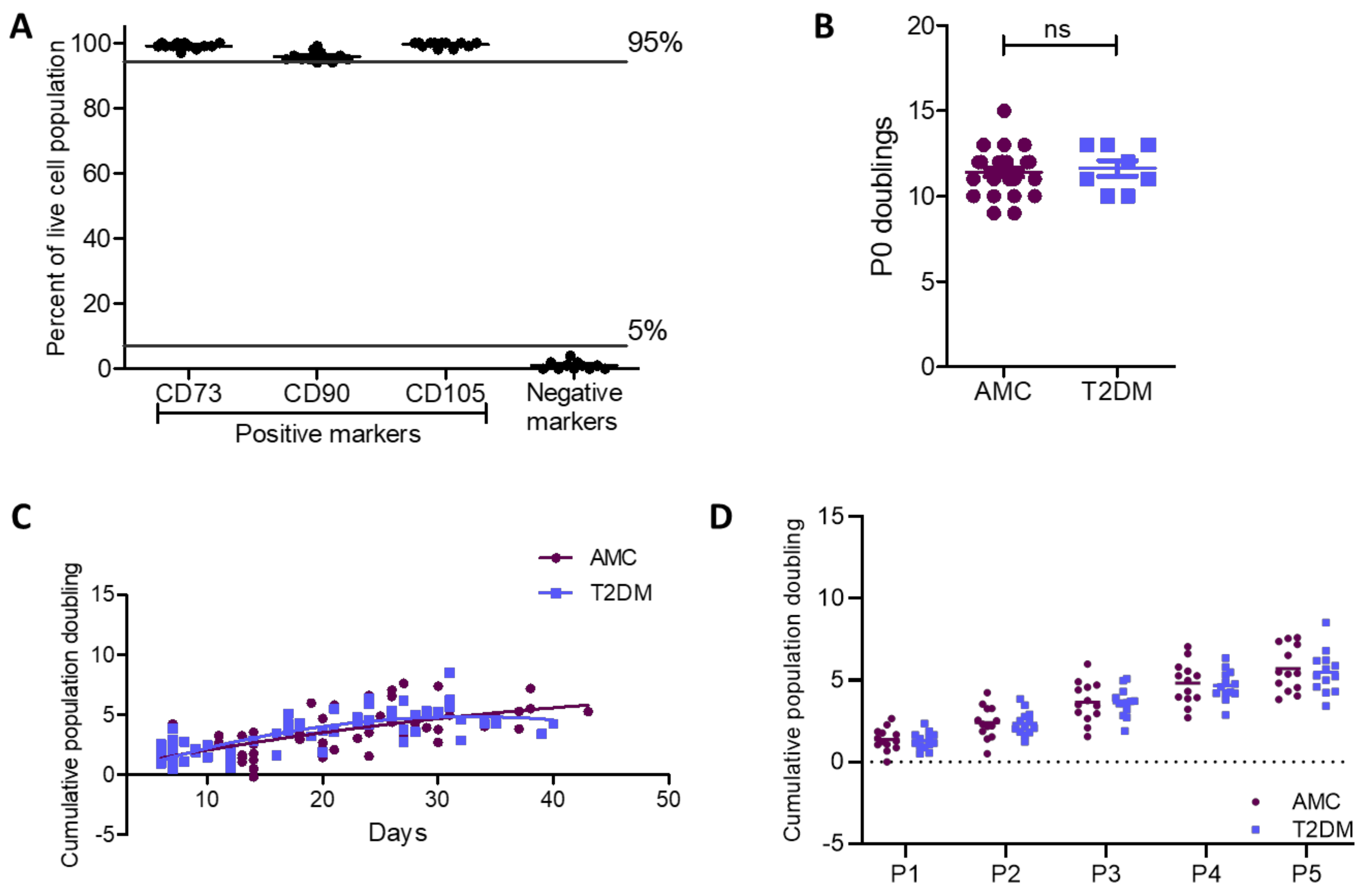
2.2. Cell Surface Characterisation and Cumulative Population Doublings of MSCs Unaffected by T2DM
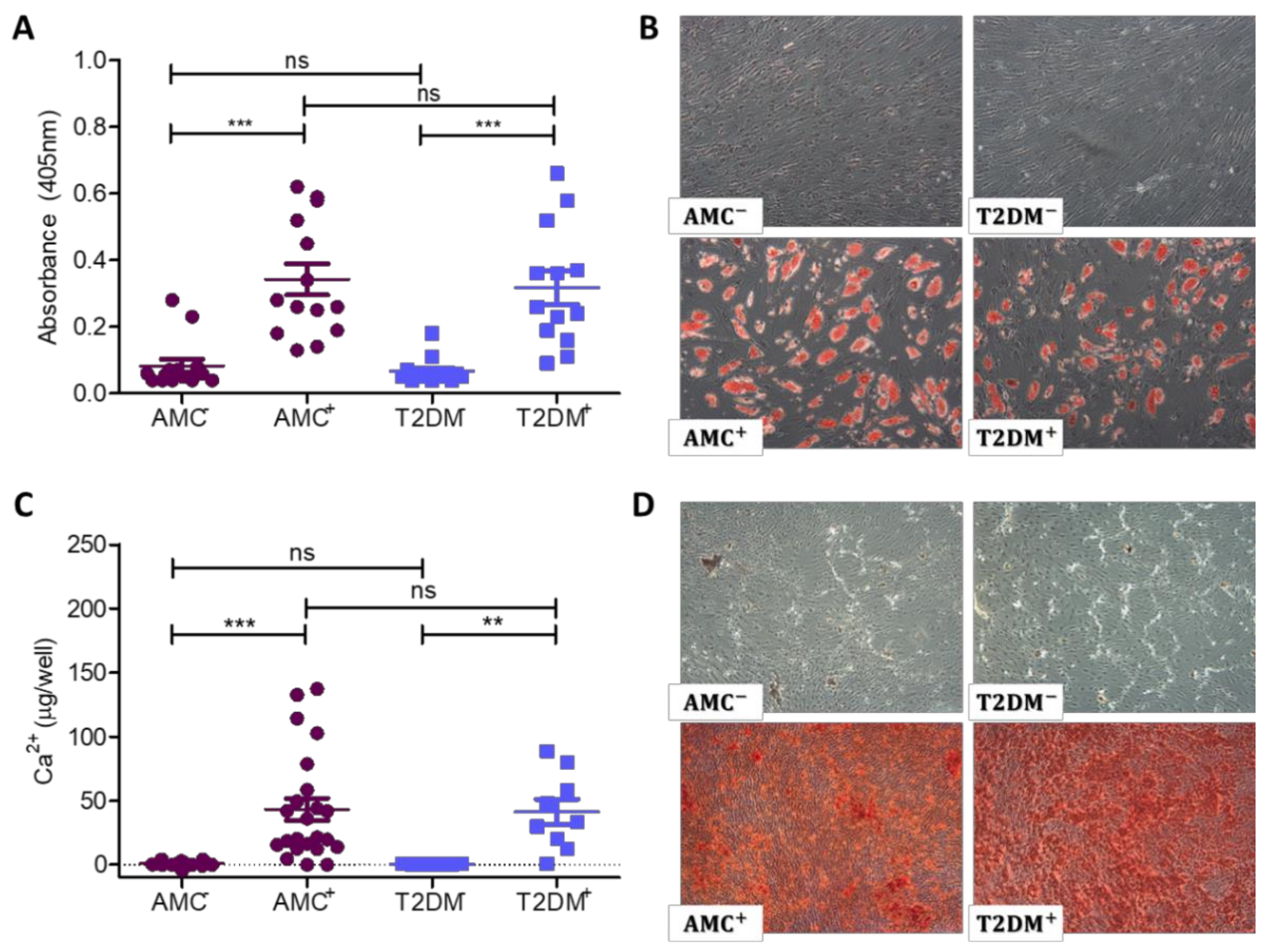
2.3. Adipogenic and Osteogenic Differentiation Capacity not Permanently Unbalanced by T2DM
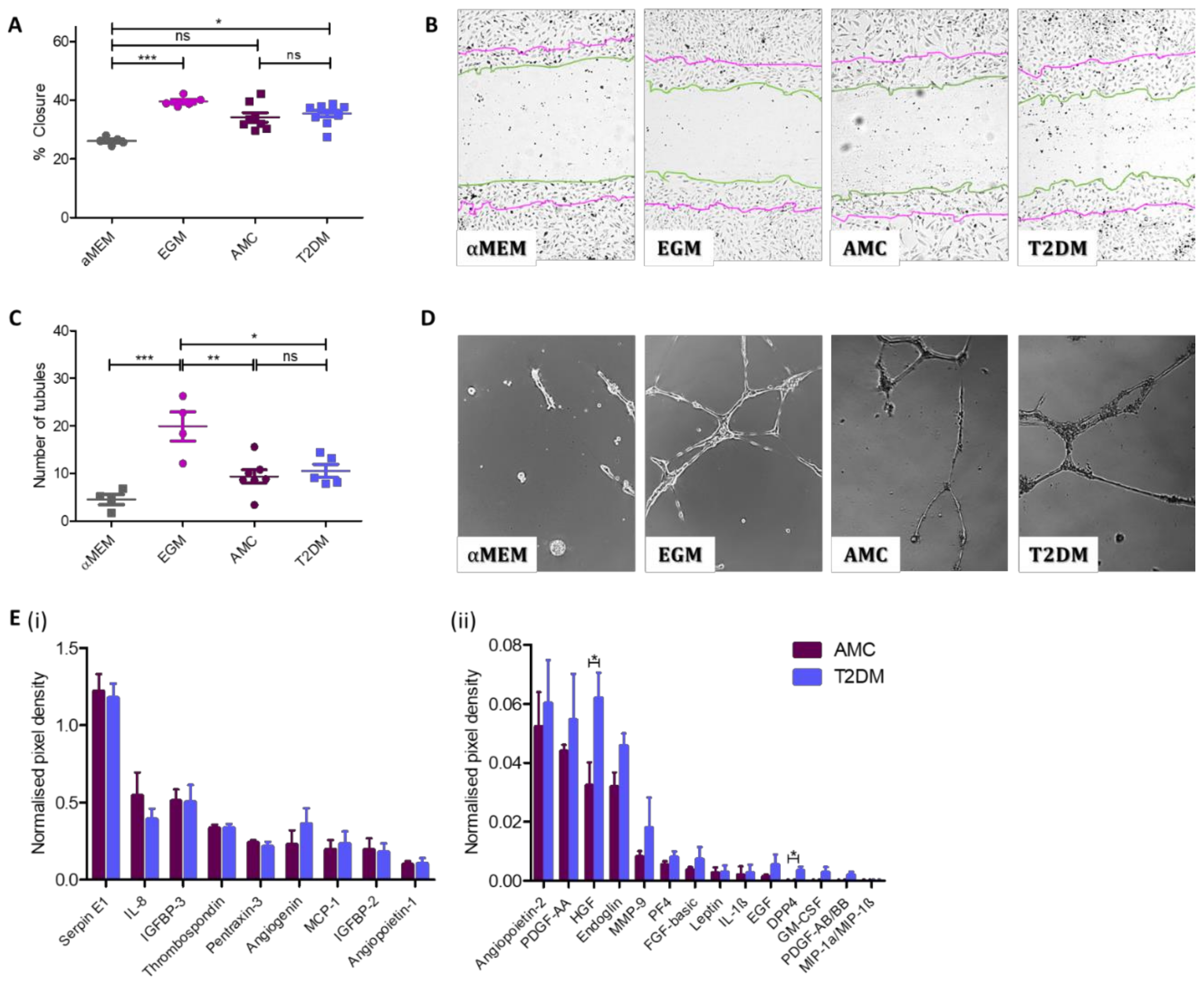
2.4. Angiogenic Capacity Comparable in MSCs Isolated From Donors With or Without T2DM
2.5. Comparable Self-Mobility in T2DM and AMC Cohorts
3. Discussion
4. Materials and Methods
4.1. Bone Marrow Donors
4.2. Mesenchymal Stromal Cell Isolation
4.3. Colony Forming Unit Assays
4.4. Flow Cytometry
4.5. Cumulative Population Doublings
4.6. Osteogenic and Adipogenic Differentiation
4.7. Scratch Assay
4.8. Matrigel Assays
4.9. Chemiarray Analysis of Secreted Proteins
4.10. Transwell Migration Assay
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | Advanced glycation end products |
| AMC | Age-matched controls |
| BMI | Body mass index |
| BMD | Bone mineral density |
| CD | Cluster of differentiation |
| CFU-F | Colony-forming unit fibroblast |
| CFU-O | Colony-forming unit osteoblast |
| CO2 | Carbon dioxide |
| DMEM | Dulbecco’s modified eagle medium |
| DM | Diabetes mellitus |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| EGM | Endothelial growth medium |
| FBS | Fetal bovine serum |
| FGF | Fibroblast growth factor |
| GDPR | General data protection regulation (EU) |
| HGF | Hepatocyte growth factor |
| HLA-DR | Human leukocyte antigen—DR isotype |
| HUVEC | Human umbilical vein endothelial cell |
| ISCT | International Society of Cell and Gene Therapy |
| MEM | Minimum essential medium |
| MIX | 3-Isobutyl-1-Methyl-Xanthine |
| MNC | Mononuclear cell |
| MSC | Mesenchymal stromal cell |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NBT/BCIP | p-nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate |
| P | Passage |
| PBS | Phosphate buffered saline |
| PE | Phycoerythrin |
| PS | Penicillin-Streptomycin |
| RAGE | Receptor for advanced glycation end products |
| SEM | Standard error of the mean |
| T2DM | Type 2 diabetes mellitus |
References
