Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy

1
Department of Biochemistry and Molecular Biology, College of Medicine, Yanan University, Yanan 716000, China
2
Division of Oncology, University of Saskatchewan, Saskatoon, SA S7N 4H4, Canada
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(8), 2793; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082793
Submission received: 16 December 2019
/
Revised: 9 April 2020
/
Accepted: 15 April 2020
/
Published: 17 April 2020
(This article belongs to the Section Biochemistry)
Abstract
:The arrival of the monoclonal antibody (mAb) technology in the 1970s brought with it the hope of conquering cancers to the medical community. However, mAbs, on the whole, did not achieve the expected wonder in cancer therapy although they do have demonstrated successfulness in the treatment of a few types of cancers. In 1990, another technology of making biomolecules capable of specific binding appeared. This technique, systematic evolution of ligands by exponential enrichment (SELEX), can make aptamers, single-stranded DNAs or RNAs that bind targets with high specificity and affinity. Aptamers have some advantages over mAbs in therapeutic uses particularly because they have little or no immunogenicity, which means the feasibility of repeated use and fewer side effects. In this review, the general properties of the aptamer, the advantages and limitations of aptamers, the principle and procedure of aptamer production with SELEX, particularly the undergoing studies in aptamers for cancer therapy, and selected anticancer aptamers that have entered clinical trials or are under active investigations are summarized.
1. Introduction
The advent of the technology of using B lymphocyte hybridomas to produce monoclonal antibodies (mAbs) in the 1970s gave the medical world a ray of hope to conquer cancers because mAbs are capable of binding to target molecules, such as tumor antigens specifically, and carrying cytotoxic agents selectively to cancer cells, which means effective destruction of cancer cells with minimum damage to normal cells. In the years that followed, mAbs demonstrated their exceptional usefulness particularly in biomedical researches and also in clinical diagnoses and treatment of a few types of cancers; nevertheless, because of a number of reasons, mAbs did not achieve the expected wonder in cancer therapy on the whole.
The 1990s of the last century witnessed another technology of making biomolecules capable of specific binding. The technique was dubbed systematic evolution of ligands by exponential enrichment (SELEX) and the molecules it produced were named aptamers [1,2]. Aptamers are single-stranded nucleic acids (either DNA or RNA) that can recognize targets through three-dimensional complementarity (rather than base pairing) and bind to targets with high specificity and affinity; hence, they have been described as nucleic acid antibodies [3]. Similarly to mAbs, aptamers can be used in laboratory research and in clinical diagnosis and treatment, but aptamers have some advantages over mAbs particularly in in vivo applications, one of which is that they have little or no immunogenicity and thus do not elicit immunological rejections, meaning the feasibility of repeated use and also fewer side effects [4]. There has already been an aptamer called Macugen (Pegaptanib Sodium Injection) being approved by the US Food and Drug Administration (FDA) for therapeutic application. This aptamer targets vascular endothelial growth factor (VEGF) and is used for the treatment of age-related macular degeneration (AMD) [5].
This paper summarizes the general properties of the aptamer, the advantages and limitations of aptamers compared with mAbs, the principle and procedure of aptamer production with SELEX, (particularly) the undergoing investigations into aptamer’s applications in cancer treatment, and selected aptamers that have entered clinical trials or are under active investigation for cancer therapy.
2. General Properties of the Aptamer
Single-stranded nucleic acid molecules can bind to complementary sequences via base pairings, but they can also form multiple secondary structures (such as stem-loops, hairpins, bugle, pseudoknots, etc.) and further form unique three-dimensional structures. Thus, they can bind to a great variety of targets including proteins, cells, bacteria, virions, and various small organic and inorganic molecules through closely matched spatial complementarity and by means of electrostatic/ionic interaction, hydrogen bonding, van der Waals’ force, as well as hydrophobic interactions [6]. Exploiting the latter properties, Gold lab and Szostak lab of the United States in the early 1990s independently established a kind of in vitro evolution technique that can generate oligonucleotides with high specificity and binding affinity [1,2]. The Szostak group gave the technique the name “in vitro selection” while the Gold group named it systematic evolution of ligands by exponential enrichment or SELEX; later literature all adopted the latter term. The Szostak group first called the molecule aptamer and the name has been accepted universally. Aptamers are single-stranded DNA (ssDNA) or single-stranded RNA (ssRNA) oligonucleotides of variable lengths (usually 20–80 nt residues). The sequence in the central region of the molecules, generated during chemical synthesis and selection, is highly random and can bind to various targets, while the flanking sequences, predesigned by the researcher, are constant and are used for aptamer selection or production (the sequences on both sides can be truncated or trimmed after the final selection). Aptamers can form diverse three-dimensional structures and can thus bind to a variety of targets specifically and tightly. The binding affinities (dissociation constants) of aptamers are usually in the range of low nanomolar to high picomolar but may reach femtomolar [7,8]. The recognition is highly exquisite and can discriminate structural differences between enantiomers (mirror images with identical chemical composition) and the presence or absence of a hydroxyl group [9].
Even though aptamers are highly specific, the issue of off-target binding must still be carefully investigated for them to meet regulations and enter clinical use. To date, quite a few aptamers have been shown to have no or negligible target toxicity (not to bind to off-target cells/tissue) [10,11,12,13,14,15,16,17].
3. Advantages and Limitations of Aptamers
Aptamers can bind to targets with high specificity and affinity, similar to mAbs, and they are also similar to mAbs in terms of biomedical applications. However, aptamers have a significant advantage over mAbs in medications because they have no or little immunogenicity and will not cause immune rejection. Experiments using monkeys demonstrated that there was no or only negligible immune response even when the dose was 1000 times higher than the normal therapeutic dose [18,19]. Moreover, aptamers can conjugate with and deliver various therapeutics into cells in addition to affecting target molecules directly [20]. Refer to Table 1 for a comparison between aptamers and mAbs.
Aptamers have other advantages as well: (1) Aptamers have a broader spectrum of targets than mAbs—they can be raised against non-immunogenic substances such as small organic and inorganic molecules and even metal ions; they can be made against strong bio-toxins because they do not need animal immunization for production [25]. (2) The manufacture of aptamers is less time consuming because they are made in vitro thus avoiding the long time animal immunization; in addition, the affinity optimization and manufacturability improvement for the production of mAbs may require even more time. [20]. (3) Aptamers can easily be modified to increase their applications and stability. Stability is especially important for RNA aptamers because RNA aptamers are liable to degradation by nucleases when used in vivo [26]. (4) Aptamers have less batch-to-batch variations [27,28,29,30]. (5) The cost of making aptamers is lower than that of making mAbs [24]. (6) Aptamers can be cloned into vectors and stored for a very long time, and can be produced any time in need by bacterial expression and/or PCR amplification. Alternatively, after the sequence of aptamers have been obtained, they can be chemically synthesized and PCR amplified at any time. (7) Aptamers are not sensitive to temperature: they can tolerate 80 °C while mAbs will generally denature at this temperature. Even if they are denatured, aptamers can easily be renatured to their original three-dimensional structure [27]. (8) Aptamers have a smaller molecular weight (~20,000 Da vs. ~150,000 Da for mAbs) and are easy to penetrate tissues such as tumors when used for therapeutic purposes [27].
The main limitations of aptamers lie in their susceptibility to degradation by nucleases (this can be overcome by aptamer modification and is discussed in Section 4.3 of the text), the difficulty to select aptamers against some targets, and the rapid renal clearance [31]. In theory, aptamers can be produced against any type of targets, from small inorganic and organic molecules to cells and to whole organisms such parasites, but in practical situations, it is sometimes difficult to obtain high-specific and high-affinity aptamers to some targets [32]. For instance, it is difficult to select aptamers against negatively charged targets because oligonucleotide strands have negative charges on their backbone [33]. The low molecular weight of aptamers is both advantageous and disadvantageous in terms of in vivo application. The small size makes them easier to penetrate tissue, an advantage for therapy as well as in vivo imaging; it also makes them faster to be cleared from the kidney, which means shorter half-life and is disadvantageous for therapy but advantageous for in vivo imaging [34]. Although the disadvantage of short circulating half-life can be overcome by conjugating aptamers with an inert molecule such as polyethylene glycol or cholesterol, this may meanwhile retard tissue uptake; hence, there is a paradox and the designer of aptamers should take all these into consideration and make a good balance between them [26,35,36,37].
4. Principle and Procedure of SELEX
The principle of aptamer production using SELEX includes first the chemical synthesis of a DNA pool containing about 1014–17 different oligonucleotide molecules. Because the pool is huge, there must be some chains that can bind to a specific target with high affinity. The task of the ensuing work is to screen out and amplify these specific oligonucleotides, which usually requires several cycles of selection and amplification. With the progression of the selection process and the increased stringency of selection conditions, the affinity of the selected oligonucleotides to the target increases gradually. In addition, because the PCR amplifications are not accurate (this may be done intentionally by using low fidelity Taq polymerase), “point mutations” may take place, which results in the generation of new oligonucleotide chains, and the binding affinity and specificity of the oligonucleotides may improve or be enriched in the course of selection. The finally resulting DNA or RNA oligonucleotides that bind to the target very tightly and specifically are the desired aptamers for the target [29,38,39,40]. The typical procedure of SELEX includes the following steps (see Figure 1).
4.1. Preparation of DNA Oligonucleotide Pool
Whether you are to make DNA aptamers or RNA aptamers, the initial step is the preparation of a DNA oligonucleotide pool by chemical synthesis. While the random central sequences of the oligonucleotides are generated during the random chemical synthesis, the constant sequences on both ends must be predesigned. The constant regions contain primer binding sequences for PCR amplification and are commonly 20–30 nucleotides in length. The constant regions may also include restriction sites for aptamer cloning. If RNA aptamers are to be made, the binding site for RNA polymerase (such as bacteriophage T7 promoter) must also be included in a constant region [41].
The length of the central region should be considered carefully in designing an oligonucleotide pool. Shorter chains are easier to control chemically, however, if the central region is too short, it cannot form enough secondary structures to bind to the target stably and specifically, and there may not be enough kinds of three-dimensional structures in the pool for screening (that is, the sequence space of the pool is not large enough). In theory, the kinds of oligonucleotide chains that can be formed in the random oligonucleotide pool are four to the power of n (4n), and n is the base number of the random nucleotides in the central region. For example, an oligonucleotide pool can have approximately 1015 different chains if the central region has 25 nucleotides (425 ≈ 1015) [41].
Generally, the sequence space of the oligonucleotide pool increases with the length of the central region (i.e., the available three-dimensional structures in the pool increase with central region length). However, when the length reaches a certain degree, the kinds of three-dimensional structures that can be formed do not increase significantly with the increase of length. For example, the types of oligonucleotide chains that can be formed in the oligonucleotide pool containing 30 random nucleotides are only about 1/1000 of the calculated number. Therefore, the central random sequence is typically designed to be 24–40 nucleotides long, which can already form diverse enough three-dimensional conformations to bine almost all kinds of target molecules in nature [41]. Once the sequence has been designed with satisfaction, the oligonucleotide pool can be synthesized by a DNA synthesizer or by a commercial company.
4.2. Selection and Enrichment of Aptamers
After the oligonucleotide pool has been chemically synthesized, it must be amplified using PCR and then converted to ssDNA oligonucleotides before any selection begins. Several selection methods based on SELEX are available at present; these include affinity chromatography, nitrocellulose membrane filtration, magnetic bead separation, capillary electrophoresis, microfluidic selection, microarray method, etc. Besides, semi-automatic or automated SELEX screening systems have also been established [42].
The screening and enrichment program of DNA aptamers begins with incubating the single-stranded oligonucleotide pool with target molecules or cells under proper selection conditions; then, the unbound or loosely bound oligonucleotides are washed out. Next, the bound oligonucleotides are separated from target molecules and collected, and the collected oligonucleotides are PCR amplified, which completes the first round of selection. The PCR product is then used to carry out the second round of selection, and so on. Generally, 6–14 cycles of screening and enrichment are required to obtain the desired aptamer. For the production of RNA aptamers, the initial DNA oligonucleotide pool must be in vitro transcribed into an RNA oligonucleotide pool before screening; the selected RNA oligonucleotides must be reverse transcribed into DNA by RT–PCR (the number of the molecules are amplified in the course) and then be in vitro transcribed into RNA molecules for the next round of screening and enrichment. Owing to the low fidelity of DNA polymerase used in PCR, some variants will be introduced in each PCR cycle; as a result, the binding capacity of oligonucleotide pool gradually increases in the screening and amplification process [43,44].
