TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings

 ,
, 

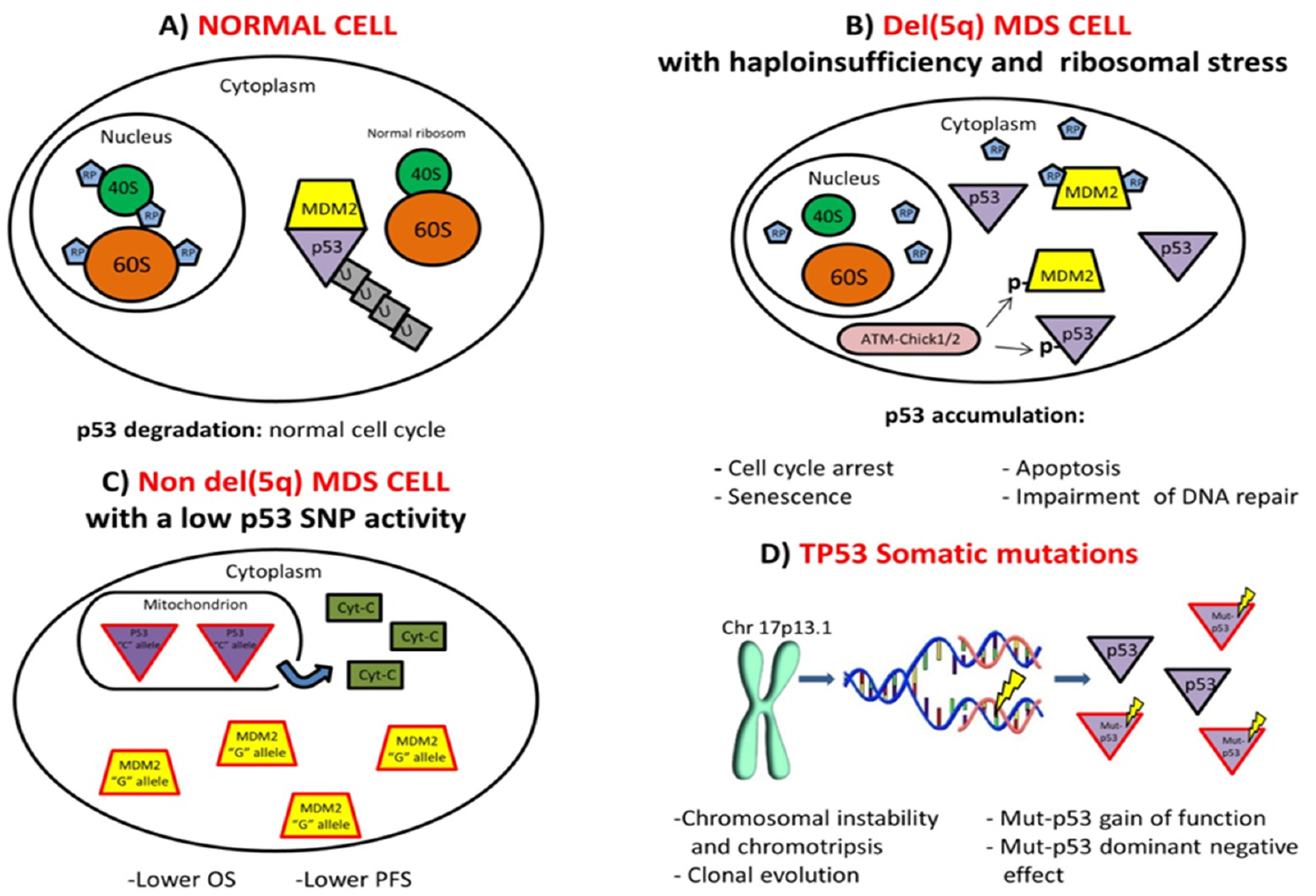{kind=link}
Abstract
:1. Introduction
2. Biological and Molecular Aspects
2.1. Molecular Pathways Activation
2.2. TP53 Allelic State and Mutational Burden
2.3. TP53 Germline Mutations and Familial MDS Predisposition
2.4. p53 Protein Expression
2.5. TP53 and Karyotype Aberrations
2.6. Therapy-Related MDS
3. Clinical Implications
3.1. Prognosis and Clinical Outcome
3.2. Conventional Therapeutic Approaches
3.3. Emerging Therapeutic Strategies
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| allo-HSCT | allogeneic hematopoietic stem cell transplantation |
| AML | acute myeloid leukemia |
| ATO | arsenic trioxide |
| AZA | azacytidine |
| BM | bone marrow |
| CDR | commonly deleted region |
| CHIP | clonal hemopoiesis of indeterminate potential |
| CK | complex karyotypes |
| CLL | chronic lymphocytic leukemia |
| cn-LOH | copy-neutral loss of heterozygosity |
| CR | complete remission |
| DAC | decitabine |
| DBD | DNA-binding domain |
| DDR | DNA damage responses |
| DNE | dominant negative effect |
| DSB | double-strand breaks |
| GOF | gain of function |
| H3K27me3 | trimethylation of lysine 27 of histone H3 |
| HMAs | hypomethylating agents |
| HSC | hematopoietic stem cell |
| HSPC | hematopoietic stem and progenitor cell |
| IHC | immunohistochemistry |
| IPSS | International Prognostic Scoring System |
| IPSS-R | revised International Prognostic Scoring System |
| LFS | Li-Fraumeni syndrome |
| LOH | loss of heterozygosity |
| MDM2 | Murine Double Minute-2 |
| MDS | myelodysplastic syndromes |
| MK | Monosomal karyotype |
| MM | multiple myeloma |
| MQ | methylene quinuclidinone |
| NGS | next generation sequencing |
| ORR | overall response rate |
| OS | overall survival |
| PFS | progression-free survival |
| PRC2 | Polycomb repressive complex 2 |
| RFS | relapse-free survival |
| RPs | ribosomal proteins |
| SNP | single-nucleotide polymorphism |
| t-AML | therapy-related acute myeloid leukemia |
| t-MDS | therapy-related myelodysplastic syndromes |
| t-MNs | therapy-related myeloid neoplasms |
| TP53 | tumor protein p53 |
| TPII | topoisomerase II |
| VAF | variant allele frequency |
| WPSS | WHO-based Prognostic Scoring System |
| WT | wild-type |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.; Walter, M.J. Genetics of myelodysplastic syndromes: New insights. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Galili, N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat. Rev. Cancer 2012, 12, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; McGraw, K.L.; Sallman, D.A.; List, A.F. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: Molecular aspects and clinical implications. Leukemia Lymphoma 2017, 58, 1777–1790. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Sallman, D.A. Targeting TP53 Mutations in Myelodysplastic Syndromes. Hematol. Oncol. Clin. North Am. 2020, 34, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Hamada, M.; Fujiwara, T.; Hizuta, A.; Gochi, A.; Naomoto, Y.; Takakura, N.; Takahashi, K.; Roth, J.A.; Tanaka, N.; Orita, K. The p53 gene is a potent determinant of chemosensitivity and radiosensitivity in gastric and colorectal cancers. J. Cancer Res. Clin. Oncol. 1996, 122, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Grimm, E.A.; Mukhopadhyay, T.; Zhang, W.W.; Owen-Schaub, L.B.; Roth, J.A. Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res. 1994, 54, 2287–2291. [Google Scholar] [PubMed]
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gäken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B.; et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef]
- Kaneko, H.; Misawa, S.; Horiike, S.; Nakai, H.; Kashima, K. TP53 mutations emerge at early phase of myelodysplastic syndrome and are associated with complex chromosomal abnormalities. Blood 1995, 85, 2189–2193. [Google Scholar] [CrossRef] [Green Version]