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Consentius, C.; Mirenska, A.; Jurisch, A.; Reinke, S.; Scharm, M.; Zenclussen, A.C.; Hennig, C.; Volk, H.-D. In situ detection of CD73+ CD90+ CD105+ lineage: Mesenchymal stromal cells in human placenta and bone marrow specimens by chipcytometry. Cytom. Part A 2018, 93, 889–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Feng, X. Genetic contribution to mesenchymal stem cell dysfunction in systemic lupus erythematosus. Stem Cell Res. Ther. 2018, 9, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennert, R.C.; Sorkin, M.; Januszyk, M.; Duscher, D.; Kosaraju, R.; Chung, M.T.; Lennon, J.; Radiya-Dixit, A.; Raghvendra, S.; Maan, Z.N.; et al. Diabetes impairs the angiogenic potential of adipose-derived stem cells by selectively depleting cellular subpopulations. Stem Cell Res. Ther. 2014, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Han, J.W.; Lee, J.Y.; Choi, Y.J.; Sohn, Y.-D.; Song, M.; Yoon, Y.-S. Diabetic Mesenchymal Stem Cells Are Ineffective for Improving Limb Ischemia Due to Their Impaired Angiogenic Capability. Cell Transplant. 2015, 24, 1571–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, A.V. Diabetes Mellitus: Does it Affect Bone? Calcif. Tissue Int. 2003, 73, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Räkel, A.; Sheehy, O.; Rahme, E.; LeLorier, J. Osteoporosis among patients with type 1 and type 2 diabetes. Diabetes Metab. 2008, 34, 193–205. [Google Scholar] [CrossRef]
- Ma, L.; Oei, L.; Jiang, L.; Estrada, K.; Chen, H.; Wang, Z.; Yu, Q.; Zillikens, M.C.; Gao, X.; Rivadeneira, F. Association between bone mineral density and type 2 diabetes mellitus: A meta-analysis of observational studies. Eur. J. Epidemiol. 2012, 27, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, Y.; Fritton, J.C.; Yakar, S.; Epstein, S.; Schaffler, M.B.; Jepsen, K.J.; LeRoith, D. Type 2 diabetic mice demonstrate slender long bones with increased fragility secondary to increased osteoclastogenesis. Bone 2009, 44, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Stolzing, A.; Sellers, D.; Llewelyn, O.; Scutt, A. Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs 2010, 191, 453–465. [Google Scholar] [CrossRef]