A counter selection or negative selection is usually necessary before the SELEX selection, whether it is for DNA aptamer or RNA aptamer production. The purpose of the counter selection is to remove any oligonucleotides that will bind to the immobilizer, the matrix/material used for immobilization of the target molecules, such as the magnetic beads or nitrocellulose membrane. In a counter selection, the DNA or RNA pool is first incubated together with the supporting matrix/immobilizing material and then the bound oligonucleotides are discarded and the unbound oligonucleotides are collected and used for the (positive) SELEX selection [45,46].
4.3. Aptamer Sequencing, Characterization, and Modification
When aptamers have been successfully selected, they should be cloned into vectors, their base sequence determined, and their possible secondary structure, target-binding affinity, stability, and some other characteristics analyzed [47].
Therapeutic aptamers, particularly RNA aptamers, frequently require modifications because they are sensitive to nucleases and are easily degraded in vivo, which shortens their half-life and limits their applications. Modification methods include: (1) Using chemically modified bases—this is done by adding nucleotide triphosphates with chemically modified bases (such as five-position modified uridine) to the reaction system [48]. (2) Modifying the phosphate–sugar backbone—this is usually done by adding 2′-modified nucleotide triphosphates (such as the 2′-position modification of fluorine, methoxy, or amino group) to the reaction system [49,50,51]. The above modifications depend on the ability of RNA polymerase to incorporate modified nucleotide triphosphates to the growing chain. (3) Conjugating high molecular mass moieties such as polyethylene glycol or fatty acid to the 5′ end or the 3′ end of the RNA aptamer [52,53]. (4) Producing mirror-image aptamers—this process involves creating a chemical mirror image (an enantiomer or optical isomer) of the target molecule, selecting aptamers against this mirror image, and, finally, creating (chemically synthesizing) a mirror image of the aptamer. These types of aptamers are called Spiegelmers (Spiegel is the German word for mirror) [54,55,56,57,58]. The above modifications can all increase the half-life of the aptamers by making them insensitive to nucleases, whereas the third method, conjugating high molecular mass moiety, can, meanwhile, increase the renal retention time of the molecules; the third method can be conducted in combination with other modifications [59].
5. Aptamers in Cancer Therapy
Taking together all of the aforementioned advantages, it could be seen that aptamers have promising therapeutic potential in cancer treatment. Aptamers can be used in cancer therapy either by directly inhibiting the activities of target molecules through binding to the targets such as growth factors or oncoproteins, or through guiding and delivering anticancer agents such as chemotherapeutics or siRNAs into cancer cells; aptamers can also block or stimulate immune receptors in lymphocytes to relieve immune suppression or boost immune response against cancer [60,61,62].
5.1. Targeted Inhibition
Aptamers can bind to ligands such as growth factors and chemokines and interrupt their interaction with cell receptors, or alternatively, they can bind to cell receptors to block ligand binding [63,64]. When transported into cells, they can also interfere with the functions of cytoplasmic targets [65].
5.1.1. Targeting Platelet-Derived Growth Factor (PDGF)
Platelet-derived growth factor (PDGF) regulates blood vessel formation (angiogenesis), mesenchymal cell mitogenesis, as well as chemotaxis. Overexpression of PDGF has been linked to malignancies. PDGFs function as disulfide-linked homodimers PDGF-AA, PDGF-BB, PDGF-CC, PDGF-DD, or heterodimer PDGF-AB. Their receptors are PDGFR-α and PDGFR-β (PDGF-A, PDGF-B, and PDGF-C bind PDGFR-α, whereas PDGF-B and PDGF-D bind PDGFR-β). Upon PDGF binding, the two receptor isoforms dimerize into three possible combinations: αα, ββ, or αβ. PDGFRs are classified as receptor tyrosine kinase; following PDGF activation, they are switched on by auto-phosphorylation of tyrosine residues on their intracellular tyrosine kinase domain [66].
Angiogenesis is necessary for tumor growth and is a prerequisite for cancer metastasis via blood vessels. Inhibition of angiogenesis has been considered as one of the promising measures to treat cancers. DNA aptamers against PDGFs were first developed by Green et al. [67]. These oligonucleotides bind to PDGF-AB and PDGF-BB with much higher affinity (Kd ≈ 10−10 M) than to PDGF-AA (Kd > 10−8 M), which indicates that the aptamers have a specific recognition of the PDGF-B-chain in the context of PDGF-AB heterodimer or PDGF-BB homodimer. The aptamers were truncated to determine the minimal sequence length necessary for high-affinity binding. Representative minimal oligonucleotides were shown to be able to inhibit the binding of PDGF-BB but not of PDGF-AA to PDGF receptors PDGFR-α or PDGFR-β in porcine aortic endothelial cells. These aptamers also potently inhibited PDGF-BB-dependent [3H] thymidine incorporation in the porcine aortic endothelial cells expressing the PDGFR-β receptor. Therefore, the PDGF aptamer has high promise to be used as an effective and specific inhibitor of PDGF in cancer therapy. Overexpression of PDGF-BB is associated with the development of colorectal cancer. Sae-Lim et al. [68] recently used the above-mentioned PDGF aptamer in their study to inhibit the proliferation of colorectal cancer cells. The results showed that cells treated with the aptamer proliferated slower than control. Western blot revealed a reduced phosphorylation level of ERK1/2 (a key component in the PDGF signaling pathway). It was concluded that the DNA aptamer against PDGF-BB blocked the binding of PDGF-BB to its receptor and inhibited colorectal cancer cell proliferation partly through downregulating the Ras/Raf/MEK/ERK signaling pathway.
Sennino et al. [69] examined the effects of the DNA aptamer AX102, a modified version of the aforementioned aptamer that binds PDGF-B chain selectively, on tumor vasculature. Treatment with AX102 caused the progressive loss of pericytes; reduction of pericytes and endothelial cells further led to empty basement membrane sleeves that gradually became invisible. The end result was inhibited tumor angiogenesis. The magnitude of tumor vessel regression caused by PDGF-B blockade was tumor-specific. Lu et al. [70] tested the efficacy of AX102 aptamer in pericyte regulation in an ovarian cancer model, with or without targeting endothelial cells by Bevacizumab (a VEGF-A neutralizing mAb). The results indicated that dual targeting of pericytes and endothelial cells achieved a much better anti-angiogenesis effect in ovarian carcinoma, resulting in a 76%–88% inhibition of tumor growth. Lately, Falcon et al. [71] found that inhibition of PDGF-B signaling in Lewis lung carcinomas by the DNA aptamer AX102 enhanced the transport and effectiveness of the chemotherapeutic drug cyclophosphamide by augmenting the efficiency of tumor blood vessels; combined application of cyclophosphamide and AX102 also showed synergistic effects on tumor cell proliferation in RIP-Tag2 tumors.
5.1.2. Targeting PDGFR
Platelet-derived growth factor receptor-β (PDGFR-β) is a transmembrane receptor tyrosine kinase. Overexpression of PDGFR-β in endothelial and tumor-associated stromal cells occurs in different human cancers. PDGFR-β expression in certain cancers has been linked with aggressiveness, resistance to therapy and recurrence [72]. Camorani et al. [73] reported that a PDGFRβ-specific RNA aptamer named Gint4.T could specifically bind to the human PDGFRβ ectodomain and cause strong inhibition of receptor activation and of downstream signaling both in continuous cell lines and in primary cultures of human glioblastoma. In addition, the Gint4.T aptamer could significantly inhibit cell migration and proliferation, induce cell differentiation, and impede tumor growth in vivo.
Tumors exhibiting interstitial hypertension have reduced uptake of chemotherapeutics, and PDGFR signaling has been shown to mediate interstitial hypertension. Pietras et al. [74] reported that inhibition of PDGFR signaling by combined treatment with PDGF-B inhibitory aptamers and PDGFR tyrosine kinase inhibitor lowered tumor interstitial hypertension and enhanced drug uptake by the tumor cells.
Triple-negative breast cancers (TNBCs) do not express estrogen receptor, progesterone receptor, and HER2; these cancers are difficult to treat because of their lack of targetable molecules. Cell-SELEX was exploited to find alternative targets and PDGFR has been suggested as one of them [75]. It has been suggested that PDGFR-β may mark breast cancer cells with stem-like characteristics and/or epithelial-mesenchymal transition. Camorani et al. [76] found the expression of PDGFR-β in a subgroup of mesenchymal tumors with invasive and stem-like phenotype, and suggested a role of PDGFR-β in driving TNBC invasiveness and metastases. They showed that PDGFR-β aptamers inhibited the invasive growth of TNBC cells in three-dimensional culture and blocked migration and invasion of mesenchymal TNBC cells and prevented lung metastases of TNBC cells.
In a separate study, the above research group demonstrated that bone marrow-derived mesenchymal stem cells (BM-MSCs) increased the aggressiveness of TNBC cells as indicated by their ability of migration, invasion, and acquisition of stemness markers. They also showed that treatment of the BM-MSCs with an RNA aptamer against PDGFR-β led to the inhibition of receptor-dependent signaling pathways and thus significantly blocked BM-MSC recruitment towards TNBC cells as well as BM-MSC’s trans-differentiation into carcinoma-associated fibroblast-like cells [77].
5.1.3. Targeting Chemokine
C-X-C chemokine ligand 12 (CXCL12), also known as stromal cell-derived factor 1 (SDF1), is a CXC subfamily chemokine and is the ligand for chemokine receptors CXCR4 and CXCR7. CXCL12 is widely expressed in many tissues and cell types and is strongly chemotactic for lymphocytes. During embryogenesis, CXCL12 directs the migration of hematopoietic cells from fetal liver to bone marrow and is responsible for large blood vessel formation. In adulthood, CXCL12 plays an essential role in angiogenesis by recruiting endothelial progenitor cells from the bone marrow through activating CXCR4 [78]. CXCL12 signaling has also been observed in CXCR4 expressing cancers and is involved in cancer cell invasion and metastasis [79]. The blockade of the CXCL12/CXCR4 passage has, therefore, emerged as a potential way of targeted cancer therapy [80].
In chronic lymphocytic leukemia (CLL), the homing and retention of cancer cells into the lymph nodes and bone marrow is critical to disease development. CLL cells are attracted to the bone marrow through activation of CXCR4 expressed on CLL cells by CXCL12 secreted by stromal cells. CLL cells localized in the bone marrow are protected from anticancer drugs; this can result in the persistence of the residual disease after conventional treatment and favor cancer recurrence [81].
NOX-A12 is a mirror-image RNA aptamer, an L-RNA oligonucleotide or the mirror image of naturally occurring D-RNA molecule (Spiegelmer), developed by the biotechnology company Noxxon Pharma in Berlin, Germany [82]. NOX-A12 aptamer is a CXCL12 antagonist that binds to the chemokine and disrupts the homing and the accumulation of CLL cells in the bone marrow, sensitizing these cells to cytotoxic drugs [81]. NOX-A12 molecules are highly resistant to degradation by nucleases owing to their L-configuration. Hoellenriegel et al. [83] inspected the effect of NOX-A12 on CXCL12-induced CLL cell migration and drug resistance; in the experiment, CLL cells were allowed to migrate toward CXCL12 concentration gradient (a chemotactic response) in the absence or presence of NOXA12. They found that NOX-A12 significantly inhibited CXCL12-mediated migration of CLL cells at a very low concentration of 3 nM. They also demonstrated that CXCL12-mediated chemotaxis of two other lymphoid cell lines (Jurkat and Nalm-6) could be reduced effectively in the presence of NOX-A12.
5.1.4. Targeting HER2
Several aptamers targeting human epidermal growth factor receptor 2 (HER2) have been developed but most of them were used to deliver anticancer therapeutics such as cytotoxic agents, therapeutic RNAs, and nanoparticles into cancer cells instead of inhibiting HER2 function directly and so will not be discussed here; readers can refer to the relevant sections and to Table 2.
5.2. Targeted Delivery
Not only can aptamers be selected to target and inhibit cancer-specific molecules, but they can also be selected to recognize the surface structures of specific cell types and some of them can be endocytosed or taken in by the cells. The latter properties can be employed to deliver therapeutic agents such as cytotoxic drugs and therapeutic RNAs selectively into cancer cells to exert their activities while avoiding damaging normal cells [114,115,116,117]. Aptamers used for drug delivery mostly target cell membrane receptors that can internalize upon ligand binding, and successful deliveries of a variety of cargoes targeting different receptors have been reported to date [118] (see Figure 2).