- Solé, F.; Espinet, B.; Sanz, G.F.; Cervera, J.; Calasanz, M.J.; Luño, E.; Prieto, F.; Granada, I.; Hernández, J.M.; Cigudosa, J.C.; et al. Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. Grupo Cooperativo Español de Citogenética Hematológica. Br. J. Haematol. 2000, 108, 346–356. [Google Scholar] [CrossRef]
- Ebert, B.L. Deletion 5q in myelodysplastic syndrome: A paradigm for the study of hemizygous deletions in cancer. Leukemia 2009, 23, 1252–1256. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef]
- Ebert, B.L. Molecular Dissection of the 5q Deletion in Myelodysplastic Syndrome. Semin. Oncol. 2011, 38, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.K.; Schenone, M.; Ferreira, M.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Germing, U.; Platzbecker, U.; et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Wei, S.; Chen, X.; McGraw, K.; Zhang, L.; Komrokji, R.; Clark, J.; Caceres, G.; Billingsley, D.; Sokol, L.; Lancet, J.; et al. Lenalidomide promotes p53 degradation by inhibiting MDM2 auto-ubiquitination in myelodysplastic syndrome with chromosome 5q deletion. Oncogene 2013, 32, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, E.C.; Ebert, B.L. The novel mechanism of lenalidomide activity. Blood 2015, 126, 2366–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.h.; List, A.; Sallman, D.A. Molecular pathogenesis of myelodysplastic syndromes with deletion 5q. Eur. J. Haematol. 2019, 102, 203–209. [Google Scholar] [CrossRef] [Green Version]
- McGraw, K.L.; Zhang, L.M.; Rollison, D.E.; Basiorka, A.A.; Fulp, W.; Rawal, B.; Jerez, A.; Billingsley, D.L.; Lin, H.-Y.; Kurtin, S.E.; et al. The relationship of TP53 R72P polymorphism to disease outcome and TP53 mutation in myelodysplastic syndromes. Blood Cancer J. 2015, 5, e291. [Google Scholar] [CrossRef]
- McGraw, K.L.; Cluzeau, T.; Sallman, D.A.; Basiorka, A.A.; Irvine, B.A.; Zhang, L.; Epling-Burnette, P.K.; Rollison, D.E.; Mallo, M.; Sokol, L.; et al. TP53 and MDM2 single nucleotide polymorphisms influence survival in non-del(5q) myelodysplastic syndromes. Oncotarget 2015, 6, 34437–34445. [Google Scholar] [CrossRef]
- Liu, Y.C.; Hsiao, H.H.; Yang, W.C.; Liu, T.C.; Chang, C.S.; Yang, M.Y.; Lin, P.M.; Hsu, J.F.; Lee, C.P.; Lin, S.F. MDM2 promoter polymorphism and p53 codon 72 polymorphism in chronic myeloid leukemia: The association between MDM2 promoter genotype and disease susceptibility, age of onset, and blast-free survival in chronic phase patients receiving imatinib. Mol. Carcinog. 2014, 53, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.R.; Breccia, M.; Abruzzese, E.; Castagnetti, F.; Luciano, L.; Gozzini, A.; Annunziata, M.; Martino, B.; Stagno, F.; Cavazzini, F.; et al. Outcome of 82 chronic myeloid leukemia patients treated with nilotinib or dasatinib after failure of two prior tyrosine kinase inhibitors. Haematologica 2013, 98, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Lutzmann, M.; Bernex, F.; da Costa de Jesus, C.; Hodroj, D.; Marty, C.; Plo, I.; Vainchenker, W.; Tosolini, M.; Forichon, L.; Bret, C.; et al. MCM8- and MCM9 Deficiencies Cause Lifelong Increased Hematopoietic DNA Damage Driving p53-Dependent Myeloid Tumors. Cell Rep. 2019, 28, 2851–2865.e4. [Google Scholar] [CrossRef] [PubMed]
- Salari, A.; Thomay, K.; Himmler, K.; Vajen, B.; Schienke, A.; Hagedorn, M.; Ebersold, J.; Kreipe, H.-H.; Krüger, A.; Schambach, A.; et al. Establishing a murine xenograft-model for long-term analysis of factors inducing chromosomal instability in myelodysplastic syndrome: Pitfalls and successes. Cancer Genet. 2016, 209, 258–266. [Google Scholar] [CrossRef]