- Park, D.; Spencer, J.A.; Koh, B.I.; Kobayashi, T.; Fujisaki, J.; Clemens, T.L.; Lin, C.P.; Kronenberg, H.M.; Scadden, D.T. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012, 10, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.-C.; Chuang, P.-Y.; Yang, T.-Y.; Huang, T.-W.; Chang, S.-F. Hyperglycemia inhibits osteoblastogenesis of rat bone marrow stromal cells via activation of the Notch2 signaling pathway. Int. J. Med. Sci. 2019, 16, 696–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, M.; Abu-Shahba, N.; Azmy, O.; El-Badri, N. Impact of Diabetes Mellitus on Human Mesenchymal Stromal Cell Biology and Functionality: Implications for Autologous Transplantation. Stem Cell Rev. Rep. 2019, 15, 194–217. [Google Scholar] [CrossRef] [PubMed]
- Phadnis, S.M.; Ghaskadbi, S.M.; Hardikar, A.A.; Bhonde, R.R. Mesenchymal stem cells derived from bone marrow of diabetic patients portrait unique markers influenced by the diabetic microenvironment. Rev. Diabet. Stud. 2009, 6, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Z.; Liu, T.; Xue, X.; Jiang, H.; Huang, J.; Wang, H. Impaired cardioprotective function of transplantation of mesenchymal stem cells from patients with diabetes mellitus to rats with experimentally induced myocardial infarction. Cardiovasc. Diabetol. 2013, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Ferland-Mccollough, D.; Maselli, D.; Spinetti, G.; Sambataro, M.; Sullivan, N.; Blom, A.; Madeddu, P.; Masseli, D.; Spinetti, G.; Sambataro, M. MCP-1 Feedback Loop Between Adipocytes and Mesenchymal Stromal Cells Causes Fat Accumulation and Contributes to Hematopoietic Stem Cell Rarefaction in the Bone Marrow of Patients with Diabetes. Diabetes 2018, 67, 1380–1394. [Google Scholar] [CrossRef] [Green Version]
- Rezaie, J.; Nejati, V.; Khaksar, M.; Oryan, A.; Aghamohamadzadeh, N.; Shariatzadeh, M.A.; Rahbarghazi, R.; Mehranjani, M.S. Diabetic sera disrupted the normal exosome signaling pathway in human mesenchymal stem cells in vitro. Cell Tissue Res. 2018, 374, 555–565. [Google Scholar] [CrossRef]
- Siegel, G.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schäfer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Fossett, E.; Khan, W.S.; Longo, U.G.; Smitham, P.J. Effect of age and gender on cell proliferation and cell surface characterization of synovial fat pad derived mesenchymal stem cells. J. Orthop. Res. 2012, 30, 1013–1018. [Google Scholar] [CrossRef]