5.2.1. Delivery of Cytotoxic Agents
Chemotherapeutics or other cytotoxic or anticancer drugs can be linked either covalently or non-covalently to cell internalizing aptamers and be transported into cancer cells. Human prostate-specific membrane antigen (PSMA) is a class II membrane glycoprotein that resides mainly in the extracellular space and is highly expressed in normal prostate tissue and prostate cancers; PSMA has an internalizing signal that allows internalization of the bound ligands into the cell [119]. Two PSMA-targeting aptamers, called xPSM-A9 and xPSM-A10 (A9 and A10), were first developed by Lupold et al. [91] early in 2002; they share no consensus sequences and each binds to a unique extracellular epitope of PSMA. The aptamers were subjected to 3′-truncation in their central random regions to identify the minimum required binding elements. One of the aptamers, A10, when the central region was truncated from 40 nt to 25 nt, could still bind to the PSMA-positive LNCaP prostate cancer cells specifically but not to the PSMA-negative PC-3 prostate cancer cells [91].
Bagalkot et al. [120] investigated the targeted delivery of the chemotherapeutic drug doxorubicin (Dox) to cancer cells by using the PSMA aptamer A10. Multiple Dox molecules were non-covalently loaded (intercalated) to the double-stranded region of the aptamer A10, and when used in cell culture, resulted in the uptake of the aptamer–Dox conjugates by PSMA-expressing prostate cancer cells and the intracellular release of the Dox molecules.
Dox has also been covalently linked to a DNA aptamer named sgc8c; the drug was conjugated with the aptamer by a hydrazone linker which allows the release of Dox at pH 4.5–5.5. The Dox-sgc8c conjugate could be efficiently internalized by T-cell acute lymphoblastic leukemia (TALL) cells through binding to protein tyrosine kinase 7, a transmembrane receptor highly expressed on TALL cells, and after internalization, Dox could be cleaved from the conjugate in the acidic endosomal environment because the linkage between Dox and the aptamer had been designed to be acid-labile [121].
An aptamer developed against HER2 (the aptamer’s name being HB5) has been used to deliver Dox to breast cancer cells in vitro. The aptamer–Dox complex, which was formulated by intercalating Dox molecules into the aptamers, could selectively deliver Dox to HER2-positive breast cancer cells and retained the cytotoxicity of Dox against these cells; the aptamers preferentially target to HER2-positive breast cancer cells but not to HER2-negative cells [108].
5.2.2. Delivery of Therapeutic RNAs
Cell-specific internalizing aptamers have been used to deliver therapeutic oligonucleotides such as siRNAs, miRNAs, gRNAs, antisense RNAs, as well as protein-binding aptamers into cancer cells. Thus far, a number of researches have been successfully conducted using cell internalizing aptamers to deliver siRNAs into cancer cells. siRNAs can be conjugated with the internalizing aptamers covalently by using one strand of the siRNA, either sense or antisense, directly or indirectly by using a short linker RNA sequence; they can also be conjugated with the aptamers non-covalently by exploiting streptavidin-biotin interaction or through a short region of complementary base pairing called “sticky bridge”. Chu et al. [122] constructed an aptamer–streptavidin–siRNA conjugate (non-covalent linkage), in which two biotinylated aptamers targeting PSMA and two biotinylated siRNAs targeting lamin A/C mRNA were bound to a tetrameric streptavidin molecule. The assemblies were endocytosed by the PSMA-expressing LNCaP (a human prostate adenocarcinoma cell line) cells and efficiently inhibited the expression of the lamin A/C gene.
McNamara et al. [123] generated covalently linked aptamer–siRNA chimeric molecules to specifically deliver therapeutic siRNAs targeting PLK1 and BCL2 (two survival genes that are overexpressed in most human cancers) mRNAs into prostate cancer cells expressing the surface receptor PSMA. After internalizing the cells, the constructs were directed to the RNAi pathway, cleaved by Dicer, and silenced their cognate mRNA molecules which resulted in cell death. The aptamer–siRNA chimera was constructed by covalently linking the 3′ end of a PSMA-binding aptamer with the 5′ end of the sense strand of a gene-silencing siRNA, which was achieved by co-transcribing the aptamer and siRNA sense sequence; the antisense strand was then hybridized to the sense strand.
HER2 has been employed as a target to deliver siRNAs into HER2-positive breast cancer cells. Thiel et al. [124] selected RNA aptamers that are specific to HER2 and can be internalized upon binding. The RNA aptamers were covalently linked to siRNAs targeting Bcl2, an apoptosis suppressing gene. The HER2 aptamer–Bcl2 siRNA conjugates were specifically internalized into HER2-positive cells and silenced the expression of Bcl2 gene. In addition, the silence of the Bcl-2 gene resulted in the sensitization of these cells to the chemotherapeutic drug cisplatin. HER2 aptamers have also been used to deliver EGFR (epidermal growth factor receptor) siRNAs into HER2-positive cancer cells [125,126].
5.2.3. Delivery of Nanocarriers
Using aptamers to deliver nanoparticles loaded with drugs is a promising approach to drug delivery. Aptamers are only a few nanometers in diameter, and with this small size, they do not increase much to the overall size of the nanocarriers. Thus, the resulting nanoparticle–aptamer conjugates can relatively easily penetrate the microvasculature and the interstitial tissue of tumors [136]. In addition, aptamers are chemically synthesized and this makes it relatively easy to attach functional groups to their ends and link them covalently to nanoparticles [137].
The first example of nanoparticle–aptamer conjugates used the A10 aptamers targeting PSMA [138]. The authors generated the nanoparticles by synthesizing poly(lactic acid)-block-polyethylene glycol copolymer with a terminal carboxylic acid functional group; they then encapsulated the anticancer drug dextran within these nanoparticles. Next, they conjugated the A10 RNA aptamers that target PSMA to the nanoparticles. The authors found that these nanoparticle–aptamer conjugates could efficiently target the PSMA-positive prostate LNCaP cells and be internalized while the boosted uptake of these conjugates was not observed in cells that do not express PSMA. This nanoparticle–aptamer construction was later assessed in vivo and was demonstrated to be able to reduce the tumor size of PSMA-positive prostate carcinomas effectively [139].
Several studies used the HER2 molecule as a target to deliver nanoparticles to cancer cells for therapeutic purposes. One study conjugated HER2 aptamers with dextran-coated ferric oxide nanoparticles and applied them to induce magnetic hyperthermia in human adenocarcinoma SK-BR3 cells that overexpress HER2 receptors [140]. The HB5 aptamer was used to functionalize mesoporous silica–carbon nanoparticles loaded with DOX. The construction (MSCN–PEG–HB5/DOX) was expected to exert a chemo-photothermal combined therapeutic effect on HER2-positive breast cancer cells. The researchers found that the MSCN–PEG–HB5/DOX assembling exhibited significantly higher cellular uptake in the HER2-positive SK-BR-3 breast cancer cell line but not in the normal breast epithelial cell line. The uptake was based on the receptor-mediated mechanism that was energy-dependent. Cytotoxicity experiments showed that the chemo-photothermal combined therapy using the MSCN–PEG–HB5/DOX complex led to a much higher cytotoxic effect than either chemotherapy or photothermal therapy alone [109]. Another study conjugated an HER2 aptamer to curcumin-loaded human serum albumin nanoparticle that could be taken up by HER2-overexpressing SK-BR-3 cells, having the effect of remarkable cytotoxicity in these cells [141].
Other nanoparticle–aptamer conjugates that have been reported thus far include: conjugating the anti-CD44 aptamer to liposomes loaded with siRNA for gene silencing in CD44-expressing tumor cells in vivo [142], mucin1 aptamer-conjugated chitosan nanoparticles loaded with docetaxel and cMET siRNA that was delivered into mucin1 positive SKBR3 breast cancer cells [143], LA1 aptamer-conjugated grapefruit-derived nanovectors harboring Dox and P-glycoprotein siRNA that could be internalized into multidrug resistance LoVo colon cancer cells [144], self-assembled DNA nanostructures (DNA nanoprisms) decorated with therapeutic siRNAs targeting the GTPase Rab26 and MUC-1 aptamers that target non-small cell lung cancer [145], and paclitaxel-encapsulated PEGylated PLGA nanoparticles that were surface-functionalized with the heparanase aptamers targeting triple-negative breast cancers [146].
5.3. Immunomodulation
Up to the present time, a number of aptamers have been developed that are capable of modulating immune responses against cancer cells. These aptamers were used either as antagonists of immune checkpoints or as agonists of immuno-stimulatory receptors [147].
5.3.1. Immune Checkpoint Antagonists
The recently developed immune checkpoint inhibitors of such programmed death 1 (PD-1) inhibitors and programmed death-ligand 1 (PD-L1) inhibitors have revolutionized cancer immunotherapy [148]. Nevertheless, cancer treatment with PD-1 inhibitors and PD-L1 inhibitors benefited only a subset of patients or some type of cancers, the majority of cancer patients did not show complete responses, and adverse reactions have been observed [148]. The search for new checkpoint targets and new checkpoint inhibitors is still necessary (see Figure 3).
Currently, the most prevalent immune checkpoint receptors that have been used to manipulate the immune system for cancer immunotherapy are PD1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4); both are T-cell surface receptors. However, there are many other immune checkpoints that can be employed to modulate the immune system to enhance cancer immunity. These include T-cell immunoglobulin and mucin-domain containing-3 (TIM3), lymphocyte activating 3 (Lag3), B- and T-lymphocyte attenuator (BTLA), adenosine A2A receptor (A2AR), as well as a number of intracellular immune checkpoints such as forkhead box protein P3 (Foxp3), signal transducer and activator of transcription 3 (STAT3), casitas B-lineage lymphoma-b (Cbl-b), early growth response proteins-2 (EGR-2), Src homology region 2 domain-containing phosphatase-1 (SHP1), Src Homology region 2 domain-containing phosphatase-2 (SHP2), etc. Aptamers have been produced for most of these immune checkpoints up to now.
The first report using aptamers to manipulate immune responses against cancer by antagonizing the immune checkpoint receptor CTLA-4 was the development of RNA aptamers that could bind CTLA-4 with high affinity and specificity and inhibited CTLA-4 function in vitro and enhanced tumor immunity in mice; tetrameric assemblies of the aptamers could improve their bioactivity significantly both in vitro and in vivo [149].
DNA aptamers that bind specifically to the extracellular domain of PD-1 have been in development and blocked murine PD-1 and PD-L1 interaction; one of the aptamers functionally inhibited PD-L1 mediated suppression of interleukin-2 secretion in primary murine T cells. Its PEGylated form suppressed the growth of PD-L1 positive murine colon carcinoma cells in vivo with a potency comparable to the anti-PD-1 antibody [112]. Lai et al. [113] reported that a DNA aptamer against human PD-L1 blocked the binding between human PD-1 and PD-L1. They showed that the aptamer promoted lymphocyte proliferation in vitro and suppressed tumor growth in vivo with minimum liver and renal toxicity. Tumors treated with the aptamer showed elevated infiltration of CD4+ and CD8+ T cells and raised levels of IL-2, tumor necrosis factor-α (TNFα), interferon-γ (IFNγ), and C-X-C chemokine ligands CXCL9 and CXCL10. The CD8+ T cells in the treated tumors displayed higher CXCR3 expression than control.
TIM3 is an immune checkpoint receptor expressed on multiple immune cells that has demonstrated several unique properties and is one of the most promising immune checkpoint targets other than CTLA-4 and PD-1 [150]. TIM3 protein is a member of the TIM family that includes TIM-1, TIM-3, and TIM-4. They are type-I cell-surface glycoproteins comprising extracellular signal peptides and IgV domains, mucin-like transmembrane domains, and an intracellular cytoplasmic tail.
TIM3 was first identified as a molecule selectively expressed on CD4 + IFNγ–producing Th1 and CD8 + cytotoxic Tc1 cells that function to limit the magnitude and duration of Th1 and Tc1 responses [151]. Later studies have shown that TIM3 is expressed on numerous immune cells; in addition to Th1 and Tc1 cells, there are Tregs (regulatory T cells), Th17 cells, TILs (tumor-infiltrating lymphocytes), and innate immune cells. The observations that TIM3 marks dysfunctional phenotype of CD8+ T cells in both experimental models and cancer patients and that TIM3 + Tregs are found solely in tumor tissue have made TIM3 pathway a very suitable target for anticancer immunotherapy based on immune checkpoint blockade [151].