- Amelio, I.; Melino, G. Context is everything: Extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Popp, H.D.; Naumann, N.; Brendel, S.; Henzler, T.; Weiss, C.; Hofmann, W.-K.; Fabarius, A. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk. Res. 2017, 57, 112–118. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Abáigar, M.; Robledo, C.; Benito, R.; Ramos, F.; Díez-Campelo, M.; Hermosín, L.; Sánchez-Del-Real, J.; Alonso, J.M.; Cuello, R.; Megido, M.; et al. Chromothripsis Is a Recurrent Genomic Abnormality in High-Risk Myelodysplastic Syndromes. PLoS ONE 2016, 11, e0164370. [Google Scholar] [CrossRef]
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? DMM Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; Van De Locht, L.T.; De Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myelodysplastic syndromes. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.J.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Makishima, H.; Kerr, C.M.; Przychodzen, B.P.; Aly, M.; Goyal, A.; Awada, H.; Asad, M.F.; Kuzmanovic, T.; Suzuki, H.; et al. Invariant patterns of clonal succession determine specific clinical features of myelodysplastic syndromes. Nat. Commun. 2019, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Lodé, L.; Ameur, A.; Coste, T.; Ménard, A.; Richebourg, S.; Gaillard, J.-B.; Le Bris, Y.; Béné, M.-C.; Lavabre-Bertrand, T.; Soussi, T. Single-molecule DNA sequencing of acute myeloid leukemia and myelodysplastic syndromes with multiple TP53 alterations. Haematologica 2018, 103, e13–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, Dose, and Mutation-type Specificity Dictate Mutant p53 Functions In Vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storlazzi, C.T.; Albano, F.; Locunsolo, C.; Lonoce, A.; Funes, S.; Guastadisegni, M.C.; Cimarosto, L.; Impera, L.; D’Addabbo, P.; Panagopoulos, I.; et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene. Leukemia 2006, 20, 2190–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. bioRxiv 2019. bioRxiv:2019.12.19.868844. [Google Scholar]
- Sallman, D.A.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef]
- Belickova, M.; Vesela, J.; Jonasova, A.; Pejsova, B.; Votavova, H.; Merkerova, M.D.; Zemanova, Z.; Brezinova, J.; Mikulenkova, D.; Lauermannova, M.; et al. TP53 mutation variant allele frequency is a potential predictor for clinical outcome of patients with lower-risk myelodysplastic syndromes. Oncotarget 2016, 7, 36266–36279. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, B.K.; Rybicki, L.; Hirsch, C.; Przychodzen, B.; Nazha, A.; Gerds, A.T.; Hanna, R.; Kalaycio, M.; Sekeres, M.A.; Sobecks, R.; et al. Mutation clonal burden and allogeneic hematopoietic cell transplantation outcomes in acute myeloid leukemia and myelodysplastic syndromes. Bone Marrow Transplant. 2019, 54, 1281–1286. [Google Scholar] [CrossRef]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Nanopore Sequencing in Blood Diseases: A Wide Range of Opportunities. Front. Genet. 2020, 11, 76. [Google Scholar] [CrossRef]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Brunetti, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Casieri, P.; Coccaro, N.; Tota, G.; et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Orsini, P.; Minervini, C.F.; Cumbo, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Tota, G.; Casieri, P.; Impera, L.; et al. Design and MinION testing of a nanopore targeted gene sequencing panel for chronic lymphocytic leukemia. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes 2019, 10, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumbo, C.; Impera, L.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Coccaro, N.; Tota, G.; Minervini, A.; Casieri, P.; et al. Genomic BCR-ABL1 breakpoint characterization by a multistrategy approach for “personalized monitoring” of residual disease in chronic myeloid leukemia patients. Oncotarget 2018, 9, 10978–10986. [Google Scholar] [CrossRef] [Green Version]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Impera, L.; Coccaro, N.; Brunetti, C.; Minervini, A.; Casieri, P.; et al. Mutational analysis in BCR-ABL1 positive leukemia by deep sequencing based on nanopore MinION technology. Exp. Mol. Pathol. 2017, 103, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A. The epidemiology of myelodysplastic syndromes. Hematol. Oncol. Clin. North Am. 2010, 24, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Does, M.; Raza, A.; Mayne, S.T. Myelodysplastic syndromes: Incidence and survival in the United States. Cancer 2007, 109, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://seer.cancer.gov/archive/csr/1975_2011/results_merged/sect_30_mds.pdf (accessed on 31 March 2020).