- Bara, J.J.; Richards, R.G.; Alini, M.; Stoddart, M.J. Concise Review: Bone Marrow-Derived Mesenchymal Stem Cells Change Phenotype Following In Vitro Culture: Implications for Basic Research and the Clinic. Stem Cells 2014, 32, 1713–1723. [Google Scholar] [CrossRef]
- Yang, Y.-H.K.; Ogando, C.R.; See, C.W.; Chang, T.-Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, N.; Rojewski, M.T.; Fürst, D.; Kreja, L.; Ignatius, A.; Dausend, J.; Schrezenmeier, H. GMP-compliant isolation and large-scale expansion of bone marrow-derived MSC. PLoS ONE 2012, 7, e43255. [Google Scholar] [CrossRef] [PubMed]
- Fekete, N.; Rojewski, M.T.; Lotfi, R.; Schrezenmeier, H. Essential Components for Ex Vivo Proliferation of Mesenchymal Stromal Cells. Tissue Eng. Part C Methods 2014, 20, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Lech, W.; Figiel-Dabrowska, A.; Sarnowska, A.; Drela, K.; Obtulowicz, P.; Noszczyk, B.H.; Buzanska, L.; Domanska-Janik, K. Phenotypic, Functional, and Safety Control at Preimplantation Phase of MSC-Based Therapy. Stem Cells Int. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Li, D.; Fu, J.; Shi, Q.; Lu, Y.; Ju, X. Comparison of biological properties of umbilical cord-derived mesenchymal stem cells from early and late passages: Immunomodulatory ability is enhanced in aged cells. Mol. Med. Rep. 2015, 11, 166–174. [Google Scholar] [CrossRef]
- Qu, C.; Brohlin, M.; Kingham, P.J.; Kelk, P. Evaluation of growth, stemness, and angiogenic properties of dental pulp stem cells cultured in cGMP xeno-/serum-free medium. Cell Tissue Res. 2020, 380, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Nogués, C.; O’Brien, T. In vitro models for assessing therapeutic angiogenesis. Drug Discov. Today 2016, 21, 1495–1503. [Google Scholar] [CrossRef]
- Mangialardi, G.; Madeddu, P. Bone Marrow-Derived Stem Cells: A Mixed Blessing in the Multifaceted World of Diabetic Complications. Curr. Diab. Rep. 2016, 16, 43. [Google Scholar] [CrossRef] [Green Version]
- Loder, R.T. The influence of diabetes mellitus on the healing of closed fractures. Clin. Orthop. Relat. Res. 1988, 232, 210–216. [Google Scholar] [CrossRef]