Aptamers have been developed to antagonize TIM3, one of which, TIM3Apt, has been shown to bind to the extracellular motives of TIM3 with high affinity and specificity and its monomeric form displayed a potent antagonist capacity in TIM3-positive lymphocytes. Combinational treatment with TIM3Apt and PD-L1 inhibitor showed a synergistic effect in colon carcinoma-bearing mice [152]. A trimeric form of TIM3 aptamer blocked the interaction of TIM3 with Galectin-9, boosted cell proliferation and cytokine secretion, and reduced cell death in vitro. In tumor-bearing mice, the trimeric aptamer delayed tumor growth and enhanced the survival of the tumor-bearing animals used either independently or in combination with a PD-1 inhibitor. In particular, the trimeric aptamer displayed better activity than the currently commercially available monoclonal antibody, RMT3-23, both in vitro and in vivo [153].
5.3.2. Immune Stimulation Agonists
The anti-tumor immunity can also be boosted by providing artificial stimulatory or costimulatory signals to immune cells. Most costimulatory receptors in lymphocytes exert their function through crosslinking their cytoplasmic domains, thus the desired immuno-stimulatory agents need to be able to bring together the receptors to initiate the activation signaling. Antibodies are well suited to crosslinking receptors because of their bivalent antigen-binding construction. To meet that requirement, the first immuno-stimulatory aptamer was designed as a dimer [154]. The aptamer was developed as a murine 4-1BB costimulatory receptor agonist. 4-1BB, also known as CD137 and tumor necrosis factor receptor superfamily member 9 (TNFRSF9), is a type 2 transmembrane glycoprotein receptor belonging to the TNF superfamily. 4-1BB is an important costimulatory receptor expressed on activated T Lymphocytes and supports the survival and expansion of activated T lymphocytes.
The 4-1BB agonistic aptamer could bind 4-1BB expressed on the surface of activated murine T lymphocytes and stimulate the activation of the T cells in vitro and mediate tumor rejection in mice [154]. To reduce the non-specific immuno-cytotoxic damage to normal tissue or cells, several investigations constructed bi-specific aptamers that bind to 4-1BB as well as to a tumor-specific target either on tumor cells or in tumor stroma. Pastor and others [155,156] conjugated an agonistic 4-1BB aptamer to a PSMA aptamer, which resulted in enhanced antitumor immunity in subcutaneously implanted tumors and lung metastasis in mice. Berezhnoy et al. [157,158] conjugated an agonistic 4-1BB aptamer to an siRNA targeting mTOR complex 1 and found that systemic administration of the conjugates to mice downregulated mTOR complex 1 activity in CD8 + T cells and led to a potent memory response that showed cytotoxic effectors and boosted vaccine-induced immunity in tumor-bearing mice. Schrand et al. [159,160] conjugated an agonistic 4-1BB aptamer to an aptamer that binds to VEGF that is widely expressed in tumor stroma; systemic administration of the conjugates in preclinical murine tumor models exhibited potent antitumor immunity against multiple unrelated tumors and an enhanced therapeutic index. 4-1BB aptamer has been used to conjugate to an siRNA against IL-2Rα whose signaling negatively regulates activated CD8+ T cells. Systemic administration of 4-1BB aptamer-IL-2Rα siRNA conjugates downregulated IL-2Rα mRNA in 4-1BB-positive CD8+ T cells and promoted their differentiation into memory cells [161]. 4-1BB aptamer has also been used in conjugation with an siRNA against Smad4 in the TGFβ signaling pathway, the signaling of which mediates immune suppression at the tumor microenvironment. The 4-1BB aptamer-Smad4 siRNA conjugates rendered T cell resistant to TGFβ inhibition, and systemic administration of the conjugates to tumor-bearing mice boosted irradiation- and vaccine-induced antitumor immunity [162].
Other aptamers generated as agonists to activate costimulatory receptors include the dimeric aptamer against murine OX40 [163], the dimeric aptamers against CD28 [164], the dimeric aptamers against CD40 [165], the aptamer against ICOS [166], and the aptamer against human OX40 [167]. These aptamers were used independently or in combination with immune checkpoint inhibitors.
6. Cancer Therapy Aptamers in Clinical Trials
In addition to Macugen, which was approved by the FDA more than 10 years ago for the treatment of ADM, there have been a number of aptamers that have entered clinical trials. Table 2 shows the selected aptamers that are under clinical trials or in active laboratory investigation for cancer therapy.
7. Conclusions
Aptamers are single-stranded RNA or DNA oligonucleotides generated with SELEX technology. Aptamers can form numerous specified three-dimensional structures and thus can bind diverse target molecules with high affinity and specificity. Like mAbs, aptamers have promising potential applications in clinical diagnosis and therapy particularly in cancer treatment, but aptamers have advantages over mAbs in therapeutic uses mainly because they have little or no immunogenicity, which means the feasibility of repeat prescriptions and fewer side effects. Researches on aptamers are still in the early stage, and a deeper understanding of aptamer–target interactions and pharmacokinetics is still needed to prepare for better clinical use or marketing. So far, although a number of aptamers have been developed and some of them have entered clinical trials, only one aptamer has been approved by the FDA for clinical use. The progress towards effective therapies tends to be slow, but it never ceases and the best results are yet to come. With regard to cancer treatment, in addition to their advantage of little or no immunogenicity, aptamers are relatively easy to penetrate solid tumor tissue because of their smaller molecular weight and are comparatively less costly than mAbs since they are produced in vitro. Therefore, more efforts should be devoted to the discovery of new anticancer aptamers, to the in-depth characterization of the current aptamers, and to speeding up the process for the entry of existing anticancer aptamers into clinical trials.
Author Contributions
Z.F. wrote the manuscript. J.X. checked the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by NSFC, grant number 81760732.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| A2AR | adenosine A2A receptor |
| AMD | age-related macular degeneration |
| BTLA | B- and T-lymphocyte attenuator |
| Cbl-b | casitas B-lineage lymphoma-b |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | C-X-C chemokine ligand |
| CXCR | C-X-C chemokine receptor |
| Dox | doxorubicin |
| EGFR | epidermal growth factor receptor |
| EGR-2 | early growth response proteins-2 |
| Foxp3 | forkhead box protein P3 |
| HER2 | human epidermal growth factor receptor 2 |
| IFNγ | interferon-γ |
| Lag3 | lymphocyte activating 3 |
| mAb | monoclonal antibody |
| PD-1 | programmed death 1 |
| PD-L1 | programmed death-ligand 1 |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptor |
| PSMA | prostate-specific membrane antigen |
| SELEX | systematic evolution of ligands by exponential enrichment |
| SHP1 | Src homology region 2 domain-containing phosphatase-1 |
| STAT3 | signal transducer and activator of transcription 3 |
| TALL | T-cell acute lymphoblastic leukemia |
| TIM3 | mucin-domain-containing-3 |
| TNFα | tumor necrosis factor-α |
| Treg | regulatory T cell |
| VEGF | vascular endothelial growth factor |
References
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2016, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. Extended Duration Vascular Endothelial Growth Factor Inhibition in the Eye: Failures, Successes, and Future Possibilities. Pharmaceutics 2018, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An Emerging Class of Therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef]
- Moreno, M.; Fernandez-Algar, M.; Fernandez-Chamorro, J.; Ramajo, J.; Martinez-Salas, E.; Briones, C. A Combined ELONA-(RT)qPCR Approach for Characterizing DNA and RNA Aptamers Selected against PCBP-2. Molecules 2019, 24, 1213. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Qi, X.; Liu, Y.; Zhang, F.; Yan, H. DNA-Nanoscaffold-Assisted Selection of Femtomolar Bivalent Human α-Thrombin Aptamers with Potent Anticoagulant Activity. ChemBioChem 2019, 20, 2494–2503. [Google Scholar] [CrossRef]
- Röthlisberger, P.; Hollenstein, M. Aptamer chemistry. Adv. Drug Deliv. Rev. 2018, 134, 3–21. [Google Scholar] [CrossRef]
- Zhao, N.; You, J.; Zeng, Z.; Li, C.; Zu, Y. An Ultra pH-Sensitive and Aptamer-Equipped Nanoscale Drug-Delivery System for Selective Killing of Tumor Cells. Small 2013, 9, 3477–3484. [Google Scholar] [CrossRef] [Green Version]
- Dassie, J.P.; I Hernandez, L.; Thomas, G.S.; Long, M.; Rockey, W.; A Howell, C.; Chen, Y.; Hernandez, F.J.; Liu, X.Y.; Wilson, M.E.; et al. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol. Ther. 2014, 22, 1910–1922. [Google Scholar] [CrossRef]