- Kuendgen, A.; Strupp, C.; Aivado, M.; Hildebrandt, B.; Haas, R.; Gattermann, N.; Germing, U. Myelodysplastic syndromes in patients younger than age 50. J. Clin. Oncol. 2006, 24, 5358–5365. [Google Scholar] [CrossRef]
- Garber, J.E.; Dreyfus, M.G.; Li, F.P.; Dreyfus, M.G.; Fraumeni, J.F.; Li, F.P. Follow-up Study of Twenty-four Families with Li-Fraumeni Syndrome. Cancer Res. 1991, 51, 6094–6097. [Google Scholar]
- Babushok, D.V.; Bessler, M.; Olson, T.S. Genetic predisposition to myelodysplastic syndrome and acute myeloid leukemia in children and young adults. Leuk. Lymphoma 2016, 57, 520–536. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Achatz, M.I.; Brugieres, L.; Frebourg, T.; Garber, J.E.; Greer, M.L.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [Green Version]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.N.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.C.; Druley, T.E.; Erez, A.; Kuiper, R.P.; Onel, K.; Schiffman, J.D.; Schneider, K.W.; Scollon, S.R.; Scott, H.S.; Strong, L.C.; et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin. Cancer Res. 2017, 23, e14–e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keel, S.B.; Scott, A.; Bonilla, M.S.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.C.; Walsh, T.; Shimamura, A. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 2016, 101, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawana, K.; Drazer, M.W.; Churpek, J.E. Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: Are we there yet? Leukemia 2018, 32, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- McGraw, K.L.; Nguyen, J.; Komrokji, R.S.; Sallman, D.; Al Ali, N.H.; Padron, E.; Lancet, J.E.; Moscinski, L.C.; List, A.F.; Zhang, L. Immunohistochemical pattern of p53 is a measure of TP53 mutation burden and adverse clinical outcome in myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica 2016, 101, e320–e323. [Google Scholar] [CrossRef] [PubMed]
- Kerns, B.J.M.; Jordan, P.A.; Moore, M.B.H.; Humphrey, P.A.; Berchuck, A.; Kohler, M.F.; Bast, R.C.; Iglehart, J.D.; Marks, J.R. p53 overexpression in formalin-fixed, paraffin-embedded tissue detected by immunohistochemistry. J. Histochem. Cytochem. 1992, 40, 1047–1051. [Google Scholar] [CrossRef]
- Iggo, R.; Bartek, J.; Lane, D.; Gatter, K.; Harris, A.L.; Bartek, J. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet 1990, 335, 675–679. [Google Scholar] [CrossRef]
- Chang, H.; Jiang, A.M.; Qi, C.X.Y. Aberrant nuclear p53 expression predicts hemizygous 17p (TP53) deletion in chronic lymphocytic leukemia. Am. J. Clin. Pathol. 2010, 133, 70–74. [Google Scholar] [CrossRef]
- Chang, H.; Yeung, J.; Qi, C.; Xu, W. Aberrant nuclear p53 protein expression detected by immunohistochemistry is associated with hemizygous P53 deletion and poor survival for multiple myeloma. Br. J. Haematol. 2007, 138, 324–329. [Google Scholar] [CrossRef]
- Chen, M.H.; Qi, C.X.Y.; Saha, M.N.; Chang, H. p53 nuclear expression correlates with hemizygous TP53 deletion and predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with lenalidomide. Am. J. Clin. Pathol. 2012, 137, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Jädersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Göhring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Müller-Thomas, C.; Rudelius, M.; Rondak, I.C.; Haferlach, T.; Schanz, J.; Huberle, C.; Schmidt, B.; Blaser, R.; Kremer, M.; Peschel, C.; et al. Response to azacitidine is independent of p53 expression in higher-risk myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica 2014, 99, e179–e181. [Google Scholar] [CrossRef] [PubMed]