- Mushahary, D.; Spittler, A.; Kasper, C.; Weber, V.; Charwat, V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytom. Part A 2018, 93, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Schafer, A.L. Diabetes and Bone Marrow Adiposity. Curr. Osteoporos. Rep. 2016, 14, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabr, M.M.; Zakaria, M.M.; Refaie, A.F.; Ismail, A.M.; Abou-El-Mahasen, M.A.; Ashamallah, S.A.; Khater, S.M.; El-Halawani, S.M.; Ibrahim, R.Y.; Uin, G.S.; et al. Insulin-producing cells from adult human bone marrow mesenchymal stem cells control streptozotocin-induced diabetes in nude mice. Cell Transplant. 2013, 22, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.-C.; Hsieh, T.-Y.; Lai, H.-S.; Young, T.-H. High glucose-induced reactive oxygen species generation promotes stemness in human adipose-derived stem cells. Cytotherapy 2016, 18, 371–383. [Google Scholar] [CrossRef]
- Nawrocka, D.; Kornicka, K.; Szydlarska, J.; Marycz, K. Basic Fibroblast Growth Factor Inhibits Apoptosis and Promotes Proliferation of Adipose-Derived Mesenchymal Stromal Cells Isolated from Patients with Type 2 Diabetes by Reducing Cellular Oxidative Stress. Oxid. Med. Cell. Longev. 2017, 2017, 1–22. [Google Scholar] [CrossRef]
- Stolzing, A.; Coleman, N.; Scutt, A. Glucose-Induced Replicative Senescence in Mesenchymal Stem Cells. Rejuvenation Res. 2006, 9, 31–35. [Google Scholar] [CrossRef]
- Dentelli, P.; Barale, C.; Togliatto, G.; Trombetta, A.; Olgasi, C.; Gili, M.; Riganti, C.; Toppino, M.; Brizzi, M.F. A diabetic milieu promotes OCT4 and NANOG production in human visceral-derived adipose stem cells. Diabetologia 2013, 56, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Policha, A.; Zhang, P.; Chang, L.; Lamb, K.; Tulenko, T.; DiMuzio, P. Endothelial differentiation of diabetic adipose-derived stem cells. J. Surg. Res. 2014, 192, 656–663. [Google Scholar] [CrossRef]
- Cramer, C.; Freisinger, E.; Jones, R.K.; Slakey, D.P.; Dupin, C.L.; Newsome, E.R.; Alt, E.U.; Izadpanah, R. Persistent High Glucose Concentrations Alter the Regenerative Potential of Mesenchymal Stem Cells. Stem Cells Dev. 2010, 19, 1875–1884. [Google Scholar] [CrossRef]