- Zhu, H.; Li, J.; Zhang, X.; Ye, M.; Tan, W. ChemInform Abstract: Nucleic Acid Aptamer-Mediated Drug Delivery for Targeted Cancer Therapy. ChemMedChem 2015, 46. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M. Aptamers as Both Drugs and Drug-Carriers. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Pei, S.-N.; Qi, J.; Zeng, Z.; Iyer, S.; Lin, P.; Tung, C.-H.; Zu, Y. Oligonucleotide aptamer-drug conjugates for targeted therapy of acute myeloid leukemia. Biomaterials 2015, 67, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Liu, B.; Yu, B.; Zhong, W.; Lu, Y.; Zhang, J.; Liao, J.; Liu, J.; Pu, Y.; Qiu, L.; et al. Advances in the development of aptamer drug conjugates for targeted drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2016, 9, e1438. [Google Scholar] [CrossRef] [Green Version]
- Kratschmer, C.; Levy, M. Targeted Delivery of Auristatin-Modified Toxins to Pancreatic Cancer Using Aptamers. Mol. Ther. Nucleic Acids 2018, 10, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Tung, C.H.; Zu, Y. Aptamer-Equipped Protamine Nanomedicine for Precision Lymphoma Therapy. Cancers (Basel) 2020, 12, 780. [Google Scholar] [CrossRef] [Green Version]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef] [Green Version]
- Eyetech Study Group. Preclinical and phase 1a clinical evaluation of an anti-vegf pegylated aptamer (eye001) for the treatment of exudative age-related macular degeneration. Retina (Phila. Pa) 2002, 22, 143–152. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Leifer, A.; Fang-Yen, C.; Gershow, M.; Alkema, M.J.; Samuel, A.D. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat. Methods 2011, 8, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calarco, J.A.; Samuel, A.D.T. Imaging whole nervous systems: Insights into behavior from worms to fish. Nat. Methods 2019, 16, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Fiala, A.; Suska, A.; Schluter, O.M.; Beroun, A. Optogenetic Approaches in Neuroscience. Curr. Boil. 2010, 20, R897–R903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.H.; Elsherbiny, M.; Emara, M. Updates on Aptamer Research. Int. J. Mol. Sci. 2019, 20, 2511. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer Therapeutics in Cancer: Current and Future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Ge, Z. Chemical Modifications of Nucleic Acid Aptamers for Therapeutic Purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Gilbert, J.C.; Jilma, B. Pharmacokinetics, pharmacodynamics and safety of aptamers. Adv. Drug Deliv. Rev. 2018, 134, 36–50. [Google Scholar] [CrossRef]
- Hassanzadeh, L.; Chen, S.; Veedu, R.N. Radiolabeling of Nucleic Acid Aptamers for Highly Sensitive Disease-Specific Molecular Imaging. Pharmaceuticals (Basel Switz.) 2018, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, C.; Larcher, L.M.; Barrero, R.A.; Veedu, R.N. Three decades of nucleic acid aptamer technologies: Lessons learned, progress and opportunities on aptamer development. Biotechnol. Adv. 2019, 37, 28–50. [Google Scholar] [CrossRef]
- Dhiman, A.; Anand, A.; Malhotra, A.; Khan, E.; Santra, V.; Kumar, A.; Sharma, T. Rational truncation of aptamer for cross-species application to detect krait envenomation. Sci. Rep. 2018, 8, 17795. [Google Scholar] [CrossRef] [Green Version]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Al Bawab, A.; Ismail, S.I. Ismail Aptamers Chemistry: Chemical Modifications and Conjugation Strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Yan, J.H.; Xiong, H.; Liu, Y.; Peng, D.; Liu, Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 2018, 143, 5317–5338. [Google Scholar] [CrossRef] [PubMed]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Rossi, J.J. Emerging cancer-specific therapeutic aptamers. Curr. Opin. Oncol. 2017, 29, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Hassel, S.K.; Mayer, G. Aptamers as Therapeutic Agents: Has the Initial Euphoria Subsided? Mol. Diagn. Ther. 2019, 23, 301–309. [Google Scholar] [CrossRef]
- Tan, Y.; Li, Y.; Tang, F. Nucleic Acid Aptamer: A Novel Potential Diagnostic and Therapeutic Tool for Leukemia. Oncotargets Ther. 2019, 12, 10597–10613. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Drolet, D.W.; Wolk, S.K.; Waugh, S.M.; Rohloff, J.C.; Carter, J.D.; Mayfield, W.S.; Otis, M.R.; Fowler, C.R.; Suzuki, T.; et al. Pharmacokinetic Properties of DNA Aptamers with Base Modifications. Nucleic Acid Ther. 2017, 27, 345–353. [Google Scholar] [CrossRef]
- Gopinath, S.C. Methods developed for SELEX. Anal. Bioanal. Chem. 2007, 387, 171–182. [Google Scholar] [CrossRef]
- Kulbachinskiy, A.V. Methods for selection of aptamers to protein targets. Biochem. Biokhimiia 2007, 72, 1505–1518. [Google Scholar] [CrossRef]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef]
- Hall, B.; Micheletti, J.M.; Satya, P.; Ogle, K.; Pollard, J.; Ellington, A.D. Design, synthesis, and amplification of DNA pools for in vitro selection. Curr. Protoc. Nucleic Acid Chem. 2009. [Google Scholar] [CrossRef] [Green Version]
- Piasecki, S.K.; Hall, B.; Ellington, A.D. Nucleic acid pool preparation and characterization. Methods Mol. Biol. (Cliftonnj) 2009, 535, 3–18. [Google Scholar]
- Jeong, S.; Lee, H.K.; Kim, M.Y. Use of RNA Aptamers for the Modulation of Cancer Cell Signaling. In Advanced Structural Safety Studies; Humana Press: Totowa, NJ, USA, 2009; Volume 542, pp. 363–377. [Google Scholar]
- Long, Y.; Ersoy, F.; Mayer, G.; Schrøder, T.D.; Ozalp, V.C.; Olsen, L.F. Selection of Aptamers for Metabolite Sensing and Construction of Optical Nanosensors. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: New York, NY, USA, 2016; Volume 1380, pp. 3–19. [Google Scholar]
- Piganeau, N. Selecting allosteric ribozymes. Methods Mol. Biol. (Clifton Nj) 2012, 848, 317–328. [Google Scholar]
- Berg, K.; Magbanua, E.; Hahn, U. SELEX of Cell-Specific RNA Aptamers. Methods Mol. Biol. (Clifton Nj) 2016, 1380, 21–32. [Google Scholar]
- Breitsprecher, D.; Schlinck, N.; Witte, D.; Duhr, S.; Baaske, P.; Schubert, T. Aptamer Binding Studies Using MicroScale Thermophoresis. In Advanced Structural Safety Studies; Humana Press: New York, NY, USA, 2016; Volume 1380, pp. 99–111. [Google Scholar]
- Minagawa, H.; Onodera, K.; Fujita, H.; Sakamoto, T.; Akitomi, J.; Kaneko, N.; Shiratori, I.; Kuwahara, M.; Horii, K.; Waga, I. Selection, Characterization and Application of Artificial DNA Aptamer Containing Appended Bases with Sub-nanomolar Affinity for a Salivary Biomarker. Sci. Rep. 2017, 7, 42716. [Google Scholar] [CrossRef] [Green Version]
- Hoshika, S.; Minakawa, N.; Matsuda, A. Synthesis and physical and physiological properties of 4’-thioRNA: Application to post-modification of RNA aptamer toward NF-kappaB. Nucleic Acids Res. 2004, 32, 3815–3825. [Google Scholar] [CrossRef]
- Virgilio, A.; Petraccone, L.; Scuotto, M.; Vellecco, V.; Bucci, M.; Mayol, L.; Varra, M.; Esposito, V.; Galeone, A. 5-Hydroxymethyl-2′-Deoxyuridine Residues in the Thrombin Binding Aptamer: Investigating Anticoagulant Activity by Making a Tiny Chemical Modification. ChemBioChem 2014, 15, 2427–2434. [Google Scholar] [CrossRef]
- Maio, G.E.; Enweronye, O.; Zumrut, H.E.; Batool, S.; Van, N.A.; Mallikaratchy, P. Systematic Optimization and Modification of a DNA Aptamer with 2’-O-Methyl RNA Analogues. ChemistrySelect 2017, 2, 2335–2340. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, Y.; Kitadume, S.; Morihiro, K.; Kuwahara, M.; Ozaki, H.; Sawai, H.; Imanishi, T.; Obika, S. Effect of 3′-end capping of aptamer with various 2′,4′-bridged nucleotides: Enzymatic post-modification toward a practical use of polyclonal aptamers. Bioorganic Med. Chem. Lett. 2010, 20, 1626–1629. [Google Scholar] [CrossRef]
- Takafuji, Y.; Jo, J.-I.; Tabata, Y. Simple PEG modification of DNA aptamer based on copper ion coordination for tumor targeting. J. Biomater. Sci. Polym. Ed. 2011, 22, 1179–1195. [Google Scholar]
- Vater, A.; Klussmann, S. Turning mirror-image oligonucleotides into drugs: The evolution of Spiegelmer® therapeutics. Drug Discov. Today 2015, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberthür, M.; Achenbach, J.; Gabdoulkhakov, A.G.; Buchner, K.; Maasch, C.; Falke, S.; Rehders, D.; Klussmann, S.; Betzel, C. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat. Commun. 2015, 6, 6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, S.; Hoos, J.; Klussmann, S.; Vonhoff, S. RNA Aptamers and Spiegelmers: Synthesis, Purification, and Post-Synthetic PEG Conjugation. Curr. Protoc. Nucleic Acid Chem. 2011, 46, 4–46. [Google Scholar] [CrossRef] [PubMed]
- Helmling, S.; Maasch, C.; Eulberg, D.; Buchner, K.; Schröder, W.; Lange, C.; Vonhoff, S.; Wlotzka, B.; Tschöp, M.H.; Rosewicz, S.; et al. Inhibition of ghrelin action in vitro and in vivo by an RNA-Spiegelmer. Proc. Natl. Acad. Sci. USA 2004, 101, 13174–13179. [Google Scholar] [CrossRef] [Green Version]
- Purschke, W.G. A DNA Spiegelmer to staphylococcal enterotoxin B. Nucleic Acids Res. 2003, 31, 3027–3032. [Google Scholar] [CrossRef] [Green Version]
- Kratschmer, C.; Levy, M. Effect of Chemical Modifications on Aptamer Stability in Serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, X. Aptamer-based targeted therapy. Adv. Drug Deliv. Rev. 2018, 134, 65–78. [Google Scholar] [CrossRef]
- Tan, W.; Wang, H.; Chen, Y.; Zhang, X.; Zhu, H.; Yang, C.; Yang, R.; Liu, C. Molecular aptamers for drug delivery. Trends Biotechnol. 2011, 29, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef]
- Poolsup, S.; Kim, C.Y. Therapeutic applications of synthetic nucleic acid aptamers. Curr. Opin. Biotechnol. 2017, 48, 180–186. [Google Scholar] [CrossRef]