- Molteni, A.; Ravano, E.; Riva, M.; Nichelatti, M.; Bandiera, L.; Crucitti, L.; Truini, M.; Cairoli, R. Prognostic impact of immunohistochemical p53 expression in bone marrow biopsy in higher risk MDS: A pilot study. Mediterr. J. Hematol. Infect. Dis. 2019, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, S.; Ito, M.; Watarai, R.; Okuno, S.; Harada, Y.; Yamamoto, S.; Suzuki, K.; Kurahashi, S.; Iwasaki, T.; Sugiura, I. A new prognostic index to make short-term prognoses in MDS patients treated with azacitidine: A combination of p53 expression and cytogenetics. Leuk. Res. 2016, 41, 21–26. [Google Scholar] [CrossRef]
- Pich, A.; Godio, L.; Davico Bonino, L. P53 Protein Expression in Patients with Myelodysplasia Treated with Allogeneic Bone Marrow Transplantation. Mol. Clin. Oncol. 2017, 6, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Ramos, F.; Robledo, C.; Izquierdo-García, F.M.; Suárez-Vilela, D.; Benito, R.; Fuertes, M.; Insunza, A.; Barragán, E.; Del Rey, M.; De Morales, J.M.G.R.; et al. Bone marrow fibrosis in myelodysplastic syndromes: A prospective evaluation including mutational analysis. Oncotarget 2016, 7, 30492–30503. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.B.; Barbosa, M.C.; Jesus dos Santos, T.E.; Lemes, R.P.G.; Vasconcelos, J.P.; de Vasconcelos, P.R.L.; Rocha, F.D.; Zalcberg, I.; Coutinho, D.F. Bone marrow fibrosis at diagnosis is associated with TP53 overexpression and adverse prognosis in low-risk myelodysplastic syndrome. Br. J. Haematol. 2018, 181, 547–549. [Google Scholar] [CrossRef] [Green Version]
- Loghavi, S.; Al-Ibraheemi, A.; Zuo, Z.; Garcia-Manero, G.; Yabe, M.; Wang, S.A.; Kantarjian, H.M.; Yin, C.C.; Miranda, R.N.; Luthra, R.; et al. TP53 overexpression is an independent adverse prognostic factor in de novo myelodysplastic syndromes with fibrosis. Br. J. Haematol. 2015, 171, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.F.; Malik, U.A.; Sohail, M.; Hassan, I.N.; Ali, S.; Shaukat, M.H.S. Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int. J. Hematol. Stem. Cell Res. 2017, 11, 231–239. [Google Scholar]
- Meggendorfer, M.; Haferlach, C.; Kern, W.; Haferlach, T. Molecular analysis of myelodysplastic syndrome with isolated deletion of the long arm of chromosome 5 reveals a specific spectrum of molecular mutations with prognostic impact: A study on 123 patients and 27 genes. Haematologica 2017, 102, 1502–1510. [Google Scholar] [CrossRef]
- Hosono, N.; Makishima, H.; Mahfouz, R.; Przychodzen, B.; Yoshida, K.; Jerez, A.; LaFramboise, T.; Polprasert, C.; Clemente, M.J.; Shiraishi, Y.; et al. Recurrent genetic defects on chromosome 5q in myeloid neoplasms. Oncotarget 2017, 8, 6483–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossner, M.; Jann, J.C.; Nowak, D.; Platzbecker, U.; Giagounidis, A.; Götze, K.; Letsch, A.; Haase, D.; Shirneshan, K.; Braulke, F.; et al. Prevalence, clonal dynamics and clinical impact of TP53 mutations in patients with myelodysplastic syndrome with isolated deletion (5q) treated with lenalidomide: Results from a prospective multicenter study of the German MDS study group (GMDS). Leukemia 2016, 30, 1956–1959. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Høyer, S.; Docking, R.; Chan, S.; Jadersten, M.; Parker, J.; Karsan, A. Mechanisms of Resistance to Lenalidomide in Del(5q) Myelodysplastic Syndrome Patients. Blood 2015, 126, 5228. [Google Scholar] [CrossRef]
- Lodé, L.; Ménard, A.; Flet, L.; Richebourg, S.; Loirat, M.; Eveillard, M.; Le Bris, Y.; Godon, C.; Theisen, O.; Gagez, A.L.; et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 2018, 103, e143–e146. [Google Scholar] [CrossRef] [Green Version]