- Kočí, Z.; Turnovcová, K.; Dubský, M.; Baranovičová, L.; Holáň, V.; Chudíčková, M.; Syková, E.; Kubinová, Š. Characterization of human adipose tissue-derived stromal cells isolated from diabetic patient’s distal limbs with critical ischemia. Cell Biochem. Funct. 2014, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Schilling, T.; Benisch, P.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Limbert, C.; Seufert, J.; Kassem, M.; Schütze, N.; et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem. Biophys. Res. Commun. 2007, 363, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.L.; Yukata, K.; Farnsworth, C.W.; Chen, D.G.; Awad, H.; Hilton, M.J.; O’Keefe, R.J.; Xing, L.; Mooney, R.A.; Zuscik, M.J. Delayed fracture healing and increased callus adiposity in a C57BL/6J Murine model of obesity-associated type 2 diabetes mellitus. PLoS ONE 2014, 9, e0099656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Tie, G.; Wang, S.; Messina, K.E.; DiDato, S.; Guo, S.; Messina, L.M. Type 2 Diabetes Restricts Multipotency of Mesenchymal Stem Cells and Impairs Their Capacity to Augment Postischemic Neovascularization in db/db Mice. J. Am. Heart Assoc. 2012, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Cai, X.; Dong, H.; Jing, W.; Huang, Y.; Yang, X.; Wu, Y.; Lin, Y. Serum regulates adipogenesis of mesenchymal stem cells via MEK/ERK-dependent PPARγ expression and phosphorylation. J. Cell. Mol. Med. 2010, 14, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.; Elliman, S.J.; Coleman, C.M. From isolation to implantation: A concise review of mesenchymal stem cell therapy in bone fracture repair. Stem Cell Res. Ther. 2014, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.H.; Lee, J.S.; Kim, D.-W.; Yoon, E.-S.; Dhong, E.-S. Neovascular potential of adipose-derived stromal cells (ASCs) from diabetic patients. Wound Repair Regen. 2012, 20, 243–252. [Google Scholar] [CrossRef]
- Bancks, M.P.; Bielinski, S.J.; Decker, P.A.; Hanson, N.Q.; Larson, N.B.; Sicotte, H.; Wassel, C.L.; Pankow, J.S. Circulating level of hepatocyte growth factor predicts incidence of type 2 diabetes mellitus: The Multi-Ethnic Study of Atherosclerosis (MESA). Metabolism 2016, 65, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Rezaie, J.; Mehranjani, M.S.; Rahbarghazi, R.; Shariatzadeh, M.A. Angiogenic and Restorative Abilities of Human Mesenchymal Stem Cells Were Reduced Following Treatment With Serum From Diabetes Mellitus Type 2 Patients. J. Cell. Biochem. 2018, 119, 524–535. [Google Scholar] [CrossRef]