- Camorani, S.; Crescenzi, E.; Fedele, M.; Cerchia, L. Oligonucleotide aptamers against tyrosine kinase receptors: Prospect for anticancer applications. Biochim. Biophys. Acta (Bba) Bioenerg. 2018, 1869, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Rossi, J.J. Aptamers: Uptake mechanisms and intracellular applications. Adv. Drug Deliv. Rev. 2018, 134, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Asp. Med. 2018, 62, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Green, L.S.; Jellinek, D.; Jenison, R.; Östman, A.; Heldin, C.-H.; Janjic, N. Inhibitory DNA Ligands to Platelet-Derived Growth Factor B-Chain. Biochemistry 1996, 35, 14413–14424. [Google Scholar] [CrossRef] [PubMed]
- Sae-Lim, S.; Soontornworajit, B.; Pichayanoot, P.; Rotkrua, P. Inhibition of Colorectal Cancer Cell Proliferation by Regulating Platelet-Derived Growth Factor B Signaling with a DNA Aptamer. Asian Pac. J. Cancer Prev. 2019, 20, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Falcon, B.L.; McCauley, D.; Le, T.; McCauley, T.; Kurz, J.C.; Haskell, A.; Epstein, D.M.; McDonald, D.M. Sequential loss of tumor vessel pericytes and endothelial cells after inhibition of platelet-derived growth factor B by selective aptamer AX102. Cancer Res. 2007, 67, 7358–7367. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.S.M.; Moreno-Smith, M.; Lin, Y.G.; Jennings, N.B.; Allen, J.K.; Landen, C.N.; Mangala, L.S.; Armaiz-Pena, G.N.; Schmandt, R.; Nick, A.M.; et al. Targeting pericytes with a PDGF-B aptamer in human ovarian carcinoma models. Cancer Biol. Ther. 2010, 9, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.L.; Pietras, K.; Chou, J.; Chen, D.; Sennino, B.; Hanahan, U.; McDonald, D.M. Increased Vascular Delivery and Efficacy of Chemotherapy after Inhibition of Platelet-Derived Growth Factor-B. Am. J. Pathol. 2011, 178, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, K.; Pomeroy, H.; Marneros, A. Targeting Platelet-Derived Growth Factor Receptor β + Scaffold Formation Inhibits Choroidal Neovascularization. Am. J. Pathol. 2016, 186, 1890–1899. [Google Scholar] [CrossRef] [Green Version]
- Camorani, S.; Esposito, C.L.; Rienzo, A.; Catuogno, S.; Iaboni, M.; Condorelli, G.; de Franciscis, V.; Cerchia, L. Inhibition of receptor signaling and of glioblastoma-derived tumor growth by a novel PDGFRbeta aptamer. Molecular therapy. J. Am. Soc. Gene Ther. 2014, 22, 828–841. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Rubin, K.; Sjöblom, T.; Buchdunger, E.; Sjöquist, M.; Heldin, C.-H.; Östman, A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002, 62, 5476–5484. [Google Scholar] [PubMed]
- Camorani, S.; Fedele, M.; Zannetti, A.; Cerchia, L. TNBC Challenge: Oligonucleotide Aptamers for New Imaging and Therapy Modalities. Pharmaceuticals 2018, 11, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camorani, S.; Hill, B.S.; Collina, F.; Gargiulo, S.; Napolitano, M.; Cantile, M.; Di Bonito, M.; Botti, G.; Fedele, M.; Zannetti, A.; et al. Targeted imaging and inhibition of triple-negative breast cancer metastases by a PDGFRβ aptamer. Theranostics 2018, 8, 5178–5199. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Hill, B.S.; Fontanella, R.; Greco, A.; Gramanzini, M.; Auletta, L.; Gargiulo, S.; Albanese, S.; Lucarelli, E.; Cerchia, L.; et al. Inhibition of Bone Marrow-Derived Mesenchymal Stem Cells Homing Towards Triple-Negative Breast Cancer Microenvironment Using an Anti-PDGFRβ Aptamer. Theranostics 2017, 7, 3595–3607. [Google Scholar] [CrossRef]
- Janssens R, Struyf S, Proost P: Pathological roles of the homeostatic chemokine CXCL12. Cytokine Growth Factor Rev. 2018, 44, 51–68. [CrossRef]
- De Nigris, F.; Schiano, C.; Infante, T.; Napoli, C. CXCR4 inhibitors: Tumor vasculature and therapeutic challenges. Recent Pat. Anti-Cancer Drug Discov. 2012, 7, 251–264. [Google Scholar] [CrossRef]
- Cho, B.S.; Kim, H.J.; Konopleva, M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248–257. [Google Scholar] [CrossRef]
- Marasca, R.; Maffei, R. NOX-A12: Mobilizing CLL away from home. Blood 2014, 123, 952–953. [Google Scholar] [CrossRef] [Green Version]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Vater, A.; Frömming, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2’-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Canny, M.D.; De Erkenez, A.; Krilleke, D.; Ng, Y.-S.; Shima, D.T.; Pardi, A.; Jucker, F. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc. Natl. Acad. Sci. USA 2005, 102, 18902–18907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinores, S.A. Pegaptanib in the treatment of wet, age-related macular degeneration. Int. J. Nanomed. 2006, 1, 263–268. [Google Scholar]
- Cheng, Y.; Zhao, G.; Zhang, S.; Nigim, F.; Zhou, G.; Yu, Z.; Song, Y.; Chen, Y.; Li, Y. AS1411-Induced Growth Inhibition of Glioma Cells by Up-Regulation of p53 and Down-Regulation of Bcl-2 and Akt1 via Nucleolin. PLoS ONE 2016, 11, e0167094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Reyes, E.M.; Teng, Y.; Bates, P.J. A New Paradigm for Aptamer Therapeutic AS1411 Action: Uptake by Macropinocytosis and Its Stimulation by a Nucleolin-Dependent Mechanism. Cancer Res. 2010, 70, 8617–8629. [Google Scholar] [CrossRef] [Green Version]
- E Lupold, S.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar]
- Lupold, S.E. Aptamers and apple pies: A mini-review of PSMA aptamers and lessons from Donald S. Coffey. Am. J. Clin. Exp. Urol. 2018, 6, 78–86. [Google Scholar]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Zhang, L.; Teply, B.A.; Mann, N.; Wang, A.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, F.; Langer, R.; Farokhzad, O.C. Formulation/preparation of functionalized nanoparticles for in vivo targeted drug delivery. Methods Mol. Biol. (Clifton NJ) 2009, 544, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.M.; Valencia, P.M.; Zhang, L.; Langer, R.; Farokhzad, O.C. Polymeric nanoparticles for drug delivery. Methods Mol. Biol. (Clifton NJ) 2010, 624, 163–175. [Google Scholar] [CrossRef]
- Zhang, H. Thermally cross-linked superparamagnetic iron oxide nanoparticle-A10 RNA aptamer-doxorubicin conjugate. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Kolishetti, N.; Dhar, S.; Valencia, P.M.; Lin, L.Q.; Karnik, R.; Lippard, S.J.; Langer, R.; Farokhzad, O.C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 17939–17944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Jo, H.; Song, K.; Cho, M.; Chun, Y.S.; Jon, S.; Kim, W.J.; Ban, C. Dual-aptamer-based delivery vehicle of doxorubicin to both PSMA (+) and PSMA (-) prostate cancers. Biomaterials 2011, 32, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ding, B.; Gao, J.; Wang, H.; Fan, W.; Wang, X.; Zhang, W.; Wang, X.; Ye, L.; Zhang, M.; et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int. J. Nanomed. 2011, 6, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Taghdisi, S.M.; Danesh, N.M.; Sarreshtehdar Emrani, A.; Tabrizian, K.; Zandkarimi, M.; Ramezani, M.; Abnous, K. Targeted delivery of Epirubicin to cancer cells by PEGylated A10 aptamer. J. Drug Target. 2013, 21, 739–744. [Google Scholar] [CrossRef]
- Hao, Z.; Fan, W.; Hao, J.; Wu, X.; Zeng, G.Q.; Zhang, L.J.; Nie, S.F.; Wang, X.D. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. 2016, 23, 874–881. [Google Scholar] [CrossRef]
- Tang, L.; Tong, R.; Coyle, V.J.; Yin, Q.; Pondenis, H.; Borst, L.B.; Cheng, J.; Fan, T.M. Targeting tumor vasculature with aptamer-functionalized doxorubicin-polylactide nanoconjugates for enhanced cancer therapy. ACS Nano 2015, 9, 5072–5081. [Google Scholar] [CrossRef]
- Diao, Y.; Liu, J.; Ma, Y.; Su, M.; Zhang, H.; Hao, X. A specific aptamer-cell penetrating peptides complex delivered siRNA efficiently and suppressed prostate tumor growth in vivo. Cancer Biol 2016, 17, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.C.; Wang, A.; Ye, K.; Jin, S. A RNA-DNA Hybrid Aptamer for Nanoparticle-Based Prostate Tumor Targeted Drug Delivery. Int. J. Mol. Sci. 2016, 17, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wang, Y.; Wang, Y.; Zhang, M.; Luo, Y.; Tang, J.; Wang, Z.; Wang, D.; Hao, L.; Wang, Z. Paclitaxel-loaded and A10-3.2 aptamer-targeted poly(lactide-co-glycolic acid) nanobubbles for ultrasound imaging and therapy of prostate cancer. Int. J. Nanomed. 2017, 12, 5313–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Zhao, H.; Guo, L.; Wang, Y.; Song, J.; Zhao, X.; Li, C.; Hao, L.; Wang, D.; Tang, J. Ultrasound-mediated nanobubble destruction (UMND) facilitates the delivery of A10-3.2 aptamer targeted and siRNA-loaded cationic nanobubbles for therapy of prostate cancer. Drug Deliv. 2018, 25, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Duan, J.-H.; Song, Y.-M.; Ma, J.; Wang, F.-D.; Lü, X.; Yang, X.-D. Novel HER2 Aptamer Selectively Delivers Cytotoxic Drug to HER2-positive Breast Cancer Cells in Vitro. J. Transl. Med. 2012, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yao, H.; Meng, Y.; Wang, Y.; Yan, X.; Huang, R. Specific aptamer-conjugated mesoporous silica-carbon nanoparticles for HER2-targeted chemo-photothermal combined therapy. Acta Biomater. 2015, 16, 196–205. [Google Scholar] [CrossRef]
- Liang, T.; Yao, Z.; Ding, J.; Min, Q.; Jiang, L.; Zhu, J.-J. Cascaded Aptamers-Governed Multistage Drug-Delivery System Based on Biodegradable Envelope-Type Nanovehicle for Targeted Therapy of HER2-Overexpressing Breast Cancer. ACS Appl. Mater. Interfaces 2018, 10, 34050–34059. [Google Scholar] [CrossRef]