- Scharenberg, C.; Giai, V.; Pellagatti, A.; Saft, L.; Dimitriou, M.; Jansson, M.; Jädersten, M.; Grandien, A.; Douagi, I.; Neuberg, D.S.; et al. Progression in patients with low- and intermediate-1-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 2017, 102, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [Green Version]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Benton, C.B.; Class, C.A.; Chien, K.S.; Sasaki, K.; Naqvi, K.; Alvarado, Y.; Kadia, T.M.; Ravandi, F.; et al. Genomic context and TP53 allele frequency define clinical outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv. 2020, 4, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Wang, J.; Zhang, H.; Mei, C.; Ye, L.; Luo, Y.; Zhou, X.; Zhu, S.; Jiang, L.; Wang, L.; et al. TP53 mutations are associated with very complex karyotype and suggest poor prognosis in newly diagnosed myelodysplastic syndrome patients with monosomal karyotype. Asia. Pac. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Xing, R.; Li, C.; Gale, R.P.; Zhang, Y.; Xu, Z.; Qin, T.; Li, B.; Fang, L.; Zhang, H.; Pan, L.; et al. Monosomal karyotype is an independent predictor of survival in patients with higher-risk myelodysplastic syndrome. Am. J. Hematol. 2014, 89, E163–E168. [Google Scholar] [CrossRef]
- McQuilten, Z.K.; Sundararajan, V.; Andrianopoulos, N.; Curtis, D.J.; Wood, E.M.; Campbell, L.J.; Wall, M. Monosomal karyotype predicts inferior survival independently of a complex karyotype in patients with myelodysplastic syndromes. Cancer 2015, 121, 2892–2899. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Grau, J.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. Monosomal karyotype in MDS: Explaining the poor prognosis. Leukemia 2013, 27, 1988–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcárcel, D.; Ademà, V.; Solé, F.; Ortega, M.; Nomdedeu, B.; Sanz, G.; Luño, E.; Cañizo, C.; de la Serna, J.; Ardanaz, M.; et al. Complex, not monosomal, karyotype is the cytogenetic marker of poorest prognosis in patients with primary myelodysplastic syndrome. J. Clin. Oncol. 2013, 31, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Idossa, D.; Lasho, T.L.; Mudireddy, M.; Finke, C.; Shah, S.; Nicolosi, M.; Patnaik, M.M.; Pardanani, A.; Gangat, N.; et al. Mutations and karyotype in myelodysplastic syndromes: TP53 clusters with monosomal karyotype, RUNX1 with trisomy 21, and SF3B1 with inv(3)(q21q26.2) and del(11q). Blood Cancer J. 2017, 7, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Routbort, M.J.; Tang, Z.; Ok, C.Y.; Patel, K.P.; Daver, N.; Garcia-Manero, G.; Medeiros, L.J.; Wang, S.A. Characterization of TP53 mutations in low-grade myelodysplastic syndromes and myelodysplastic syndromes with a non-complex karyotype. Eur. J. Haematol. 2017, 99, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Ganser, A.; Heuser, M. Therapy-related myeloid neoplasms. Curr. Opin. Hematol. 2017, 24, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.E.; Boice, J.D.; Stovall, M.; Bernstein, L.; Greenberg, R.S.; Flannery, J.T.; Schwartz, A.G.; Weyer, P.; Moloney, W.C.; Hoover, R.N. Risk of Leukemia after Chemotherapy and Radiation Treatment for Breast Cancer. N. Engl. J. Med. 1992, 326, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Cowell, I.G.; Austin, C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health 2012, 9, 2075–2091. [Google Scholar] [CrossRef]
- Chua, C.C.; Fleming, S.; Wei, A.H. Clinicopathological aspects of therapy-related acute myeloid leukemia and myelodysplastic syndrome. Best Pract. Res. Clin. Haematol. 2019, 32, 3–12. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Fu, B.; Tang, G.; Goswami, M.; Singh, R.; Kanagal-Shamanna, R.; Pierce, S.A.; et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk. Res. 2015, 39, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldoss, I.; Pham, A.; Li, S.M.; Gendzekhadze, K.; Afkhami, M.; Telatar, M.; Hong, H.; Padeganeh, A.; Bedell, V.; Cao, T.; et al. Favorable impact of allogeneic stem cell transplantation in patients with therapy-related myelodysplasia regardless of TP53 mutational status. Haematologica 2017, 102, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, A.; Schinke, C.D.; Georgiev, G.; da Silva Ferreira, M.; Thiruthuvanathan, V.; MacArthur, I.; Bhagat, T.D.; Choudhary, G.S.; Aluri, S.; Chen, J.; et al. Stem cell mutations can be detected in myeloma patients years before onset of secondary leukemias. Blood Adv. 2019, 3, 3962–3967. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.L.; Yoder, S.J.; Balasis, M.E.; Mesa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017, 18, 112–121. [Google Scholar] [CrossRef]