- Serena, C.; Keiran, N.; Ceperuelo-Mallafre, V.; Ejarque, M.; Fradera, R.; Roche, K.; Nuñez-Roa, C.; Vendrell, J.; Fernández-Veledo, S. Obesity and Type 2 Diabetes Alters the Immune Properties of Human Adipose Derived Stem Cells. Stem Cells 2016, 34, 2559–2573. [Google Scholar] [CrossRef]
- Amable, P.R.; Teixeira, M.V.T.; Carias, R.B.V.; Granjeiro, J.M.; Borojevic, R. Protein synthesis and secretion in human mesenchymal cells derived from bone marrow, adipose tissue and Wharton’s jelly. Stem Cell Res. Ther. 2014, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Nicholson, L.; Al Harbi, N.; Mancuso, E.; Jones, E.; Dickinson, A.; Wang, X.N.; Dalgarno, K. Osteogenic potential of heterogeneous and CD271-enriched mesenchymal stromal cells cultured on apatite-wollastonite 3D scaffolds. BMC Biomed. Eng. 2019, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cassidy, F.C.; Shortiss, C.; Murphy, C.G.; Kearns, S.R.; Curtin, W.; De Buitléir, C.; O’Brien, T.; Coleman, C.M. Impact of Type 2 Diabetes Mellitus on Human Bone Marrow Stromal Cell Number and Phenotypic Characteristics. Int. J. Mol. Sci. 2020, 21, 2476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072476
Cassidy FC, Shortiss C, Murphy CG, Kearns SR, Curtin W, De Buitléir C, O’Brien T, Coleman CM. Impact of Type 2 Diabetes Mellitus on Human Bone Marrow Stromal Cell Number and Phenotypic Characteristics. International Journal of Molecular Sciences. 2020; 21(7):2476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072476
Chicago/Turabian StyleCassidy, Féaron C., Ciara Shortiss, Colin G. Murphy, Stephen R. Kearns, William Curtin, Ciara De Buitléir, Timothy O’Brien, and Cynthia M. Coleman. 2020. "Impact of Type 2 Diabetes Mellitus on Human Bone Marrow Stromal Cell Number and Phenotypic Characteristics" International Journal of Molecular Sciences 21, no. 7: 2476. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072476