- Gijs, M.; Penner, G.; Blackler, G.; Impens, N.; Baatout, S.; Luxen, A.; Aerts, A. Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals 2016, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.; Cydzik, M.; Gariepy, J. Targeting the PD-1/PD-L1 Immune Evasion Axis With DNA Aptamers as a Novel Therapeutic Strategy for the Treatment of Disseminated Cancers. Mol. Therapy. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef]
- Lai, W.Y.; Huang, B.T.; Wang, J.W.; Lin, P.Y.; Yang, P.C. A Novel PD-L1-targeting Antagonistic DNA Aptamer With Antitumor Effects. Mol. Therapy. Nucleic Acids 2016, 5, e397. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, D.-M.; Kim, K.-S.; Jung, W.; Kim, D.-E. Applications of Cancer Cell-Specific Aptamers in Targeted Delivery of Anticancer Therapeutic Agents. Molecules 2018, 23, 830. [Google Scholar] [CrossRef] [Green Version]
- Alshaer, W.; Hillaireau, H.; Fattal, E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv. Drug Deliv. Rev. 2018, 134, 122–137. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.E.B.; Alves, L.N.; Rocha, H.F.; Cabral-Neto, J.B.; Missailidis, S.; Paulino, E.T. Aptamer delivery of siRNA, radiopharmaceutics and chemotherapy agents in cancer. Int. J. Pharm. 2017, 525, 334–342. [Google Scholar] [CrossRef]
- Catuogno, S.; Esposito, C.L.; De Franciscis, V. Aptamer-Mediated Targeted Delivery of Therapeutics: An Update. Pharmaceuticals 2016, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Franciscis, V. Challenging cancer targets for aptamer delivery. Biochimie 2018, 145, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18. [Google Scholar]
- Bagalkot, V.; Farokhzad, O.C.; Langer, R.; Jon, S. An Aptamer–Doxorubicin Physical Conjugate as a Novel Targeted Drug-Delivery Platform. Angew. Chem. Int. Ed. 2006, 45, 8149–8152. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Shangguan, D.; Liu, H.; Phillips, J.A.; Zhang, X.; Chen, Y.; Tan, W. Molecular Assembly of an Aptamer-Drug Conjugate for Targeted Drug Delivery to Tumor Cells. ChemBioChem 2009, 10, 862–868. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.C.; Twu, K.Y.; Ellington, A.D.; Levy, M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006, 34, e73. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.; Andrechek, E.; Wang, Y.; Viles, K.D.; E Rempel, R.; Gilboa, E.; A Sullenger, B.; Giangrande, P.H.; Ii, J.O.M. Cell type–specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Thiel, K.W.; Hernandez, L.I.; Dassie, J.P.; Thiel, W.; Liu, X.; Stockdale, K.R.; Rothman, A.M.; Hernandez, F.J.; McNamara, J.; Giangrande, P.H. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012, 40, 6319–6337. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ghamande, S.; Liu, H.; Xue, L.; Zhao, S.; Tan, W.; Zhao, L.; Tang, S.C.; Wu, D.; Korkaya, H.; et al. Targeting EGFR/HER2/HER3 with a Three-in-One Aptamer-siRNA Chimera Confers Superior Activity against HER2(+) Breast Cancer. Mol. Ther. Nucleic Acids 2018, 10, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Maihle, N.J.; Yu, X.; Tang, S.-C.; Liu, H.Y. Synergistic Targeting HER2 and EGFR with Bivalent Aptamer-siRNA Chimera Efficiently Inhibits HER2-Positive Tumor Growth. Mol. Pharm. 2018, 15, 4801–4813. [Google Scholar] [CrossRef] [Green Version]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.-Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., II; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Neff, C.P.; Swiderski, P.; Li, H.; Smith, D.D.; Aboellail, T.; Remling-Mulder, L.; Akkina, R.; Rossi, J.J. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tiemann, K.; Chomchan, P.; Alluin, J.; Swiderski, P.; Burnett, J.; Zhang, X.; Forman, S.; Chen, R.; Rossi, J. Dual functional BAFF receptor aptamers inhibit ligand-induced proliferation and deliver siRNAs to NHL cells. Nucleic Acids Res. 2013, 41, 4266–4283. [Google Scholar] [CrossRef] [PubMed]
- Gilboa-Geffen, A.; Hamar, P.; Le, M.T.; Wheeler, L.A.; Trifonova, R.; Petrocca, F.; Wittrup, A.; Lieberman, J. Gene Knockdown by EpCAM Aptamer-siRNA Chimeras Suppresses Epithelial Breast Cancers and Their Tumor-Initiating Cells. Mol. Cancer Ther. 2015, 14, 2279–2291. [Google Scholar] [CrossRef] [Green Version]
- Esposito, C.L.; Nuzzo, S.; Catuogno, S.; Romano, S.; de Nigris, F.; de Franciscis, V. STAT3 Gene Silencing by Aptamer-siRNA Chimera as Selective Therapeutic for Glioblastoma. Mol. Therapy. Nucleic Acids 2018, 10, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef]
- Zhou, J.; Lazar, D.; Li, H.; Xia, X.; Satheesan, S.; Charlins, P.; O’Mealy, D.; Akkina, R.; Saayman, S.; Weinberg, M.S.; et al. Receptor-targeted aptamer-siRNA conjugate-directed transcriptional regulation of HIV-1. Theranostics 2018, 8, 1575–1590. [Google Scholar] [CrossRef]
- Nachreiner, I.; Hussain, A.F.; Wullner, U.; Machuy, N.; Meyer, T.F.; Fischer, R.; Gattenlohner, S.; Meinhold-Heerlein, I.; Barth, S.; Tur, M.K. Elimination of HER3-expressing breast cancer cells using aptamer-siRNA chimeras. Exp. Ther. Med. 2019, 18, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanotechnology and aptamers: Applications in drug delivery. Trends Biotechnol. 2008, 26, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Karp, J.M.; Langer, R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin. Drug Deliv. 2006, 3, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Jon, S.; Khademhosseini, A.; Tran, T.N.; Lavan, D.A.; Langer, R. Nanoparticle-aptamer bioconjugates: A new approach for targeting prostate cancer cells. Cancer Res. 2004, 64, 7668–7672. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [Green Version]
- Pala, K.; Serwotka, A.; Jelen, F.; Jakimowicz, P.; Otlewski, J. Tumor-specific hyperthermia with aptamer-tagged superparamagnetic nanoparticles. Int. J. Nanomed. 2014, 9, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Soudi, T.; Shojaosadati, S.A. Aptamer functionalized curcumin-loaded human serum albumin (HSA) nanoparticles for targeted delivery to HER-2 positive breast cancer cells. Int. J. Biol. Macromol. 2019, 130, 109–116. [Google Scholar] [CrossRef]
- Alshaer, W.; Hillaireau, H.; Vergnaud, J.; Mura, S.; Delomenie, C.; Sauvage, F.; Ismail, S.; Fattal, E. Aptamer-guided siRNA-loaded nanomedicines for systemic gene silencing in CD-44 expressing murine triple-negative breast cancer model. J. Control. Release Off. J. Control. Release Soc. 2018, 271, 98–106. [Google Scholar] [CrossRef]
- Majidi Zolbanin, N.; Jafari, R.; Majidi, J.; Atyabi, F.; Yousefi, M.; Jadidi-Niaragh, F.; Aghebati-Maleki, L.; Shanehbandi, D.; Soltani Zangbar, M.S.; Nayebi, A.M. Targeted Co-Delivery of Docetaxel and cMET siRNA for Treatment of Mucin1 Overexpressing Breast Cancer Cells. Adv. Pharm. Bull. 2018, 8, 383–393. [Google Scholar] [CrossRef]
- Yan, W.; Tao, M.; Jiang, B.; Yao, M.; Jun, Y.; Dai, W.; Tang, Z.; Gao, Y.; Zhang, L.; Chen, X.; et al. Overcoming Drug Resistance in Colon Cancer by Aptamer-Mediated Targeted Co-Delivery of Drug and siRNA Using Grapefruit-Derived Nanovectors. Cell. Physiol. Biochem Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 50, 79–91. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, D.; Xu, Z.; Huang, C.; Zhang, C.; He, B.; Mao, C.; Wang, G.; Qian, H. Targeted delivery of Rab26 siRNA with precisely tailored DNA prism for lung cancer therapy. Chembiochem 2019. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Xu, Z.; Sun, F.; Wang, Y.; Zhang, J.; Luo, C.; Wang, M. HPA aptamer functionalized paclitaxel-loaded PLGA nanoparticles for enhanced anticancer therapy through targeted effects and microenvironment modulation. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 117, 109121. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F. Aptamers: A New Technological Platform in Cancer Immunotherapy. Pharmaceuticals (Basel Switzerland) 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Reviews. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- Friedlaender, A.; Addeo, A.; Banna, G. New emerging targets in cancer immunotherapy: The role of TIM3. Esmo Open 2019, 4, e000497. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C. Tim-3: An Emerging Target in the Cancer Immunotherapy Landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Hervas-Stubbs, S.; Soldevilla, M.M.; Villanueva, H.; Mancheno, U.; Bendandi, M.; Pastor, F. Identification of TIM3 2′-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget 2016, 7, 4522–4530. [Google Scholar] [CrossRef]
- Gefen, T.; Castro, I.; Muharemagic, D.; Puplampu-Dove, Y.; Patel, S.; Gilboa, E. A TIM-3 Oligonucleotide Aptamer Enhances T Cell Functions and Potentiates Tumor Immunity in Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2280–2288. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig. 2008, 118, 376–386. [Google Scholar] [CrossRef]
- Pastor, F.; Kolonias, D.; McNamara, J.O., II; Gilboa, E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1878–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilboa, E.; McNamara, J., II; Pastor, F. Use of oligonucleotide aptamer ligands to modulate the function of immune receptors. Clin. Cancer Res Off. J. Am. Assoc. Cancer Res. 2013, 19, 1054–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezhnoy, A.; Rajagopalan, A.; Gilboa, E. A clinically useful approach to enhance immunological memory and antitumor immunity. Oncoimmunology 2014, 3, e28811. [Google Scholar] [CrossRef] [Green Version]