- Malcovati, L.; Germing, U.; Kuendgen, A.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Giagounidis, A.; Hildebrandt, B.; Bernasconi, P.; Knipp, S.; et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J. Clin. Oncol. 2007, 25, 3503–3510. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Nazha, A.; Narkhede, M.; Radivoyevitch, T.; Seastone, D.J.; Patel, B.J.; Gerds, A.T.; Mukherjee, S.; Kalaycio, M.; Advani, A.; Przychodzen, B.; et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016, 30, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Tsai, C.H.; Lin, C.C.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Tseng, M.H.; Peng, Y.L.; Liu, M.C.; Liu, C.W.; et al. Incorporation of mutations in five genes in the revised International Prognostic Scoring System can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Saeed, L.; Mudireddy, M.; Idossa, D.; Finke, C.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Targeted next-generation sequencing in myelodysplastic syndromes and prognostic interaction between mutations and IPSS-R. Am. J. Hematol. 2017, 92, 1311–1317. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Nicolosi, M.; Szuber, N.; Patnaik, M.M.; Pardanani, A.; Hanson, C.A.; Ketterling, R.P.; et al. Mutations and prognosis in myelodysplastic syndromes: Karyotype-adjusted analysis of targeted sequencing in 300 consecutive cases and development of a genetic risk model. Am. J. Hematol. 2018, 93, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Luo, Y.; Zhu, S.; Wang, L.; Ma, L.; Zhang, H.; Shen, C.; Yang, W.; Ren, Y.; Zhou, X.; et al. Mutation status and burden can improve prognostic prediction of patients with lower-risk myelodysplastic syndromes. Cancer Sci. 2020, 111, 580–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-mutant myelodysplastic syndrome as a distinct disease subtype—A Proposal of the International Working Group for the Prognosis of Myelodysplastic Syndromes (IWG-PM). Blood 2020. [Google Scholar] [CrossRef]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [Green Version]
- Mund, C.; Brueckner, B.; Lyko, F. Reactivation of epigenetically silenced genes by DNA methyltransferase inhibitors: Basic concepts and clinical applications. Epigenetics 2006, 1, 8–14. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Yi, L.; Sun, Y.; Levine, A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget 2014, 5, 8924–8936. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.; Samper, E.; Fraga, M.F.; González De Buitrago, G.; Esteller, M.; Serrano, M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene 2004, 23, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.K.; Zhao, Y.S.; Xu, F.; Guo, J.; Zhang, Z.; He, Q.; Wu, D.; Wu, L.Y.; Su, J.Y.; Song, L.X.; et al. TP53 mutations predict decitabine-induced complete responses in patients with myelodysplastic syndromes. Br. J. Haematol. 2017, 176, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Falconi, G.; Fabiani, E.; Piciocchi, A.; Criscuolo, M.; Fianchi, L.; Lindfors Rossi, E.L.; Finelli, C.; Cerqui, E.; Ottone, T.; Molteni, A.; et al. Somatic mutations as markers of outcome after azacitidine and allogeneic stem cell transplantation in higher-risk myelodysplastic syndromes. Leukemia 2019, 33, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Bally, C.; Adès, L.; Renneville, A.; Sebert, M.; Eclache, V.; Preudhomme, C.; Mozziconacci, M.J.; de The, H.; Lehmann-Che, J.; Fenaux, P. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk. Res. 2014, 38, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Patel, K.; Bueso-Ramos, C.; Zhang, J.; Gumbs, C.; Jabbour, E.; Kadia, T.; Andreff, M.; Konopleva, M.; Di Nardo, C.; et al. Clinical implications of TP53 mutations in myelodysplastic syndromes treated with hypomethylating agents. Oncotarget 2016, 7, 14172–14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Hilsenbeck, S.; Gazitt, Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent g1 or g2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 2003, 101, 4078–4087. [Google Scholar] [CrossRef]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Huang, L.; Liu, Z.; Jiang, H.; Li, L.; Fu, R. Decitabine shows synergistic effects with arsenic trioxide against myelodysplastic syndrome cells via endoplasmic reticulum stress-related apoptosis. J. Investig. Med. 2019, 67, 1067–1075. [Google Scholar] [CrossRef]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Koreth, J.; Pidala, J.; Perez, W.S.; Deeg, H.J.; Garcia-Manero, G.; Malcovati, L.; Cazzola, M.; Park, S.; Itzykson, R.; Ades, L.; et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: An international collaborative decision analysis. J. Clin. Oncol. 2013, 31, 2662–2670. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Gallì, A.; Bacigalupo, A.; Zibellini, S.; Bernardi, M.; Rizzo, E.; Allione, B.; Van Lint, M.T.; Pioltelli, P.; Marenco, P.; et al. Clinical effects of driver somatic mutations on the outcomes of patients with myelodysplastic syndromes treated with allogeneic hematopoietic stem-cell transplantation. J. Clin. Oncol. 2016, 34, 3627–3637. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Jung, S.H.; Hur, E.H.; Choi, E.J.; Lee, K.H.; Yim, S.H.; Kim, H.J.; Kwon, Y.R.; Jeon, Y.W.; Lee, S.H.; et al. TP53 mutation in allogeneic hematopoietic cell transplantation for de novo myelodysplastic syndrome. Leuk. Res. 2018, 74, 97–104. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and Azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, S.E.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. P53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andreń, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies—A clinical synopsis. Cell Death Dis. 2020, 11, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; Deangelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’alessandro, A.; Culp-Hill, R.; D’almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Burke, L.P.; Kukoly, C.A. Statins induce lethal effects in acute myeloblastic lymphoma cells within 72 hours. Leuk. Lymphoma 2008, 49, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/ refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for patients with relapse after allogeneic transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef]
- Sallman, D.A.; Amy, M.; Komrokji, R.S.; McGraw, K.; Geyer, S.M.; Eksioglu, E.; Al Ali, N.; Lancet, J.E.; Wei, S.; Padron, E.; et al. Immune Checkpoint Profiling of TP53 Mutant and Wild-Type Myeloid Malignancies: TP53 Mutations Direct Immune Tolerance Via an Immunosuppressive Phenotype. Blood 2017, 130, 423. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumbo, C.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. Int. J. Mol. Sci. 2020, 21, 3432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103432
Cumbo C, Tota G, Anelli L, Zagaria A, Specchia G, Albano F. TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. International Journal of Molecular Sciences. 2020; 21(10):3432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103432
Chicago/Turabian StyleCumbo, Cosimo, Giuseppina Tota, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, and Francesco Albano. 2020. "TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings" International Journal of Molecular Sciences 21, no. 10: 3432. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103432