- Schrand, B.; Berezhnoy, A.; Brenneman, R.; Williams, A.; Levay, A.; Kong, L.Y.; Rao, G.; Zhou, S.; Heimberger, A.B.; Gilboa, E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol. Res. 2014, 2, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Schrand, B.; Berezhnoy, A.; Brenneman, R.; Williams, A.; Levay, A.; Gilboa, E. Reducing toxicity of 4-1BB costimulation: Targeting 4-1BB ligands to the tumor stroma with bi-specific aptamer conjugates. Oncoimmunology 2015, 4, e970918. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, A.; Berezhnoy, A.; Schrand, B.; Puplampu-Dove, Y.; Gilboa, E. Aptamer-Targeted Attenuation of IL-2 Signaling in CD8(+) T Cells Enhances Antitumor Immunity. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Puplampu-Dove, Y.; Gefen, T.; Rajagopalan, A.; Muheramagic, D.; Schrand, B.; Gilboa, E. Potentiating tumor immunity using aptamer-targeted RNAi to render CD8(+) T cells resistant to TGFbeta inhibition. Oncoimmunology 2018, 7, e1349588. [Google Scholar] [CrossRef] [Green Version]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; de Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 aptamers as powerful immune response modulators. Mol. Therapy. Nucleic Acids 2013, 2, e98. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Bendandi, M.; Inoges, S.; Lopez-Diaz de Cerio, A.; Pastor, F. 2-fluoro-RNA oligonucleotide CD40 targeted aptamers for the control of B lymphoma and bone-marrow aplasia. Biomaterials 2015, 67, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Soldevilla, M.M.; Villanueva, H.; Meraviglia-Crivelli, D.; Menon, A.P.; Ruiz, M.; Cebollero, J.; Villalba, M.; Moreno, B.; Lozano, T.; Llopiz, D.; et al. ICOS Costimulation at the Tumor Site in Combination with CTLA-4 Blockade Therapy Elicits Strong Tumor Immunity. Mol. Ther. J. Am. Soc. Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, OX40. Nucleic Acid 2013, 23, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Selection of DNA aptamer using SELEX. Note: For the selection of RNA aptamer, the DNA oligonucleotide pool must be in vitro transcribed into RNA oligonucleotide pool before selection, and the collected oligonucleotides must be reverse transcribe-amplified with TR-PCR into DNA and then be in vitro transcribed into RNA for the next round of selection. Abbreviations: OG, oligonucleotide; PCR, polymerase chain reaction, SELEX, systematic evolution of ligands by exponential enrichment.
Figure 1.
Selection of DNA aptamer using SELEX. Note: For the selection of RNA aptamer, the DNA oligonucleotide pool must be in vitro transcribed into RNA oligonucleotide pool before selection, and the collected oligonucleotides must be reverse transcribe-amplified with TR-PCR into DNA and then be in vitro transcribed into RNA for the next round of selection. Abbreviations: OG, oligonucleotide; PCR, polymerase chain reaction, SELEX, systematic evolution of ligands by exponential enrichment.

Figure 2.
Conjugation of aptamers with anticancer therapeutics for targeted delivery.

Figure 3.
Aptamers as immune checkpoint inhibitors. Note: The binding of programmed death-ligand 1 (PD-L1), the immune checkpoint receptor on T cells, with PD-L1, the ligand on cancer cells, will transduce an inhibitory signal to the T cell. Either programmed death 1 (PD-1) aptamer or PD-L1 aptamer can block the binding.
Figure 3.
Aptamers as immune checkpoint inhibitors. Note: The binding of programmed death-ligand 1 (PD-L1), the immune checkpoint receptor on T cells, with PD-L1, the ligand on cancer cells, will transduce an inhibitory signal to the T cell. Either programmed death 1 (PD-1) aptamer or PD-L1 aptamer can block the binding.


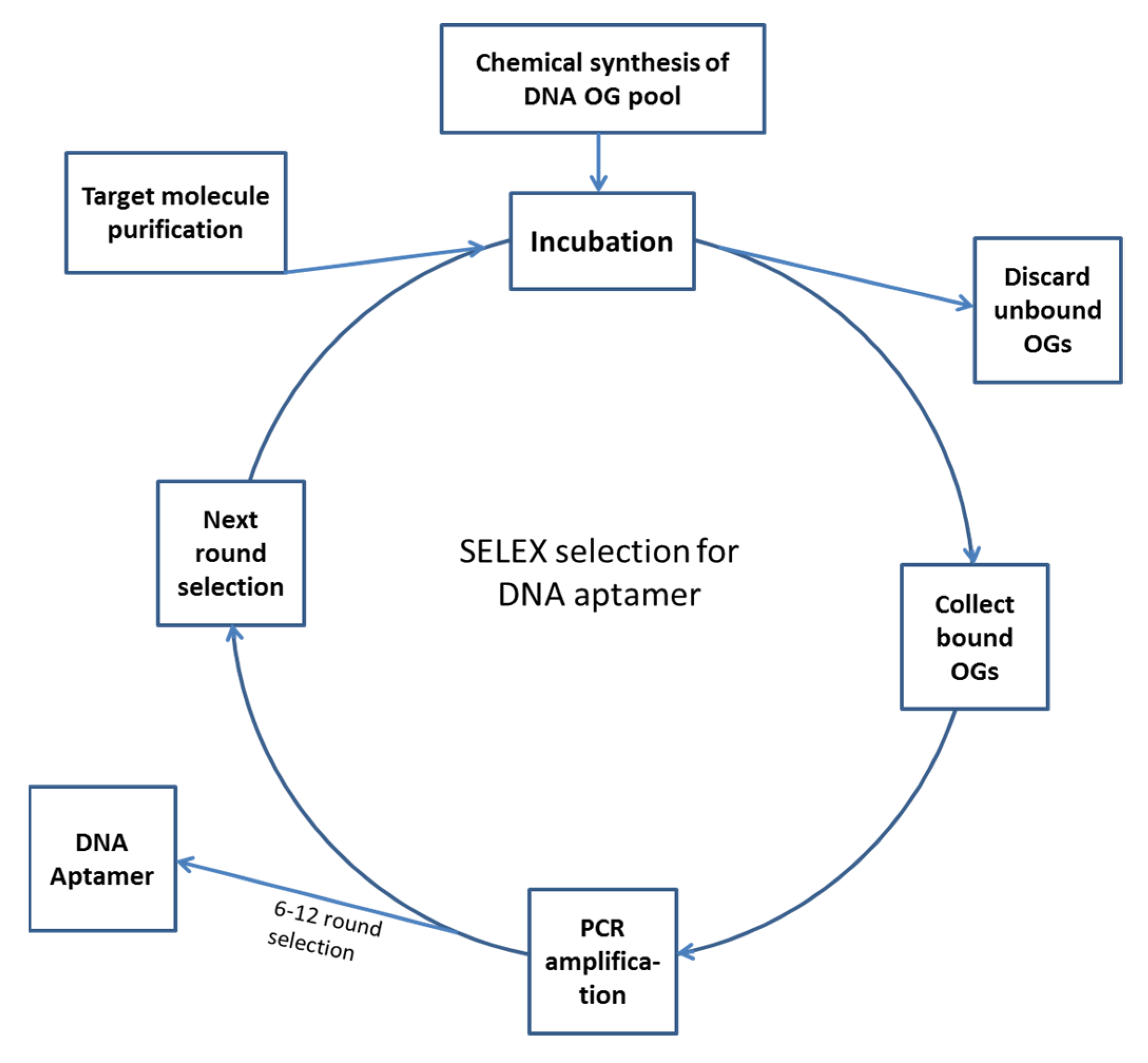{kind=link}
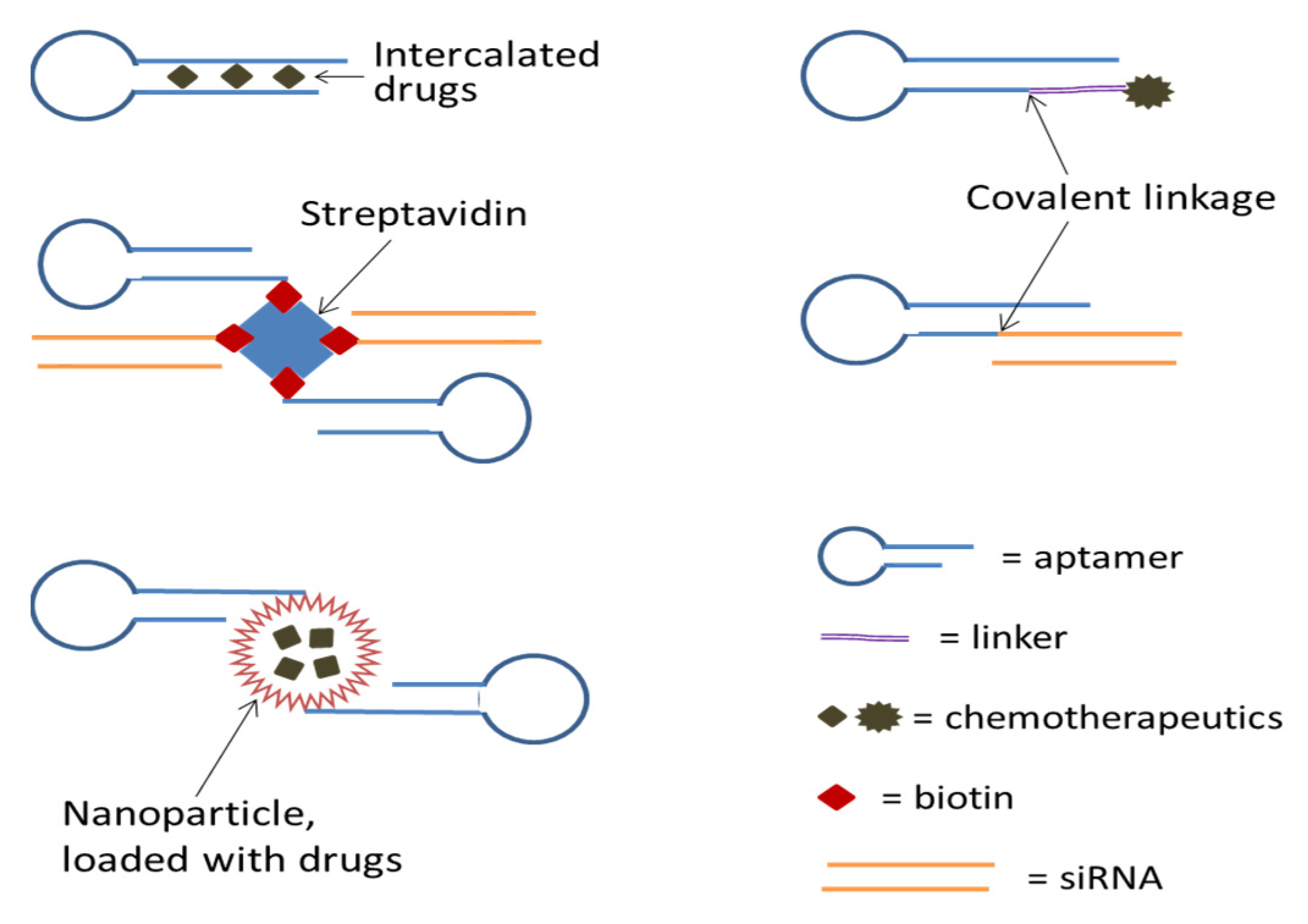{kind=link}
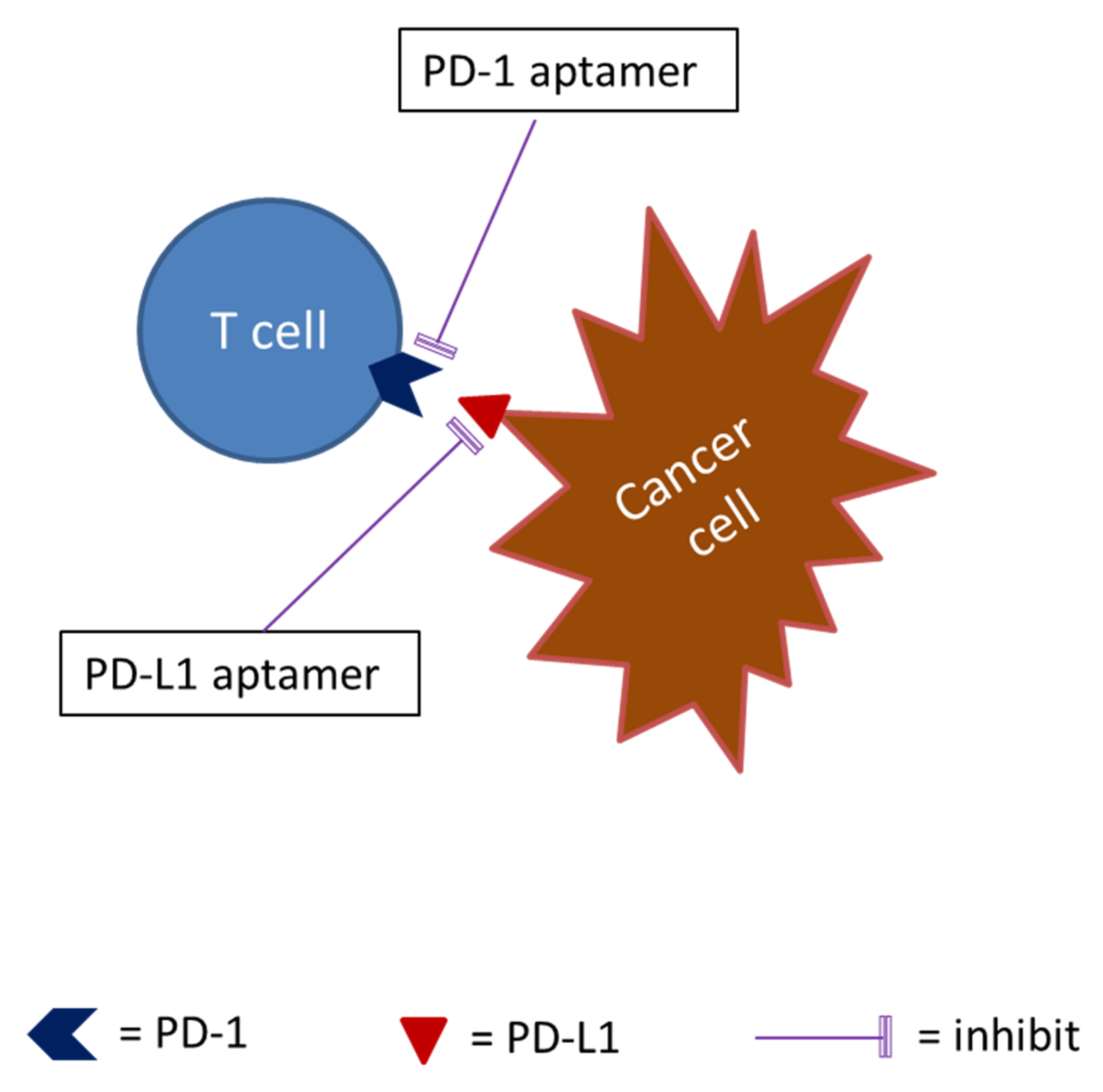{kind=link}
Table 1.
A comparison between aptamers and monoclonal antibodies (mAbs).
| Criteria | Aptamers | mAbs |
|---|---|---|
| Chemical composition | Nucleic acid (DNA or RNA) | Protein |
| Molecular weight | 10–50 kDa | 140–700 kDa |
| Animal immunization for preparation? | No | Yes (except for genetically engineered Ab) |
| In vitro preparation | Yes | No |
| Targets [21,22,23] | Multiple, including cells, viruses, proteins, peptide, polysaccharides, nucleic acids, nucleotides, amino acids, other small organic molecules and inorganic molecules, etc. | Proteins mainly, but include cells, viruses, polysaccharides, and nucleic acids |
| Specificity to target | Yes | Yes |
| Binding affinity | Nano-molar~pico-molar, maybe femto-molar | Nano-molar~pico-molar, maybe femto-molar |
| Molecular forces involved in target binding | Electrostatic forces, hydrogen bonds, hydrophobic interactions, and van der Waals forces | Electrostatic forces, hydrogen bonds, hydrophobic interactions, and van der Waals forces |
| Stability | Stable at 80 °C | denatured at 80 °C |
| Reannealing if denatured | Yes | No |
| batch-to-batch variations | Low | High |
| Shelf life | Long | Short |
| Cost | Lower | Higher |
| In vivo half-life [24] (clearance rate) | Short (~20 min) | Long (~one month) |
| Immunogenicity (causing allergy) | No | Yes (unless humanized) |
| Internalization | Higher possibility | Difficult |
| Diagnostic usage | Yes | Yes |
| Therapeutic usage | Yes | Yes, but may cause allergy if not humanized |
Table 2.
Selected aptamers under clinical trials or active laboratory investigation for cancer therapy.
Table 2.
Selected aptamers under clinical trials or active laboratory investigation for cancer therapy.
| Aptamer | Selection | DNA/RNA | Kd | Target | INT | Function | Application/Mechanism | Status | References |
|---|---|---|---|---|---|---|---|---|---|
| Pegaptanib sodium (Macugen) | SELEX, 10 rounds | RNA (28 nt) | 50 pM | VEGF165 | No | Antagonism | 1. Age-related macular degeneration (AMD). 2. Potential therapeutic application for solid cancers with extensive angiogenesis. | Approved by FDA for treatment of AMD | [84,85,86,87] |
| AS1411 | Designed and chemically synthesized | DNA (26 nt), guanosine rich quartets | 55 nM | Nucleolin | Yes | Internalization or delivery | 1. Binding cell-surface nucleolin and internalization, leading to DNA replication inhibition. 2. Drug delivery. | Phase II clinical trial | [18,88,89,90] |
| NOX-A12 | Spiegelmer * technology | L-RNA (45 nt) | 200 pM | CXCL12 | No | Antagonism | Disrupting the homing and the accumulation of CLL cells in the bone marrow, sensitizing these cells to cytotoxic drugs. | Phase II clinical trial | [54,81,83] |
| AX102 | SELEX, 12 rounds | DNA (34 nt) | 100 pM | PDGF-B | No | Antagonism | 1. Inhibition of tumor angiogenesis. 2. Promotion of tumor blood vessel efficiency, resulting in increased anticancer drug delivery. | Pre-clinical | [67,69,70,71] |
| xPSM-A10 (A10) | SELEX, 6 rounds | RNA (72 nt) | 1.5 nM | PSMA | Yes | Internalization and delivery | # Delivery of (1) chemotherapeutics, (2) therapeutic RNAs, and (3) nanoparticles to PSMA-positive prostate cancer cells. | Pre-clinical | [91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107] |
| HB5 | SELEX, multiple rounds | DNA (86) | 18.9 nM | HER2 | Yes | Internalization and delivery | Delivery of (1) chemotherapeutics, (2) nanoparticles to HER2-positive breast cancer cells. | Pre-clinical | [108,109,110] |
| HeA2_3 | Whole-cell SELEX | DNA | 6.2 nM | HER2 | Yes | Internalization | Binding with high specificity to HER2- positive cells and tumor tissue and great potential for the treatment of HER2- overexpressing cancers. | Pre-clinical | [111] |
| MP7 | SELEX, 5 rounds | DNA | 167 nM | Murine PD-1 | No | Antagonism | Blocking murine PD-1 and PD-L1 interaction so as to restore T cell function. | Pre-clinical | [112] |
| aptPD-L1 | SELEX, 8 rounds | DNA | 4.7 nM | Human PD-L1 | No | Antagonism | Blocking the binding between human PD-1 and PD-L1 so as to restore T cell function. | Pre-clinical | [113] |
Abbreviations: INT (internalization); AMD (age-related macular degeneration); CLL (chronic lymphocytic leukemia); HER2 (human epidermal growth factor receptor 2); CXCL12 (C-X-C Chemokine Ligand 12); Kd (dissociation constant), PD-1 (programmed death 1); PD-L1 (programmed death-ligand 1); PDGF-B (platelet-derived growth factor receptor-B); PSMA (prostate-specific membrane antigen); SELEX (systematic evolution of ligands by exponential enrichment); VEGF165 (165-amino-acid isoform of vascular endothelial growth factor). * See main text for the explanation of Spiegelmer. # Including a truncated version of A10 (A10-3.2).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fu, Z.; Xiang, J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 2793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082793
AMA Style
Fu Z, Xiang J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. International Journal of Molecular Sciences. 2020; 21(8):2793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082793
Chicago/Turabian StyleFu, Zhaoying, and Jim Xiang. 2020. "Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy" International Journal of Molecular Sciences 21, no. 8: 2793. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082793
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.